
Abstract
The enantiomer of natural “d-cellulose” (= “l-cellulose”), which consists of l-glucose, was synthesized from l-glucose via cationic ring-opening polymerization. l-Glucose (1L) was converted to 3-O-benzyl-2,6-di-O-pivaloyl-β-l-glucose 1,2,4-orthopivalate (6L) by five reaction steps. l-Glucose and its derivatives showed almost the same reactivity as d-glucose and its derivatives during the synthesis of compound 6L. Cationic ring-opening polymerization of compound 6L under atmospheric pressure proceeded smoothly to give 3-O-benzyl-2,6-di-O-pivaloyl-β-l-glucopyranan (7L) with a degree of polymerization (DPn) of 32.8 (Mw/Mn = 2.19). Removal of the benzyl and pivaloyl groups of compound 7L and subsequent acetylation gave acetylated β-l-glucopyranan. 1H and 13C NMR spectra of the acetylated β-l-glucopyranan had the same profiles as those of commercial cellulose triacetate prepared from natural cellulose, while its specific rotation was opposite, indicating the successful synthesis of l-cellulose. The synthesized l-cellulose had a cellulose II crystal structure. This is the first reported synthesis of l-cellulose, an l-polysaccharide that consists of an l-monosaccharide.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In nature, d-sugars are generally found in natural products because of the low abundance of l-sugars. Cellulose, which is the main constituent of the plant cell wall and the most abundant polymer in nature, is a linear homopolymer consisting of d-anhydroglucose units (hereafter “d-cellulose”). Much attention has been paid to high-value added d-cellulose utilization although d-cellulose is widely used in our daily life. One important property of d-cellulose for such utilization is chirality. Indeed, d-cellulose derivatives for chiral separation (Francotte 1994, Okamoto and Yashima 1998, Yamamoto and Okamoto 2004, D’Orazio et al. 2018, Chankvetadze 2020), chiral nematic d-cellulose liquid crystalline (Nishio et al. 2016), and d-cellulose-based organocatalysts for asymmetric synthesis (Yasukawa et al. 2015; Ranaivoarimanana et al. 2019) have been proposed and put to partial practical use. However, the effect of the chirality of d-cellulose on the applications was not well understood.
l-Glucose, an enantiomer of d-glucose, can be obtained by chemical synthesis (Sowa 1969; Weymouth-Wilson et al. 2009; Martínez et al. 2014; Xia et al. 2014). Therefore, there is theoretically an enantiomer of d-cellulose, that is, a homopolymer consisting of l-glucose (hereafter “l-cellulose”) (Fig. 1).

Chemical structure of d-Cellulose and l-Cellulose
To the best of our knowledge, l-cellulose is not found in nature, and has not been synthesized, although it is essential for basic research of the chirality of d-cellulose and its derivatives. There are currently two synthetic methods for d-cellulose, that is, enzymatic polymerization (Kobayashi et al. 1991; Egusa et al. 2007) and cationic ring-opening polymerization (Nakatsubo et al. 1996; Kamitakahara et al. 1996; Adelwöhrer et al. 2009). However, the former might not be suitable for the synthesis of l-cellulose because of the substrate specificity of the enzyme used for the preparation. Therefore, the latter method was applied to the synthesis of l-cellulose (Scheme 1). This paper describes the first synthesis of l-cellulose using cationic ring-opening polymerization.

Synthetic routes for the preparation of d-cellulose (8D) and l-cellulose (8L) [Reaction conditions; a Acetone/H2SO4. b Benzyl bromide/NaH/DMF. c DoweX 50-W/H2O–EtOH (3:1, v/v). d Pivaloyl chloride/Bu2SnO/pyridine/toluene. e: Benzenesulfonyl chloride/Et3N/CH2Cl2 f BF3 · Et2O/CH2Cl2. g (i) H2/Pd–C/THF-AcOH (1:1, v/v) and (ii) NaOMe/THF–MeOH (10:1, v/v)]
Experimental
Materials
l-Glucose (1L) was purchased from Tokyo Kasei Industry Co. (Tokyo, Japan) and dried in a vacuum desiccator under high vacuum (less than 1 hPa) at 70 °C for 16 h before use. Commercial d-cellulose triacetate (D-CTA) (DPn = 366, Mw/Mn = 1.21) was kindly supplied by Daicel Co. (Osaka, Japan). Powdered molecular sieves 4 Å was activated in a Shibata glass tube oven GTO-2000 (Shibata Scientific Technology Ltd, Tokyo, Japan) under high vacuum (less than 1 hPa) at 300 °C for 3 h and kept at 105 °C in an Masuda drying oven SA46 (Masuda Co., Osaka, Japan) before use. All other chemicals were purchased from Nacalai Tesque (Kyoto, Japan) and FUJIFILM Wako Pure Chemical Co. (Osaka, Japan) and used without further purification. Compound purifications were performed by silica gel chromatography using Wakogel® C-200 (FUJIFILM Wako Pure Chemical Co.).
Measurements
Melting points were measured using a micro melting point apparatus (Yanagimoto Seisakusho, Kyoto, Japan). Specific rotations were recorded on a JASCO P-2200 polarimeter (JASCO, Hachioji, Japan), and were determined as the average values of five measurements. 1H and 13C NMR spectra were recorded on a Varian 500 NMR spectrometer (500 MHz, Agilent Technologies, Santa Clara, CA,USA) using tetramethylsilane (TMS) as an internal reference standard in CDCl3 as the solvent if not otherwise stated. Chemical shifts (δ) and coupling constants (J) are reported in ppm (parts per million) and Hz, respectively. Matrix-assisted laser desorption/ionization time-of-flight mass (MALDI-TOF MS) spectra were recorded on a Bruker MALDI-TOF MS REFLEX III (Bruker, Billerica, MA, USA) in the positive and linear ion modes. A nitrogen laser was used for the ionization of the samples. All spectra were measured with 2,5-dihydroxybenzoic acid as the matrix. Fourier-transform infrared (FT-IR) spectra were recorded in KBr pellets with a Shimadzu IRPrestige-21 spectrophotometer (Shimadzu, Kyoto, Japan). Gel permeation chromatography (GPC) was performed on a Shimadzu LC-10 system (Shimadzu) equipped with a Shimadzu UV–vis detector (SPD-10AVp) and a Shimadzu RI detector (RID-10A) under the following conditions: columns: Shodex K-802, K-802.5, and K-805 (Showa Denko K.K., Tokyo, Japan) in series; eluent: CHCl3; temperature: 40 °C; flow rate: 1.0 mL/min; standards: polystyrene standards (Showa Denko K.K.). X-ray diffractograms were recorded on a Rigaku RINT-Ultima IV (Rigaku, Tokyo, Japan).
1,2;5,6-Di-O-isopropylidene-α-l-glucofuranose (2L)
l-Glucose (1L, 2.00 g, 11.1 mmol) was stirred in dry acetone (100 mL) with anhydrous FeCl3 (600 mg, 3.67 mmol) at 60 °C for 3 h. The reaction mixture was cooled down to room temperature (r.t.), neutralized with a saturated aqueous NaHCO3 solution and filtered and washed with ethyl acetate (EtOAc). The filtrate was concentrated in vacuo to remove acetone, and extracted with CH2Cl2 five times. The extracts were combined, washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The product was recrystallized from n-hexane to give compound 2L (2.44 g, 85% yield) as a colorless crystal.
Compound 2L: melting point: 109–110 °C; \({[\mathrm{\alpha }]}_{D}^{25}\) = + 9.9° (c = 0.1 in CHCl3); 1H NMR (CDCl3): δ 5.95 (d, 1H, J = 3.9, H-1), 4.54 (d, 1H, J = 3.6, H-2), 4.38–4.31 (m, 2H, H-5, H-3), 4.17 (dd, 1H, J = 8.7, 6.0, H-6a), 4.07 (dd, 1H, J = 7.8, 2.7, H-4), 3.99 (dd, 1H, J = 8.7, 5.4, H-6b), 2.62 (d, 1H, J = 3.6, OH-3),1.50, 1.49, 1.37, 1.32 (s, 3H, –C(CH3)2); 13C NMR (CDCl3): δ 111.8, 109.7 (–C(CH3)2), 105.2 (C-1), 85.0 (C-2), 81.0 (C-4), 75.2 (C-5), 73.5 (C-3), 67.6 (C-6), 26.8, 26.7, 26.2, 25.1 (–C(CH3)2); MALDI-TOF/MS: m/z calcd. for C12H20O6Na [M+Na]+ 283.13, found 283.06.
3-O-Benzyl-1,2;5,6-di-O-isopropylidene-α-l-glucofuranose (3L)
Compound 2L (2.00 g, 7.69 mmol) was dissolved in DMF (35 mL). NaH (60% oil suspension, 370 mg, 9.25 mmol) was added to the solution at 0 °C. After stirring at 0 °C for 30 min, the solution of benzyl bromide (1.1 mL, 9.25 mmol) in dry DMF (10 mL) was added dropwise over a period of 30 min. The reaction mixture was stirred at r.t. for 3 h. A small amount of MeOH was carefully added dropwise to the mixture at 0 °C until the foaming stopped, and then more water (105 mL) was added. The reaction mixture was twice extracted with EtOAc/n-hexane (1/4, v/v). The extracts were combined, washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluted with EtOAc/n-hexane (1/5, v/v) (Rf = 0.39) to afford compound 3L (2.71 g, 98% yield) as a colorless oil.
Compound 3L: \({[\mathrm{\alpha }]}_{D}^{25}\) = + 22.5° (c = 0.4 in CHCl3); 1H NMR (CDCl3): δ 7.36–7.26 (m, 5H, -CH2Ph), 5.90 (d, 1H, J = 3.6, H-1), 4.69 (d, 1H, J = 12.0, –CH2Ph), 4.63 (d, 1H, J = 12.0, –CH2Ph), 4.59 (d, 1H, J = 3.6, H-2), 4.37 (dt, 1H, J = 8.7, 6.0, H-5), 4.15 (dd, 1H, J = 7.8, 3.0, H-4), 4.12 (dd, 1H, J = 8.7, 6.0, H-6a), 4.02 (t, 1H, J = 2.7, H-3), 4.01 (dd, 1H, J = 8.7, 5.4, H-6b), 1.49, 1.43, 1.38, 1.31 (s, 3H, –C(CH3)2); 13C NMR (CDCl3): δ 137.7, 128.5, 127.9, 127.7 (–CH2Ph), 111.8, 109.0 (–C(CH3)2), 105.3 (C-1), 82.7 (C-2), 81.7 (C-3), 81.3 (C-5), 72.5 (C-4), 72.4 (–CH2Ph), 67.4 (C-6), 26.9, 26.8, 26.3, 25.5 (–C(CH3)2); MALDI-TOF/MS: m/z calcd. for C19H26O8Na [M+Na]+ 373.17, found 373.04.
3-O-Benzyl-l-glucopyranose (4L)
Compound 3L (3.00 g, 8.57 mmol) was dissolved in EtOH/H2O (1/3, v/v, 12 mL), and Dowex-50WX8 (Sigma-Aldrich, St. Louis, MO, USA) (1.20 g) was added to the solution. The reaction mixture was stirred under reflux for 5 h, neutralized with 28% NaOMe in MeOH, and filtered with EtOH. The filtrate was concentrated in vacuo to remove EtOH, and then lyophilized. The residue was purified by silica gel column chromatography eluted with MeOH/CH2Cl2 (15/85, v/v) (Rf = 0.27), and recrystallized from EtOAc to give compound 4L (1.92 g, 83% yield) as a colorless crystal.
Compound 4L: melting point: 124–126 °C;\({[\mathrm{\alpha }]}_{D}^{25}\) = − 33.5° (c = 0.1 in H2O); 1H NMR (D2O): δ 7.49–7.36 (m, 5H, –CH2Ph), 5.20 (d, 1H, J = 3.6, H-1α), 4.82 (d, 2H, J = 18.0, –CH2Ph), 4.63 (d, 1H, J = 7.8, H-1β), 3.84 (dd, 1H, J = 12.0, 2.1, H-6a), 3.74–3.68 (m, 1H, H-4), 3.64 (dd, 1H, J = 12.0, 5.4, H-6b), 3.53–3.40 (m, 2H, H-3, H-5), 3.31 (m, 1H, H-2); 13C NMR (D2O): δ 140.1 × 2, 131.5, 131.3, 131.0 (–CH2Ph), 98.6 (C-1β), 94.9 (C-1α), 86.6, 84.0 (C-3), 78.6, 77.8 (C-5), 77.6 (–CH2Ph), 76.6, 74.2 (C-2), 74.0, 72.0 (C-4), 63.3, 63.1 (C-6); MALDI-TOF/MS: m/z calcd. for C13H18O6Na [M+Na]+ 293.11, found 293.02.
3-O-Benzyl-2,6-di-O-pivaloyl-l-glucopyranose (5L)
Compound 4L (2.03 g, 7.51 mmol), Bu2SnO (4.06 g, 16.3 mmol) and powdered molecular sieves 4 Å (3.00 g) were combined and dried in vacuo overnight. After the addition of anhydrous toluene (60 mL), the reaction mixture was stirred at 105 °C for 20 min and cooled down to r.t. Anhydrous pyridine (1.6 mL, 19.8 mmol) was added to the mixture at r.t. The reaction mixture was further cooled to − 15 °C. Pivaloyl chloride (2.02 mL, 16.3 mmol) in anhydrous toluene (20 mL) was added dropwise to the mixture over 30 min. The reaction mixture was kept at − 15 °C for 2.5 h. MeOH (5 mL) was added at this temperature. The reaction mixture was filtered and washed with EtOAc. The filtrate was concentrated azeotropically with EtOH to give a yellow oil. The oil was purified by column chromatography using 10% K2CO3/silica gel (w/w) (Harrowven et al 2010) eluted with EtOAc/n-hexane (1/2, v/v) (Rf = 0.50 on silica gel) to afford compound 5L (2.11 g, 65% yield) as a colorless oil.
Compound 5L: \({[\mathrm{\alpha }]}_{D}^{25}\) = − 27.8° (c = 0.4 in CHCl3); 1H NMR (CDCl3): δ 7.37–7.25 (m, 5H, –CH2Ph), 5.38 (t, 1H, J = 3.3, H-1), 4.84 (d, 1H, J = 11.4, –CH2Ph), 4.80–4.67 (m, 1H, H-2), 4.77 (d, 1H, J = 11.4, –CH2Ph), 4.38–4.28 (m, 2H, H-6a and H-6b), 4.03–3.90 (m, 1H, H-5), 3.98–3.87 (m, 1H, H-3), 3.58–3.40 (m, 1H, H-4), 3.14 (d, 1H, J = 3.3, OH-1), 2.95 (d, 1H, J = 3.3, OH-4), 1.25–1.19 (m, 18H, –C(CH3)3); 13C NMR (CDCl3): δ 179.4, 179.3, 178.8, 178.0 (C=O), 138.1, 137.8, 128.5, 128.4, 128.2, 127.9, 127.7, 127.6 (–CH2Ph), 95.9, 90.1 (C-1), 81.7, 78.9 (C-3), 77.4, 77.0 (–CH2Ph), 76.6, 75.2 (C-2), 74.9, 74.2 (C-5), 70.0, 69.5 (C-4), 62.9, 62.6 (C-6), 38.9, 38.8, 38.7 (× 2) (–C(CH3)3), 27.1, 27.0, 26.9 (-C(CH3)3); MALDI-TOF/MS: m/z calcd. for C23H34O6Na [M+Na]+ 461.23, found 461.10.
3-O-Benzyl-2,6-di-O-pivaloyl-α-l-glucopyranose-1,2,4-orthopivalate (6L)
Compound 5L (1.48 g, 3.37 mmol) dried in a vacuum desiccator under high vacuum (less than 1 hPa) at r.t. for 16 h was dissolved in anhydrous CH2Cl2 (30 mL), and Et3N (0.94 mL, 6.74 mmol) and benzenesulfonyl chloride (0.46 mL, 3.59 mmol) were added. The reaction mixture was stirred at r.t. overnight, and concentrated to give a yellow oil. The oil was purified by silica gel column chromatography with CH2Cl2/n-hexane (1/2, v/v) (Rf = 0.22) and then CH2Cl2 to give white solid, which was recrystallized from n-hexane to afford compound 6L (1.15 g, 81% yield) as a colorless crystal.
Compound 6L: melting point: 73–74 °C; \({[\mathrm{\alpha }]}_{D}^{25}\) = − 30.6° (c = 0.1 in CHCl3); 1H NMR (CDCl3): δ 7.40–7.30 (m, 5H, –CH2Ph), 5.77 (d, 1H, J = 5.1, H-1), 4.63 (s, 2H, –CH2Ph), 4.50 (broad t, 1H, J = 6.6, H-5), 4.42 (dd, 1H, J = 11.1, 6.0, H-6a), 4.41 (dd, 1H, J = 5.1, 2.1, H-2), 4.34 (dd, 1H, J = 11.1, 6.0, H-6b), 4.16 (dd, 1H, J = 4.8, 2.1, H-3), 3.95 (broad dd, 1H, J = 4.8, 1.2, H-4),1.23 (s, 9H, –C(CH3)3), 1.03 (s, 9H, –C(CH3)3); 13C NMR (CDCl3): δ 178.2 (C=O), 137.4, 128.6, 128.1, 127.6, (–CH2Ph), 123.0 ((–O3)C(CH3)3), 97.5 (C-1), 75.3 (–CH2Ph), 72.2 (C-3), 72.0 (C-5), 71.4 (C-4), 71.2 (C-2), 64.4 (C-6), 38.8 (pivaloyl–C(CH3)3), 35.7 (orthopivalate–C(CH3)3), 27.2 (pivaloyl–C(CH3)3), 24.9 (orthopivalate–C(CH3)3); MALDI-TOF/MS: m/z calcd. for C23H33O7 [M+H]+ 421.21 and C23H32O7Na [M+Na]+ 443.21, found 421.08 and 443.11.
3-O-Benzyl-2,6-di-O-pivaloyl-(1→4)-β-l-glucopyranan (7L)
(Typical method 1) Compound 6L (50 mg, 0.12 mmol) was placed in a glass ampoule under high vacuum overnight on a vacuum line. CH2Cl2 (100 μL) was distilled from CaH2, degassed by three freeze/thaw cycles and transferred to a polymerization ampule on the vacuum line. BF3–Et2O (0.73 μL, 5 mol%) was added via a syringe through the sealed cap of the glass ampule. The reaction mixture was stirred under high vacuum at r.t. for 24 h, after which time it was partly solidified. After the reaction, the mixture was dissolved in CHCl3 (10 mL). The organic layer was washed with water, a saturated aqueous NaHCO3 solution, and brine, dried over anhydrous Na2SO4, and concentrated to afford compound 7L as a colorless solid in quantitative yield.
(Typical method 2) Compound 6L (200 mg, 0.48 mmol) was placed in a glass ampoule in a vacuum desiccator under high vacuum (less than 1 hPa) at r.t. for 16 h. Anhydrous CH2Cl2 (200 μL), which was distilled from CaH2, and BF3–Et2O (2.90 μL, 5 mol%) was added via a syringe through the sealed cap of a glass ampoule. The reaction mixture was stirred under atmospheric pressure at 40 °C for 24 h, after which time it was partly solidified. The work-up method was carried out by the same method described in Typical method 1 to give compound 7L as a colorless solid in quantitative yield.
Compound 7L: \({[\mathrm{\alpha }]}_{D}^{26}\) = + 7.3° (c = 0.5 in CHCl3); DPn = 32.8 (Mw/ Mn = 1.97); 1H NMR (CDCl3): δ 7.34–7.10 (m, 5H, –CH2Ph), 4.95 (broad d, 1H, J = 10.8, –CH2Ph), 4.83 (broad s, 1H, H-2), 4.41 (broad d, 1H, J = 10.8, –CH2Ph), 4.28 (broad s, 1H, H-1), 4.09 (broad s, 1H, H-6a), 3.83 (broad s, 1H, H-6b), 3.64 (broad s, 1H, H-5), 3.46 (broad s, 1H, H-4), 3.33 (broad s, 1H, H-3), 1.05–0.85 (m, 18H, –C(CH3)3); 13C NMR (CDCl3): δ 177.5, 176.3 (C=O), 138.6, 128.4, 128.0, 127.1, 126.7 (–CH2Ph), 100.1 (C-1), 80.7 (C-3), 74.7 (C-5), 73.2 (–CH2Ph), 72.2 (C-2), 62.3 (C-6), 35.8 (× 2) (–C(CH3)3), 27.1, 27.0, 26.9 ((–C(CH3)3).
(1→4)-β-l-Glucopyranan (l-Cellulose, 8L),
Compound 7L (200 mg, 0.48 mmol) was dissolved in THF/AcOH (1/1, v/v) (5 mL). Pd(OH)2 on charcoal (200 mg) was added. The reaction mixture was hydrogenated with vigorous stirring under H2 at atmospheric pressure and r.t. for 24 h, filtered through Celite® 535RVS (Nacalai Tesque) with THF/AcOH (1/1, v/v), washed with THF/AcOH (1/1, v/v) and concentrated azeotropically with EtOH to give a partially debenzylated product as a colorless solid. The product was subjected to debenzylation under the same conditions again to give a fully debenzylated product as a colorless solid. The product was dissolved in THF/MeOH (10/1, v/v) (60 mL) and 28% MeONa in MeOH solution (0.8 mL) was added. The reaction mixture was stirred at 50 °C overnight, and neutralized with 1 M HCl aqueous solution. The resulting precipitation was collected by centrifugation (5538×g), washed several times with water and MeOH, and dried in vacuo to afford compound 8L (49 mg, 64% yield) as a brownish solid.
Compound 8L: FT-IR (KBr): ν 3402, 2891, 1602, 1375, 1159, 1065, 1026, 895 cm−1.
Compound 8L was acetylated with Ac2O/pyridine at 110 °C for 24 h, and subjected to 1H and 13C NMR measurements.
Acetylated compound 8L:\({[\mathrm{\alpha }]}_{D}^{25}\) = + 8.3° (c = 0.02 in CHCl3); 1H NMR (CDCl3): δ 5.07 (H-3), 4.79 (H-2), 4.41(H-1), 4.38 (H-6a), 4.06 (H-6b), 3.71 (H-4), 3.54 (H-5), 2.12, 2.00, 1.94 (–COCH3); 13C NMR (CDCl3): δ 170.2, 169.7, 169.3 (C=O), 100.5 (C-1), 76.0 (C-4), 72.8, 72.5, 71.8 (C-2, C-3, C-5), 62.0 (C-6).
(1→4)-β-d-Glucopyranan (d-Cellulose, 8D)
d-Cellulose (8D) was also prepared from d-glucose (1D) according to previously reported methods (Nakatsubo et al. 1996; Kamitakahara et al. 1996; Adelwöhrer et al. 2009).
Compound 2D: melting point: 110–112 °C; \({[\mathrm{\alpha }]}_{D}^{25}\) = − 9.6° (c = 0.1 in CHCl3); MALDI-TOF/MS: m/z calcd. for C12H20O6Na [M+Na]+ 283.13, found 283.07.
Compound 3D: \({[\mathrm{\alpha }]}_{D}^{25}\) = − 23.7° (c = 0.4 in CHCl3); MALDI-TOF/MS: m/z calcd. for C19H26O8Na [M+Na]+ 373.17, found 372.93.
Compound 4D: melting point: 124–125 °C;\({[\mathrm{\alpha }]}_{D}^{25}\) = + 33.8° (c = 0.1 in H2O); MALDI-TOF/MS: m/z calcd. for C13H18O6Na [M+Na]+ 293.11, found 293.04.
Compound 5D: \({[\mathrm{\alpha }]}_{D}^{25}\) = + 26.8° (c = 0.3 in CHCl3); MALDI-TOF/MS: m/z calcd. for C23H34O6Na [M+Na]+ 461.23, found 461.25.
Compound 6D: melting point: 73–74 °C; \({[\mathrm{\alpha }]}_{D}^{25}\) = + 30.6° (c = 0.1 in CHCl3); MALDI-TOF/MS: m/z calcd. for C23H33O7 [M + H]+ 421.21 and C23H32O7Na [M+Na]+ 443.21, found 421.12 and 443.11.
Compound 7D: DPn of 11.9 (Mw/Mn = 1.86); \({[\mathrm{\alpha }]}_{D}^{26}\) = − 3.7° (c = 0.1 in CHCl3).
Acetylated compound 8D:\({[\mathrm{\alpha }]}_{D}^{25}\) = − 4.8° (c = 0.07 in CHCl3).
Results and discussion
Synthesis of glucose orthoester derivative 6L
Compound 6L was synthesized from l-glucose (1L) by five reaction steps using a modified method for compound 6D as previously reported (Scheme 1) (Nakatsubo et al. 1996; Kamitakahara et al. 1996; Karakawa and Nakatsubo 2002; Adelwöhrer et al. 2009). All the reaction steps proceeded smoothly to afford the final compound 6L. Indeed, the reactivity of l-glucose and its derivatives (compounds 1L to 5L) was found to be almost the same as those of the corresponding d-glucose and its derivatives (compounds 1D to 5D). Similar reactivities among l-glucose and d-glucose derivatives have been reported in the syntheses of digitoxigenin glucoside (Rathore et al. 1985) and coniferin (coniferyl alcohol glucoside) (Maeda et al 2019).
The main points of our modification are as follows. (1) Compound 2D has been reported to be prepared by the reactions of d-glucose (1D) with a dry acetone/H2SO4 system and with a dry acetone/CuSO4/H2SO4 system. Compound 2L has also been prepared from the reaction of 1-deoxy-1-nitroglucitol with a dry acetone/H2SO4 system (Hoeltgebaum Thiesen et al. 2017) and from 1,2-O-isopropylidene-α-l-glucose with a dry acetone/H2SO4 system (Weymouth-Wilson et al. 2009). Therefore, the dry acetone/H2SO4 system was applied to the synthesis of compound 2L. However, the reaction time was long, and the yield of compound 2L was not very high. After investigation of other acid catalysts, compound 2L was obtained in 85% yield from the reaction of compound 1L with dry acetone/anhydrous FeCl3 at 60 °C for 3 h. (2) Compound 4D was prepared in high yield by benzylation and the subsequent removal of isopropylidene groups (Adelwöhrer et al. 2009). In the latter reaction, the purification of compound 4D by five or six rounds of recrystallization was reported to be required to ensure the high yield of compound 5D in the next reaction step. This was laborious work and sometimes caused the yield of compound 4D to decrease. The reaction product resulting from the removal of the isopropylidene groups was purified by silica gel column chromatography eluted with 15% MeOH/CH2Cl2 followed by one recrystallization from EtOAc to afford compound 4L in 83% yield. (3) In the selective pivaloylation, compound 5D was reported to be purified by two different rounds of silica gel column chromatography because the organotin impurities negatively influenced the yield of compound 6D in the next reaction step (Karakawa and Nakatsubo 2002; Adelwöhrer et al. 2009). This was also laborious work. Harrowven et al. reported that a stationary phase composed of 10% powdered anhydrous K2CO3 and silica gel was effective for the removal of organotin impurities from reaction products (Harrowven et al 2010). Selective pivaloylation of compound 4L using Bn2SnO was conducted in anhydrous conditions according to the synthesis of compound 5D, and the product was purified using 10% anhydrous K2CO3/silica gel column chromatography to afford compound 5L in 65% yield. (4) In the synthesis of compound 6D, the reaction mixture had to be quickly transferred to a silica gel column after the reaction because compound 6D was highly sensitive to traces of acid. However, the volume of the reaction mixture applied to the column was sometimes too large to efficiently separate compound 6D. For the synthesis of compound 6L, the reaction mixture was concentrated before applying it to the silica gel column. Compound 6L was stable during the concentration process. In this way, compound 6L was obtained in 81% yield.
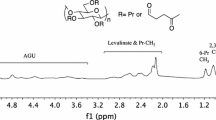
All products were characterized by their 1H and 13C NMR and MALDI-TOF MS spectra, and their specific rotation. The 1H and 13C NMR spectra of all the l-glucose derivatives had the same peak pattern as those of the corresponding d-glucose derivatives (Figures S1 and S2), but the sign of the specific rotation of the l-glucose derivatives was opposite to that of the specific rotation of the corresponding d-glucose derivatives. For example, the 1H and 13C NMR spectra of compounds 6L and 6D are shown in Fig. 2. The spectra of compound 6L were the same as those of compound 6D. The measurement m/z values of [M+H]+ and [M+Na]+ for compound 6L from the MALDI-TOF–MS spectrum also corresponded to its calculated values. The specific rotations of compounds 6L and 6D were − 30.6° and + 30.6°, respectively. These results clearly indicate that compound 6L was an enantiomer of compound 6D.

1H and 13C NMR spectra of compounds 6L and 6D
Cationic ring-opening polymerization of glucose orthoester derivative 6L
First, cationic ring-opening polymerization under vacuum of compound 6L was performed according to the polymerization method of compound 6D to afford compound 7L in quantitative yield. However, the polymerization under vacuum required a special reaction apparatus (vacuum line) and complicated operations. By comparison, the ring-opening polymerization of compound 6L under atmospheric pressure was carried out to give compound 7L in quantitative yield. Compound 7D was also prepared by the ring-opening polymerization of compound 6D as a control. Figure 3 shows the 1H and 13C NMR spectra of compounds 7L and 7D. Both spectra of compound 7L had the same profile as those of compound 7D. In particular, the signal derived from C-1 at 100.1 ppm was only found in the 13C NMR spectrum of compound 7L, suggesting that only β-bonds were formed in the polymerization of compound 6L. The DPn of compound 7L was 32.8 (Mw/Mn = 1.97). It has been reported that the positive sign of the specific rotation of compound 6D changed to a negative sign for compound 7D after the ring-opening polymerization of compound 6D in two previous papers (Kamitakahara et al.1996; Adelwöhrer et al. 2009). During the ring-opening polymerization of compound 6L, the negative specific rotation sign of compound 6L (− 30.6°) changed to a positive sign for compound 7L (+ 7.3°), and that of compound 7D with a DPn of 11.9 (Mw/Mn = 1.86), which was synthesized in this study, was negative (− 3.7°). Please note that the specific rotation seemed to be influenced by the DPn. These results indicated that compound 7L was the expected β-1,4-l-glucopyranan derivative.

1H and 13C NMR spectra of compounds 7L and 7D
Conversion of polymer 7L into (1→4)-β-l-glucopyranan (8L)
Debenzylation of compound 7D has been reportedly performed under high pressure with H2 for 24 h (Adelwöhrer et al. 2009). In an effort to simplify the operation, debenzylation of compound 7L was done under atmospheric pressure with H2 for 24 h. However, the debenzylation did not proceed completely. Then, the partially debenzylated product was further debenzylated under atmospheric pressure with H2 for 24 h to give a fully debenzylated product. By comparison, the debenzylation of compound 7L under atmospheric pressure with H2 for 48 h did not proceed completely. The repetition of the benzylation was effective for the full removal of the benzyl groups of compound 7L. The fully debenzylated product was treated with 28% NaOMe–MeOH at 50 °C overnight to give compound 8L in 64% yield (based on compound 7L).
Figure 4 shows the FT-IR spectra of compound 7L, the debenzylated product and compound 8L. The bands from the benzyl groups around 698 cm−1 and the pivaloyl groups around 1736 cm−1 disappeared in the FT-IR spectrum of compound 8L, suggesting that the protective groups were completely removed from compound 7L. Furthermore, compound 8L was treated with Ac2O/pyridine at 110 °C for 24 h to fully afford acetylated compound 8L for characterization.

FT-IR spectra of compound 7L (a); debenzylated 7L (b); compound 8L (c)
Figure 5 shows the 1H and 13C NMR spectra of acetylated compound 8L and commercial cellulose triacetate (D-CTA from natural cellulose). The 1H and 13C NMR spectra of acetylated compound 8L were completely identical with those of authentic D-CTA. The specific rotation of acetylated compound 8L was positive (+ 8.3°), whereas that of authentic D-CTA was negative ( − 23.4°). These results suggested that acetylated compound 8L was an enantiomer of D-CTA (L-CTA), and thus compound 8L was the expected l-cellulose.

1H and 13C NMR spectra of acetylated compound 8L and commercial CTA (D-CTA)
X-ray diffractograms of Whatman cellulose CF-11, mercerized Whatman cellulose CF-11, and compound 8L are shown in Fig. 6. The representative diffractions at 12°, 20°, and 22° derived from cellulose II were observed for compound 8L as well as for mercerized Whatman cellulose. These results indicated that compound 8L had a cellulose II crystal structure similar to that previously reported for compound 8D (Nakatsubo et al.1996).

X-ray diffractograms of Whatman cellulose CF11, mercerized Whatman cellulose CF11 and compound 8L
Conclusion
l-Cellulose (8L) was synthesized from l-glucose (1L) by a modified synthetic method for d-cellulose (8D) using cationic ring-opening polymerization (Scheme 1). l-Sugars showed almost the same reactivities as d-sugars during the synthesis. The availability of l-cellulose (8L) is largely expected to pave the way for new research concerning the chirality of cellulose in the future. This is also the first synthesis of a polysaccharide that consists of an l-monosaccharide (l-polysaccharide).
References
Adelwöhrer C, Takano T, Nakatsubo F, Rosenau T (2009) Synthesis of 13C-perlabeled cellulose with more than 99% isotopic enrichment by a cationic ring-opening polymerization approach. Biomacromol 10:2817–2822. https://doi.org/10.1021/bm9006612
Chankvetadze B (2020) Recent trends in preparation, investigation and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. TrAC Trends Anal Chem 122:115709. https://doi.org/10.1016/j.trac.2019.115709
D’Orazio G, Asensio-Ramos M, Fanali C (2018) Enantiomers separation by capillary electrochromatography using polysaccharide-based stationary phases. J Sep Sci 42:360–384. https://doi.org/10.1002/jssc.201800798
Egusa S, Kitaoka T, Goto M, Wariishi H (2007) Synthesis of cellulose in vitro by using a cellulase/surfactant complex in a nonaqueous medium. Angew Chem Int Ed 46:2063–2065. https://doi.org/10.1002/anie.200603981
Francotte E (1994) Contribution of preparative chromatographic resolution to the investigation of chiral phenomena. J Chromatogr A 666:565–601. https://doi.org/10.1016/0021-9673(94)80419-2
Harrowven DC, Curran DP, Kostiuk SL, Wallis-Guy IL, Whiting S, Stenning KJ, Tang B, Packard E, Nanson L (2010) Potassium carbonate-silica: a highly effective stationary phase for the chromatographic removal of organotin impurities. Chem Commun 46:6335–6337. https://doi.org/10.1039/c0cc01328e
Hoeltgebum Thiesen LJ, Cabral N, Joselice Silva M, Bezerra G, Doboszewski B (2017) Larger laboratory scale synthesis of 5-methyluridine and formal synthesis of its l-enantiomer. Arch Org Chem 4:249–264. https://doi.org/10.24820/ark.5550190.p010.101
Kamitakahara H, Hori M, Nakatsubo F (1996) Substituent effect on ring-opening polymerization of regioselectively acylated α-d-glucopyranose 1,2,4-orthopivalate derivatives. Macromolecules 29:6126–6131. https://doi.org/10.1021/ma960488h
Karakawa M, Nakatsubo F (2002) An improved method for the preparation of 3-O-benzyl-6-O-pivaloyl-α-d-glucopyranose 1,2,4-orthopivalate. Carbohydr Res 337:951–954. https://doi.org/10.1016/S0008-6215(02)00085-X
Kobayashi S, Kashiwa K, Kawasaki T, Shoda S (1991) Novel method for polysaccharide synthesis using an enzyme: the first in vitro synthesis of cellulose via a nonbiosynthetic path utilizing cellulase as catalyst. J Am Chem Soc 113:3079–3084. https://doi.org/10.1021/ja00008a042
Maeda H, Tsuyama T, Takabe K, Kamitakahara H, Takano T (2019) Preparation and properties of a coniferin enantiomer. J Wood Sci. https://doi.org/10.1186/s10086-019-1813-5
Martínez RF, Liu Z, Glawar AFG, Yoshihara A, Izumori K, Fleet GWJ, Jenkinson SF (2014) Short and sweet: d-glucose to l-glucose and l-glucuronic acid. Angew Chem Int Ed 53:1160–1162. https://doi.org/10.1002/anie.201309073
Nakatsubo F, Kamitakahara H, Hori M (1996) Cationic ring-opening polymerization of 3,6-Di-O-benzyl-α-d-glucose 1,2,4-orthopivalate and the first chemical synthesis of cellulose. J Am Chem Soc 118:1677–1681. https://doi.org/10.1021/ja953286u
Nishio Y, Sato J, Sugimura K (2016) Liquid crystals of cellulosics: fascinating ordered structures for the design of functional material systems. Adv Polym Sci 271:241–286. https://doi.org/10.1007/12_2015_308
Okamoto Y, Yashima E (1998) Polysaccharide derivatives for chromatographic separation of enantiomers. Angew Chem Int Ed 37:1020–1043. https://doi.org/10.1002/(SICI)1521-3773(19980504)37:8%3c3C1020::AID-ANIE1020%3e3E3.0.CO;2-5
Ranaivoarimanana NJ, Kanomata K, Kitaoka T (2019) Concreted catalysis by nanocellulose and proline in oragnocatalytic michael addittions. Molecules 24:1231. https://doi.org/10.3390/molecules24071231
Rathore H, Hashimoto T, Igarashi K, Nukaya H, Fullerton DS (1985) Cardiac glycosides: 5. stereoselective syntheses of digitoxigenin α-D, β-D. α-L, β-l-glucosides. Tetrahedron 41:5427–5438. https://doi.org/10.1016/S0040-4020(01)91342-0
Sowa W (1969) Synthesis of l-glucurone. conversion of d-glucose into l-glucose. Can J Chem 47:3931–3934. https://doi.org/10.1139/v69-655
Weymouth-Wilson AC, Clarkson RA, Jones NA, Best D, Wilosn FX, Pino-González M-S, Fleet GWJ (2009) Large scale synthesis of the acetonides of l-glucuronolactone and of l-glucose: easy access to l-sugar chirons. Tetrahedron Lett 50:6307–6310. https://doi.org/10.1016/j.tetlet.2009.08.124
Xia T-Y, Li Y-B, Yin Z-J, Meng X-B, Li S-C, Li Z-J (2014) Synthesis of l-glucose and l-galactose derivatives from d-sugars. Chinese Chem Lett 25:1220–1224. https://doi.org/10.1016/j.cclet.2014.06.007
Yamamoto C, Okamoto Y (2004) Optically active polymers for chiral separation. Bull Chem Soc Jpn 77:227–257. https://doi.org/10.1246/bcsj.77.227
Yasukawa T, Miyamura H, Kobayashi S (2015) Cellulose-supported chiral rhodium nanoparticles as sustainable heterogeneous catalysts for asymmetric carbon-carbon bond-forming reactions. Chem Sci 6:6224–6229. https://doi.org/10.1039/c5sc02510a
Acknowledgments
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Challenging Research 16K14957 and 18K19230.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yagura, T., Ikegami, W., Kamitakahara, H. et al. Synthesis of an enantiomer of cellulose via cationic ring-opening polymerization. Cellulose 27, 9755–9766 (2020). https://doi.org/10.1007/s10570-020-03512-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-020-03512-z