
Abstract
Cellulose hydrogels usually have poor mechanical properties which seriously limit their applications in biomedical or industrial fields. Herein, an interpenetrating polymer network strategy was proposed to construct double network hydrogels composed of cellulose and polyacrylamide with high mechanical strength and pH responsive properties. The synergistic interactions and strong hydrogen bonds between two networks contributed to the dissipation of mechanical energy, as evidenced by the compressive stress–strain and rheology analyses. The compressive strength and compressive modulus for cellulose–polyacrylamide interpenetrating network (C–PAM IPN) hydrogels could reach up to 5.62 and 22.47 MPa, which is nearly 18 and 23 times higher than that of cellulose hydrogels. Moreover, these tough C–PAM IPN hydrogels exhibit pH sensitive properties in various pH solutions. Therefore, this paper provides a general strategy to enhance the mechanical and physical properties of cellulose-based hydrogels, which are potentially useful in developing novel cellulose-based hydrogels and expanding their applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogels, which are chemically and/or physically cross-linked three-dimensional hydrophilic polymeric networks, have attracted great attention in various applications such as agriculture (El-Rehim 2010), tissue engineering (Sivashanmugam et al. 2015), sensor (Tang et al. 2016), contact lenses (Ashtiani et al. 2018), wound dressings (Fan et al. 2014), hemostasis bandages (Qin et al. 2016) and drug delivery systems (Matricardi et al. 2013). In particular, great interest has been focused on stimuli-responsive composite hydrogel based on natural polymer such as cellulose, starch, chitosan and alginate (Qiu and Hu 2013). However, most of the individual cross-linked hydrogels had disadvantage of low mechanical strength, which hindered their further applications as soft and hydrophilic smart materials.
Recently, extensive efforts have been made to modulate the mechanical strength of hydrogel. Interpenetrating polymer network (IPN) system is one of the most efficient approaches to toughening hydrogels (Sahiner and Demirci 2017). The 3D structure of IPN is composed of two or more independently and non-covalently cross-linked polymer networks (Dragan 2014). These polymer networks held together by permanent entanglements and physical interactions so as to facilitate the construction of hydrogels with good stability unless chemical bonds are broken (Bajpai et al. 2008). To date, various multicomponent hydrogel, such as polyvinyl alcohol/sodium alginate/cellulose nanofibers (Yue et al. 2016), polyacrylic acid/quaternized cellulose/polyvinyl alcohol (Wang et al. 2017), polyacrylamide/montmorillonite (Guorong et al. 2015), based on interpenetrating polymer networks (IPNs) with improved mechanical performance have been fabricated. Naseri et al. (2016) reported a double cross-linked IPN hydrogels based on cellulose nanocrystals (CNC) in a matrix of sodium alginate/gelatin with significant improvements in the tensile strength and strain. Li et al. (2015) successfully fabricated polyacrylamide/sodium alginate (PAM/SA) IPN hydrogels from Ca2+ crosslinked SA and covalently crosslinked PAM, the fracture strength of which reaches to 2.0 MPa. Compared with the individual cross-linked hydrogels, the IPN hydrogels show better mechanical performance owing to the effective physical entanglement (Bajpai et al. 2008). In some cases, unique properties such as self-healing, high strength, high toughness and antibacterial ability can be observed in the interpenetrating polymer networks that are not shown in either of the two individual networks alone (Myung et al. 2010; Wang et al. 2017; Guorong et al. 2015).
Polyacrylamide (PAM) is considered to be the most widely explored polymers for the preparation of hydrogels due to its nontoxicity, biologically inert, long-chain lengths, and the convenient adjustability of mechanical and chemical properties (Zhou and Wu 2011). Therefore, PAM hydrogels have attracted widespread interest because of its potential applications in waste treatments (Saber-Samandari et al. 2013), drug delivery (Sabbagh and Muhamad 2017) and tissue engineering (Kong et al. 2016). Up to now, several studies have been carried out to prepare PAM IPN hydrogels via free radical polymerization (Kim and Park 2004; Singh et al. 2007; Yin et al. 2007). Ilmain et al. (1991) prepared an IPN hydrogel based on poly(acrylamide) and poly(acrylic acid), and the IPN hydrogels presented large volume transitions driven by hydrogen bonding between these two networks. The other IPN hydrogels consisting of an ionically cross-linked gellan gum and a covalently cross-linked poly(acrylamide) were synthesized by Bakarich et al. (2012). Compared with the respective single network hydrogels, the resulting IPN hydrogels with double network structure presented improved mechanical properties.
Cellulose is a biodegradable and biocompatible biopolymer which has been chosen as a good candidate for the fabrication of hydrogels (Chang et al. 2014; Huang et al. 2016). In previous reports, the aqueous alkali/urea solution was introduced to afford transparent cellulose hydrogels via regeneration due to its good dissolution ability for cellulose at low temperature (− 12 °C) (Cai et al. 2008). IPN hydrogel based on cellulose could be synthesized by the radical polymerization of monomer within the cellulose hydrogel or mixture of biopolymers and synthetic polymers with polymerizable groups (Ciolacu and Suflet 2018). Qu et al. (2015) prepared a carboxymethyl cellulose/poly(methyl acrylate) IPN hydrogel with combination of hydrophobic polymer and biopolymer via fractional step method, and the IPN hydrogel exhibited more favorable mechanical properties. Moreover, Pandey et al. (2013) synthesized a smart-swelling hydrogel by graft polymerization of acrylamide on bacterial cellulose solubilized in an NaOH/urea aqueous system. The composite hydrogel showed more uniform porosity structure distribution, a higher sensitivity towards pH and ionic solution compared to that of pure PAM hydrogel, which can be explored further for oral drug delivery. Furthermore, some other polymers such as polyethylene glycol (Liang et al. 2010), polyacrylic acid (Bajpai et al. 2013) and poly(vinyl alcohol) (Chang et al. 2010a), were used to modulate the strength of cellulose hydrogels prepared in NaOH/urea solvent system, but to the best of our knowledge, few researches on the fabrication of cellulose/PAM IPN hydrogels based on NaOH/urea solvent system with high strength and pH-responsive properties have been reported.
In this study, we attempt to improve the mechanical properties of the cellulose hydrogels by using interpenetrating polymer network (IPN) technology. The cellulose hydrogels regenerated from NaOH/urea solvent system via chemically cross-linking were employed as the first network, and the second network was constructed by in situ polymerization of acrylamide (AM) in the cellulose hydrogels, resulting in the fabrication of cellulose–polyacrylamide interpenetrating network (C–PAM IPN) hydrogels with double networks structure. The chemical structure and morphology of the C–PAM IPN hydrogels were investigated by XRD, FTIR, solid-state 13CNMR and SEM. Moreover, the effects of different monomer (AM) concentration of the second network on the mechanical, swelling and pH-responsive properties of the obtained C–PAM IPN hydrogels were discussed as well.
Experimental
Materials
Bleached bamboo pulp (α-cellulose > 96%) sheet obtained from Nanping Paper CO., Ltd. (Nanping, Fujian, China) was dried at 80 °C for 12 h and beaten for 2 min to obtain powdered cellulose by using a high efficiency pulverizer (LFP-2500A, Yongkang hongtaiyang electromechanical Co., Ltd., China) prior to use. The molecular mass distribution of bleached bamboo pulp was determined by gel permeation chromatography. The weight average molecular weight (Mw) and the number average molecular weight (Mn) of bamboo pulp were determined as 203 000 and 58 000, respectively. NaOH, and urea were obtained from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). Epichlorohydrin (ECH), acrylamide (AM), potassium persulfate (KPS) and N,N′-methylene-bis(acrylamide) (MBAA) were purchased from Aladdin Industrial Corporation (Shanghai, China). All reagents were used as received, and deionized (DI) water was used for all experiments.
Synthesis of cellulose–polyacrylamide interpenetrating network hydrogels
The cellulose–polyacrylamide interpenetrating network (C–PAM IPN) hydrogels were constructed via a two-step method. In the first step, chemically cross-linked cellulose hydrogel was synthesized as previously reported (Chang et al. 2010a, b; Zhou et al. 2007). A 96.0 g aqueous solution of 7 wt% NaOH/12 wt% urea was pre-cooled (− 25 °C) for 10 h. After frozen solution was partly thawed at ambient temperature, 4.0 g of powdered cellulose was added and stirred extensively to produce a 4.0 wt% homogeneous cellulose solutions. Subsequently, 5.0 mL epichlorohydrin was dropwise added into cellulose solution and stirred for additional 40 min to achieve a homogeneous solution. After the air bubbles were removed by ultrasonication, the cellulose solution was poured into glass plates (20 mm in diameter and 10 mm in height) and maintained in a refrigerator at − 25 °C for 24 h, leading to the gelation of the mixtures. The obtained chemical crosslinked cellulose hydrogels were purified by soaking in large amount of pure water for several days to remove residual reagent completely (Chang et al. 2010a, b, 2011). At the second step, the purified disc-shaped cellulose hydrogel was immersed in a large amount of aqueous AM solution with various concentrations ranging from 1 to 5 mol/L containing 0.1 mol% MBAA and 0.1 mol% KPS for 3 days until equilibrium was reached. The second network was subsequently polymerized in situ at 60 °C for 6 h in the first network of cellulose. The obtained C–PAM IPN hydrogels were purified by soaking in a large excess of DI water for 24 h (Gao et al. 2014; Yang et al. 2013).
The synthesized IPN hydrogels based on 1, 2, 3, 4 and 5 mol/L AM aqueous solutions were designated as C–PAM-1, C–PAM-2, C–PAM-3, C–PAM-4 and C–PAM-5, respectively. For comparison, the pure PAM hydrogel was synthesized via free-radical polymerization of 5 mol/L AM aqueous solution containing 0.1 mol% MBAA and 0.1 mol% KPS at 60 °C for 6 h. The pure cellulose and PAM hydrogels were designated as cellulose-gel and PAM-gel, respectively.
Component content measurements
The cellulose content (Wcellulose), water content (\( W_{{H_{2} O}} \)) and PAM content (WPAM) of the IPN hydrogels were calculated using Eqs. 1, 2 and 3:
where Ww is the weight of the fresh IPN hydrogels, Wd is the weight of the dried IPN hydrogels, Wc is the weight of the cellulose hydrogel before soaking into AM aqueous solution, and Cc is the mass concentration of the cellulose solution. The content of cellulose was calculated without considering the dosage of epichlorohydrin as cross-linker.
Scanning electron microscopy (SEM)
The C–PAM IPN hydrogels were frozen in liquid nitrogen and snapped, followed by freeze-drying. The fracture surfaces of the C–PAM IPN hydrogels were coated with a thin layer of gold for SEM analysis using a Hitachi SU8010 instrument (Hitachi, Japan).
X-ray diffraction (XRD)
The C–PAM IPN hydrogels were freeze-dried and then milled to powders by hand using an agate mortar and pestle. XRD patterns of these samples were performed on an X’Pert Pro MPD X-ray diffractometer (Philips-FEI, Netherlands) with Cu Kα radiation at a scanning rate of 0.02° s−1. Diffractograms were obtained in the range of 2θ = 5°–60°.
Fourier transform infrared spectroscopy (FTIR)
FTIR analysis was carried out on a Nicolet 380 spectrometer (Thermo Electron Instruments Co., Ltd., USA). Prior to analysis, the powdered freeze-dried IPN hydrogels were first mixed with KBr to prepare tablets. The spectra were recorded from 4000 to 400 cm−1 at a resolution of 4 cm−1 with an average of 32 scans.
Nuclear magnetic resonance spectroscopy analysis (NMR)
The C–PAM IPN hydrogels were freeze-dried and then milled to powders. Solid-state 13C-NMR spectra of samples were investigated by Bruker AVANCEIII 400 (13C frequency = 100.0 MHz). Experimental parameters for 13C-NMR analysis were as follows: 4000 scans, spinning rate 6.0 kHz, contact time 5.0 ms, pulse delay 5.0 s and temperature of 278 K.
Thermal gravimetric analysis (TGA)
The thermal-gravimetric curves of the C–PAM IPN hydrogels were recorded on a thermal gravimetric analyzer (NETZSCH STA 449 F3 Jupiter). The samples (about 5 mg) were placed in a clean platinum pan and heated from room temperature to 600 °C with a heating rate of 10 °C min−1 under nitrogen atmosphere (flow rate = 25 mL min−1).
Mechanical measurements
Compressive measurements were performed on the C–PAM IPN hydrogel using a universal tensile-compressive tester (CMT 6104, Shenzhen SANS Materials Testing Co., Ltd, China). The cylindrical hydrogels with a dimension of 20 mm × 10 mm (diameter × height) were tested at 5 mm min−1 of compression rate. Five specimens were tested for each sample to ensure reproducibility. The compressive modulus was calculated from the liner region of the stress–strain curves. The dissipated energy, ΔU, was calculated from the area of the hysteresis loop:
where σ is stress and ε is strain.
The dynamic modulus C–PAM IPN hydrogels were performed by using Rotational Rheometer MARS III Haake (Thermo Scientific, Germany) with a parallel plate of diameter 30 mm. The disc-shaped C–PAM IPN hydrogels (20 mm in diameter, 2 mm in thickness) were subjected to a variable strain sweeps from 0.1 to 100% at a frequency of 1.0 Hz to determine the linear viscoelastic region. Subsequently, the oscillatory frequency sweeps were investigated from 0.01 to 100 Hz at 25 °C.
Swelling behavior and pH sensitivity measurements
To determine the equilibrium swelling ratios (ESR) of the C–PAM IPN hydrogels, a small IPN hydrogel was immersed in DI water for 48 h at 25 °C and thereafter the swollen hydrogel was dried at 60 °C under vacuum of 0.05 MPa to a constant weight. The ESR can be calculated using Eq. 4
where Ws and Wd are the weight of the hydrogel with swollen and dried state.
The ESR of the C–PAM IPN hydrogels in different pH solutions (ranging from 1.0 to 14.0) at 25 °C was measured by following the same methods as that in distilled water. The pH values were adjusted by HCl and NaOH solutions and determined by a pH meter (ST3100, Ohaus Corporation, USA).
Results and discussion
Preparation and chemical structure of C–PAM IPN hydrogels
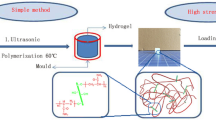
The cellulose–polyacrylamide interpenetrating network (C–PAM IPN) hydrogels were fabricated via free-radical polymerization of acrylamide (AM) in the presence of the cellulose network (the first network) to form the second network. The general procedure to synthesize C–PAM IPN hydrogels through in situ polymerization is illustrated in Fig. 1. Chemically cross-linked cellulose hydrogel was constructed as the first network, and then with AM as a monomer, MBAA as a cross-linker and KPS as an initiator were absorbed by soaking the cellulose hydrogel in their aqueous solutions. Finally, the AM monomer-swollen cellulose hydrogel was incubated at 60 °C for 6 h to achieve in situ polymerization of AM, leading to the successful fabrication of a cellulose–polyacrylamide interpenetrating network (C–PAM IPN) hydrogel. The composition of synthesized C–PAM IPN hydrogels was measured and summarized in Table 1. Obviously, the cellulose and PAM content increased as the AM concentration increased, while the water content showed the opposite trend. The decrease of water content was probably due to the dehydration of cellulose hydrogel originated from the higher osmotic pressure in the surrounding AM/MBAA/KPS solution, resulting in the formation of a denser network in the C–PAM IPN hydrogel after in situ polymerization of AM monomers. The photographs of chemical crosslinked cellulose hydrogel, C–PAM IPN hydrogel (C–PAM-5) and the possible interaction between cellulose network and PAM chains were shown in Fig. 1d. After in situ polymerization, the cellulose-based hydrogel became more opaque and elastic, which was possibly caused by the formation of a double network structure composed of cellulose networks and synthesized PAM chains. In addition, the amide groups on the PAM chains could form hydrogen bonds with the –OH groups on cellulose network to generate a mechanically flexible and tough interpenetrating network hydrogels.

Schematic illustration of the synthesis of C–PAM IPN hydrogels; (a) chemical crosslinked cellulose hydrogel (the first network), (b) monomers (AM, MBAA and KSP) onto cellulose hydrogel, (c) cellulose–polyacrylamide interpenetrating network hydrogel (C–PAM IPN hydrogel), (d) photographs of chemical crosslinked cellulose hydrogel, C–PAM IPN hydrogel (C–PAM-5) and the possible interaction between cellulose and PAM networks
The XRD patterns of C–PAM IPN hydrogels (C–PAM-5), cellulose hydrogel (cellulose-gel) and PAM-gel are shown in Fig. 2a. In the pattern of cellulose hydrogel, the wide diffraction peaks at 2θ = 20°–22° are corresponding to the characteristic peaks of cellulose. Meanwhile, the PAM hydrogel showed a broad diffraction peak around 26°, representing an amorphous phase. As for the pattern of C–PAM-5 IPN hydrogel, it can be observed that the characteristic peaks at 2θ = 20°–22° for cellulose was weakened as compared with cellulose hydrogel without PAM component, along with the decreasing of crystallinity of cellulose from 34.6 to 21.5%. These results indicated that the amorphous PAM as the second network was successfully introduced into cellulose hydrogel through the in situ chemical polymerization (Chang et al. 2010a; Liang et al. 2010).

XRD patterns (a) and FTIR spectra (b) of C–PAM IPN hydrogels, cellulose-gel and PAM-gel
Figure 2b shows the FTIR spectra of the C–PAM IPN hydrogels (C–PAM-1), cellulose-gel and PAM-gel. The cellulose-gel display a typical FTIR spectrum of cellulose at 3446 cm−1 (O–H stretching), 2923 cm−1 (C–H symmetric stretching vibration), 1642 cm−1 (H–O–H stretching vibration), 1462 cm−1 (C–H2 scissoring vibration), 1052 cm−1 (C–O stretching vibration) and 615 cm−1 (C–O–C bridge) (Zhao et al. 2016; Lin et al. 2017). As for the spectra of PAM-gel, the characteristic peaks at 3192 cm−1, 1662 cm−1 and 1606 cm−1 were due to the N–H stretching vibration, C=O stretching vibration and N–H bending vibration of the –CO–NH2 groups (Liu et al. 2012; Wang et al. 2017). The spectrum of the C–PAM-1 exhibited characteristic features of both cellulose-gel and PAM-gel. Compared with the spectra of cellulose-gel and PAM-gel, no new absorption peak was observed for C–PAM IPN hydrogel, indicating there was no chemical reaction occurred between cellulose and PAM. Moreover, the adsorption peak at 3436 cm−1 for cellulose-gel shifted to 3404 cm−1 and broadened, possibly owing to the formation of hydrogen bond between cellulose and polyacrylamide chains in the C–PAM IPN hydrogel (see Fig. 1d) (Wang et al. 2017; Zhou and Wu 2011).
The composition of hydrogel network was also investigated by the solid-state 13C-NMR spectroscopy of the C–PAM IPN hydrogels (C–PAM-5), cellulose-gel and PAM-gel (Fig. 3). As shown in Fig. 3, the spectra of C–PAM-5 showed the typical peaks at 25–50 (signal 1′, 2′) ppm assigned to methylene (CH2) and methine (CH) carbon of the PAM main chain and the peak around 180 (signal 3′) ppm relative to C=O of acrylamide group (Mahfoudhi and Boufi 2016). The characteristic peaks of the cellulose-gel were observed around 60, 70–75, 82 and 104 ppm, which can be assigned to the C6, C2–C3–C5, C4 and C1 carbons of the anhydroglucosic unit, respectively (Wang et al. 2017). Compared with the solid-state 13C-NMR spectra of the cellulose-gel and PAM-gel (Fig. 3), no new peaks were observed in the spectra of the C–PAM-5, further indicating that there were no covalent bonds between cellulose and PAM chains.

Solid-state 13C-NMR spectra of C–PAM IPN hydrogels (C–PAM-5), cellulose-gel and PAM-gel
Thermal behavior of C–PAM IPN hydrogels
The TG and DTG curves of cellulose-gel, PAM-gel and C–PAM IPN hydrogels (C–PAM-5) are demonstrated in Fig. 4. As seen from Fig. 4a, all samples exhibited a slight weight loss at around 100 °C, which can be ascribed to the evaporation of adsorbed water. In high temperature range, the weight loss of all hydrogels was different each other, and the initial thermal degradation temperature of C–PAM-5 increased from 295 to 338 °C in comparison with chemical crosslinked cellulose hydrogel. As clearly shown in Fig. 4b, the maximum degradation temperature appeared at 337 °C for the cellulose hydrogel and 364 °C for PAM-gel. As for the C–PAM-5, it increased to 408 °C, providing direct evidence that the C–PAM-5 hydrogels based on IPN networks presented better thermal stability in comparison to the hydrogels based on the two individual networks alone. This may be attributed to the synergistic improvement of IPN network formed by PAM and cellulose, resulting in more ordered structure with interconnected mesoporosity in the IPN C–PAM hydrogels as can be demonstrated by the SEM images in Fig. 5. Moreover, in Fig. 4a, the C–PAM-5 contained more char (26.6%) at the end of run at 600 °C, compared to that of PAM-gel (19.3%) and cellulose-gel (8.5%), respectively. This is probably due to the formation of more rigid networks, higher cross-linking degree and lower crystallinity of IPN hydrogel than that of cellulose hydrogel.

TG (a) and DTG (b) curves of cellulose-gel, PAM-gel and C–PAM IPN hydrogels (C–PAM-5)

SEM micrographs of the fractured cross sections of cellulose-gel (a) and C–PAM IPN hydrogels: C–PAM-1 (b), C–PAM-2 (c), C–PAM-3 (d), C–PAM-4 (e) and C–PAM-5 (f)
Morphologies of C–PAM IPN hydrogels
All the C–PAM IPN hydrogels synthesized from varied concentrations of AM aqueous solutions were subjected to freeze-drying process to prepare samples for SEM observation. The typical SEM micrographs of C–PAM IPN hydrogels were shown in Fig. 5, and the parameters were listed in Table 2. As is seen in Fig. 5a, the chemical crosslinked cellulose hydrogel without any PAM displayed relatively coarse texture with porous structure, which is similar to that in previous reports (Liu et al. 2016; Luo et al. 2016). A great impact of PAM content on the structure of the IPN hydrogels was identified: the density and average pore size were increased with PAM content rising from 0 to 42.93 wt%, and the thickness of the pore walls also increased with an increase of PAM content. The density of C–PAM-5 IPN hydrogel was 0.37 g/cm3, whereas that of cellulose-gel was only 0.023 g/cm3, indicating a more rigid network formation and higher degree of cross-linking in IPN hydrogel than cellulose-gel. Moreover, with the introduction of PAM into cellulose hydrogels, the scaffolds revealed porous inner structures and the average pore size increased from 2.60 to 22.13 μm with the increase of the PAM content. This maybe due to the fact that the thickness and stiffness of IPN networks potentially affected the cooling rates to generate larger ice crystals, which were finally represented by larger pores after freeze-dried of these IPN hydrogels (Dong et al. 2013; Hu et al. 2017). Furthermore, the thickness and stiffness IPN networks with porous structure might have significant influence on the swelling properties of C–PAM IPN hydrogels, which will be further discussed in the equilibrium swelling ratio analysis.
Mechanical properties of C–PAM IPN hydrogels
Figure 6a illustrates the mechanical behaviors of C–PAM IPN hydrogel (red) compared with pure cellulose-gel (yellow) and PAM-gel (blue). It was observed that C–PAM IPN hydrogel could withstand the compression without any obvious damages and its original shape remained intact after release loading. However, the pure cellulose-gel and PAM-gel were quite fragile and easily ruptured upon compression. The typical stress–strain curves of C–PAM IPN hydrogels were illustrated in Fig. 6b. During the stress loading process, the stress–strain curves of C–PAM IPN hydrogels exhibited a linear region at initial strains < 30% and a region with an increasing slope at strains 30–80%. The detailed values of compressive parameters were summarized in Table 3. It is obvious that as the content of PAM within C–PAM IPN hydrogels increased, the compressive strength and compressive modulus at breakage increased steadily, however, due to the extremely brittle nature of PAM-gel (Zhao et al. 2017), the fracture strain of C–PAM IPN hydrogels decreased. It is obvious that the compressive strength and compressive modulus of chemical crosslinked cellulose hydrogel is only 0.31 and 0.95 MPa, respectively, and the fracture strain is 54.75%. In contrast, the compressive strength and compressive modulus for C–PAM IPN hydrogels could reach up to 5.62 and 22.47 MPa (C–PAM-5), which was nearly 18 and 23 times higher than that of cellulose-gel, and the fracture strain was 57.69%, also much greater than that of cellulose-gel. Furthermore, C–PAM IPN hydrogels also exhibit much better mechanical properties than PAM-gel (compressive strength of 0.84 MPa, compressive modulus of 1.86 MPa and fracture strain of 36.51%). Therefore, compared with cellulose and PAM single network hydrogels, the mechanical properties of C–PAM IPN hydrogels could be improved significantly by introducing interpenetrating network (IPN) structure. Moreover, as shown in Table 4, typical cellulose-reinforcement strategies for PAM hydrogels did not result in sets of properties comparable to those of cellulose/PAM IPN hydrogels. The speculated reason for the improvement of mechanical strength of C–PAM IPN hydrogel was that the in situ polymerized PAM chains in IPN networks were intertwined with cellulose chains, contributing to the excellent energy dissipation and stress bearing upon deformations (Gong et al. 2016; Sahiner and Demirci 2017) (see Fig. 6a).

a The compression behavior of the cellulose-gel (yellow), C–PAM IPN hydrogel (red) and PAM-gel (blue) before (left), during (middle) and immediately after compression (right); b compressive stress–strain curves of C–PAM IPN hydrogels. The inset is the compressive stress–strain curves of the cellulose-gel and PAM-gel; c the cyclic loading–unloading curves of C–PAM-5 (at 50% strain); d cellulose-gel (at 50% strain); and e PAM-gel (at 30% strain) for 10 sequential test cycles without rest. (Color figure online)
Additionally, the cyclic compressive loading–unloading tests were carried out to evaluate the energy dissipation of C–PAM IPN hydrogel as presented in Fig. 6c, and the quantified data for energy dissipation of hydrogels were listed in Table 3. As illustrated in Fig. 6c, the C–PAM IPN hydrogel (C–PAM-5) exhibits a typical hysteresis loop and a dissipated energy of ~ 85.50 kJ/m3 at 50% strain in the first loading–unloading cycle. The second cycle without any pausing for tests indicates that the hysteresis loop almost maintains its original shape (~ 84.87 kJ/m3). Even after 10 cycles, the dissipated energy can still remain at 84.18 kJ/m3, which demonstrates that C–PAM IPN hydrogels exhibits good energy dissipation and fatigue resistance properties. Whereas, in the first loading–unloading cycle, the dissipated energy was only 8.45 kJ/m3 (at 50% strain) and 23.30 kJ/m3 (at 30% strain) for the single network cellulose-gel (Fig. 6d) and PAM-gel (Fig. 6e), respectively. From cycles 1 to 10, due to the permanent deformation and internal fracture of the polymer chains, the dissipated energy of cellulose-gel and PAM-gel decreased to 6.40 and 5.53 kJ/m3, respectively. These results indicated that C–PAM IPN hydrogel dissipates energy more efficiently than single network hydrogels under deformation, thus leading to high mechanical properties, in which the cooperation of cellulose and PAM chains plays a key role.
The dynamic mechanical investigations were also carried out at linear viscoelasticity region, which can provide further insight into the properties of C–PAM IPN hydrogels. The storage modulus (G′) and loss modulus (G″) for chemical crosslinked cellulose hydrogel, PAM-gel and C–PAM IPN hydrogels are illustrated in Fig. 7. It can be clearly observed from Fig. 7a, b that the G′ remains almost unchanged, much higher than G″ over the entire frequency range (0.1 to 100 Hz) for C–PAM IPN hydrogels, which indicated that the three-dimensional interpenetrating polymeric networks formed and a dominant elastic property presented in these IPN hydrogels (Gong et al. 2016). Notably, the G′ and G″ values of C–PAM IPN hydrogels ascended significantly with PAM content increasing, which was well in line with the above results of compressive stress–strain (Fig. 6b). As compared in Fig. 7b, in the case of C–PAM-5, the maximum value of 37,731 Pa (G′) and 2089 Pa (G″) can be achieved, however, the G′ and G″ values for cellulose-gel was only 993 Pa (G′) and 52 Pa (G″), respectively. Moreover, the G′ and G″ value of C–PAM-5 was 6 and 10 times higher than that of PAM-gel [5959 Pa (G′) and 202 Pa (G″)]. These results may be attributed to the synergistic interactions between the two networks of IPN hydrogels and the strong hydrogen bonds between cellulose network and PAM chains, which significantly enhanced the elasticity property of C–PAM IPN hydrogels and could effectively dissipate mechanical energy upon deformation.

a Frequency dependence of storage modulus (G′) and loss modulus (G″) with 10% strain at 25 °C for different C–PAM IPN hydrogels; b G′ and G″ with 10% strain at 25 °C for cellulose-gel, PAM-gel and C–PAM-5 hydrogel
Equilibrium swelling ratio and pH responsive properties of C–PAM IPN hydrogels
Figure 8a shows the effect of AM concentration on the equilibrium swelling ratio (ESR) of the C–PAM IPN hydrogels in deionized water at 25 °C. As is indicated, the ESR of the hydrogels sharply decreased after the combination of cellulose-gel with PAM. When the concentration of AM increased from 1 to 5 mol/L, the ESR of IPN hydrogel decreased from 24.2 to 7.1 g/g, while the ESR for cellulose-gel was 28.4 g/g. This is probably due to the fact that the dehydration of cellulose hydrogel occurred during the process of soaking in aqueous AM solution, which may increase the solid content (Table 1) as well as the thickness of the pore walls (Fig. 5) of C–PAM IPN network after in situ polymerization of AM monomers (Zhang et al. 2011).

a Equilibrium swelling ratio of the C–PAM IPN hydrogels and cellulose-gel in distilled water at 25 °C. b Effects of pH value on the swelling behaviors of C–PAM IPN hydrogels and cellulose-gel at 25 °C. Schematic illustrations of the chemical structures of IPN hydrogel under acidic (c) and alkaline (d) pH, respectively
Partially hydrolyzed PAM at a lower and higher pH contain carboxylate groups (–COO−) that either accept or release protons in response to changes in environmental pH (Qiu and Park 2001; Pandey et al. 2013; Silva and Oliveira 2007), which can render the pH-sensitive properties for C–PAM IPN hydrogels. As shown in Fig. 8b, the ESR value for pure cellulose-gel remains roughly constant in a pH value from 1.0 to 14.0, however, the ESR of all C–PAM IPN hydrogels increased gradually with the increase of pH, and reached the maximum value when pH = 13.0. The pKa value of the carboxylic acid is ~ 4.7 (Pandey et al. 2013). Under acidic pH (below the pKa of carboxylic groups), most of the carboxylate anions were protonated. Meanwhile, stronger intermolecular hydrogen bond was formed between the cellulose chains and PAM network, which induced the contraction of the IPN hydrogel to expel water molecules (Cha et al. 2012) (Fig. 8c). At higher pH (pH > 4.7), the –COOH groups were partly ionized and the electrostatic repulsive force between the charged sites (–COO−) lead to a decrease in the hydrogen bonds between the cellulose and PAM networks (Li et al. 2009) (Fig. 8d). As a result, the hydrogels displayed an expandable network structure, which could easily absorb and bind water, and the maximum ESR values were observed at pH = 13.0 for all C–PAM IPN hydrogels. However, under the strong alkaline condition with pH = 14.0, the ESR values of IPN hydrogels decreased rapidly, and this behavior is attributed to the increasing ionic strength with the increase of pH value, which would shrink the IPN hydrogels network again under this condition (Zhou et al. 2013).
Conclusions
In conclusion, a general strategy is presented to prepare a novel cellulose–polyacrylamide interpenetrating polymer network (C–PAM IPN) hydrogels with high mechanical strength and pH responsive properties through in situ polymerization. Cellulose hydrogel prepared from NaOH/urea solvent system with chemically cross-linking was employed as the first network, and the second network was constructed by in situ polymerization of acrylamide (AM) in the cellulose hydrogels, resulting in the double networks structure. The two networks co-existed without any covalent bonds and exhibited good compatibility with uniform porous network the IPN hydrogels. Compared with cellulose single network hydrogels, the mechanical properties of C–PAM IPN hydrogels were improved significantly by introducing IPN structure, which could effectively dissipate mechanical energy and transfer stress upon deformation. The compressive strength and compressive modulus for C–PAM IPN hydrogels could reach up to 5.62 and 22.47 MPa, which was nearly 18 and 23 times higher than that of cellulose-gel. Thermal analysis indicated that the thermal stability of C–PAM IPN hydrogel was obviously improved as compared to that of the individual component. Additionally, these IPN hydrogels exhibited a pH sensitive property in different pH solutions. The C–PAM IPN hydrogels with excellent mechanical strength, improved thermal stability and pH sensitive properties may expand potential application for cellulose-based hydrogels in biomedical fields.
References
Aouada FA, de Moura MR, Orts WJ, Mattoso LHC (2011) Preparation and characterization of novel micro-and nanocomposite hydrogels containing cellulosic fibrils. J Agric Food Chem 59:9433–9442
Ashtiani MK, Zandi M, Shokrollahi P, Ehsani M, Baharvand H (2018) Surface modification of poly(2-hydroxyethyl methacrylate) hydrogel for contact lens application. Polym Adv Technol 29:1227–1233
Bajpai AK, Shukla SK, Bhanu S, Kankane S (2008) Responsive polymers in controlled drug delivery. Prog Polym Sci 33:1088–1118
Bajpai SK, Bajpai M, Gautam D (2013) In situ formation of silver nanoparticles in regenerated cellulose-polyacrylic acid (RC-PAAc) hydrogels for antibacterial application. J Macromol Sci Part A Pure Appl Chem 50:46–54
Bakarich SE, Pidcock GC, Balding P, Stevens L, Calvert P, Marc IHP (2012) Recovery from applied strain in interpenetrating polymer network hydrogels with ionic and covalent cross-links. Soft Matter 8:9985–9988
Cai J, Zhang L, Liu S, Liu Y, Xu X, Chen X, Chu B, Guo X, Xu J, Cheng H (2008) Dynamic self-assembly induced rapid dissolution of cellulose at low temperatures. Macromolecules 41:9345–9351
Cha R, He Z, Ni Y (2012) Preparation and characterization of thermal/pH-sensitive hydrogel from carboxylated nanocrystalline cellulose. Carbohydr Polym 88:713–718
Chang C, Lue A, Zhang L (2010a) Effects of crosslinking methods on structure and properties of cellulose/PVA hydrogels. Macromol Chem Phys 209:1266–1273
Chang C, Zhang L, Zhou J, Zhang L, Kennedy JF (2010b) Structure and properties of hydrogels prepared from cellulose in NaOH/urea aqueous solutions. Carbohydr Polym 82:122–127
Chang C, Han K, Zhang L (2011) Structure and properties of cellulose/poly(N-isopropylacrylamide) hydrogels prepared by IPN strategy. Polym Adv Technol 22:1329–1334
Chang C, He M, Zhou J, Zhang L (2014) Swelling behaviors of pH- and salt-responsive cellulose-based hydrogels. Macromolecules 44:1642–1648
Ciolacu DE, Suflet DM (2018) 11-Cellulose-based hydrogels for medical/pharmaceutical applications. In: Popa V, Volf I (eds) Biomass as renewable raw material to obtain bioproducts of high-tech value. Elsevier, Amsterdam, pp 401–439
Dong H, Snyder JF, Tran DT, Leadore JL (2013) Hydrogel, aerogel and film of cellulose nanofibrils functionalized with silver nanoparticles. Carbohydr Polym 95:760–767
Dragan ES (2014) Design and applications of interpenetrating polymer network hydrogels. A review. Chem Eng J 243:572–590
El-Rehim HAA (2010) Characterization and possible agricultural application of polyacrylamide/sodium alginate crosslinked hydrogels prepared by ionizing radiation. J Appl Polym Sci 101:3572–3580
Fan Z, Liu B, Wang J, Zhang S, Lin Q, Gong P, Ma L, Yang S (2014) A novel wound dressing based on Ag/graphene polymer hydrogel: effectively kill bacteria and accelerate wound healing. Adv Funct Mater 24:3933–3943
Gao G, Du G, Cheng Y, Fu J (2014) Tough nanocomposite double network hydrogels reinforced with clay nanorods through covalent bonding and reversible chain adsorption. J Mater Chem B 2:1539–1548
Gong Z, Zhang G, Zeng X, Li J, Li G, Huang W, Sun R, Wong C (2016) High-strength, tough, fatigue resistant, and self-healing hydrogel based on dual physically cross-linked network. ACS Appl Mater Interfaces 8:24030–24037
Guorong G, Gaolai D, Yuanna S, Jun F (2015) Self-healable, tough, and ultrastretchable nanocomposite hydrogels based on reversible polyacrylamide/montmorillonite adsorption. ACS Appl Mater Interfaces 7:5029–5037
Hu X, Wang Y, Zhang L, Xu M, Zhang J, Dong W (2017) Dual-pH/magnetic-field-controlled drug delivery systems based on Fe3O4@SiO2-incorporated salecan graft copolymer composite hydrogels. ChemMedChem 12:1600–1609
Huang B, Qilin LU, Tang L (2016) Research progress of nanocellulose manufacture and application. J For Eng 1:1–9
Ilmain F, Tanaka T, Kokufuta E (1991) Volume transition in a gel driven by hydrogen bonding. Nature 349:400–401
Kim D, Park K (2004) Swelling and mechanical properties of superporous hydrogels of poly(acrylamide-co-acrylic acid)/polyethylenimine interpenetrating polymer networks. Polymer 45:189–196
Kong Y, Zhao Y, Ji B, Shi B, Wei S, Chen G, Zhang L, Li G, Yang Y (2016) Preparation and characterization of polyacrylamide/silk fibroin/graphene oxide composite hydrogel for peripheral nerve regeneration. J Biomater Tissue Eng 6:682–689
Li P, Kim NH, Siddaramaiah LJ (2009) Swelling behavior of polyacrylamide/laponite clay nanocomposite hydrogels: pH-sensitive property. Compos B 40:275–283
Li Y, Wang C, Zhang W, Yin Y, Rao Q (2015) Preparation and characterization of PAM/SA tough hydrogels reinforced by IPN technique based on covalent/ionic crosslinking. J Appl Polym Sci 132:41342–41348
Liang S, Wu J, Tian H, Zhang L, Xu J (2010) High-strength cellulose/poly(ethylene glycol) gels. Chemsuschem 1:558–563
Lin F, You Y, Yang X, Jiang X, Lu Q, Wang T, Huang B, Lu B (2017) Microwave-assisted facile synthesis of TEMPO-oxidized cellulose beads with high adsorption capacity for organic dyes. Cellulose 24:5025–5040
Liu R, Liang S, Tang XZ, Yan D, Li X, Yu ZZ (2012) Tough and highly stretchable graphene oxide/polyacrylamide nanocomposite hydrogels. J Mater Chem 22:14160–14167
Liu H, Wang A, Xu X, Wang M, Shang S, Liu S, Song J (2016) Porous aerogels prepared by crosslinking of cellulose with 1,4-butanediol diglycidyl ether in NaOH/urea solution. RSC Adv 6:42854–42862
Luo X, Lei X, Cai N, Xie X, Xue Y, Yu F (2016) Removal of heavy metal ions from water by magnetic cellulose-based beads with embedded chemically modified magnetite nanoparticles and activated carbon. ACS Sustain Chem Eng 4:3960–3969
Mahfoudhi N, Boufi S (2016) Poly (acrylic acid-co-acrylamide)/cellulose nanofibrils nanocomposite hydrogels: effects of CNFs content on the hydrogel properties. Cellulose 23:3691–3701
Matricardi P, Meo CD, Coviello T, Hennink WE, Alhaique F (2013) Interpenetrating polymer networks polysaccharide hydrogels for drug delivery and tissue engineering. Adv Drug Deliv Rev 65:1172–1187
Myung D, Waters D, Wiseman M, Duhamel PE, Noolandi J, Ta CN, Frank CW (2010) Progress in the development of interpenetrating polymer network hydrogels. Polym Adv Technol 19:647–657
Naseri N, Deepa B, Mathew AP, Oksman K, Girandon L (2016) Nanocellulose-based interpenetrating polymer network (IPN) hydrogels for cartilage applications. Biomacromolecules 17:3714–3723
Pandey M, Amin MCIM, Ahmad N, Abeer MM (2013) Rapid synthesis of superabsorbent smart-swelling bacterial cellulose/acrylamide-based hydrogels for drug delivery. Int J Polym Sci 10:1–10
Qin X, Labuda K, Chen J, Hruschka V, Khadem A, Liska R, Redl H, Slezak P (2016) Development of synthetic platelet-activating hydrogel matrices to induce local hemostasis. Adv Funct Mater 25:6606–6617
Qiu X, Hu S (2013) “Smart” materials based on cellulose: a review of the preparations, properties, and applications. Materials 6:738–781
Qiu Y, Park K (2001) Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev 53:321–339
Qu B, Li JR, Xiao HN, He BH, Qian LY (2015) Preparation of Sodium carboxymethylcellulose/poly(methyl acrylate) IPN hydrogels and their application for adsorption. J Appl Polym Sci 131:547–557
Sabbagh F, Muhamad II (2017) Acrylamide-based hydrogel drug delivery systems: release of Acyclovir from MgO nanocomposite hydrogel. J Taiwan Inst Chem Eng 72:182–193
Saber-Samandari S, Saber-Samandari S, Gazi M (2013) Cellulose-graft-polyacrylamide/hydroxyapatite composite hydrogel with possible application in removal of Cu(II) ions. React Funct Polym 73:1523–1530
Sahiner N, Demirci S (2017) Improved mechanical strength of p(AAm) interpenetrating hydrogel network due to microgranular cellulose embedding. J Appl Polym Sci 134:44854–44860
Silva RD, Oliveira MGD (2007) Effect of the cross-linking degree on the morphology of poly(NIPAAm–AAc) hydrogels. Polymer 48:4114–4122
Singh B, Sharma N, Chauhan N (2007) Synthesis, characterization and swelling studies of pH responsive psyllium and methacrylamide based hydrogels for the use in colon specific drug delivery. Carbohydr Polym 69:631–643
Sivashanmugam A, Kumar RA, Priya MV, Nair SV, Jayakumar R (2015) An overview of injectable polymeric hydrogels for tissue engineering. Eur Polym J 72:543–565
Tang X, Sun A, Chu C, Wang C, Liu Z, Guo J, Xu G (2016) Highly sensitive multiresponsive photonic hydrogels based on a crosslinked Acrylamide-N-isopropylacrylamide (AM-NIPAM) co-polymer containing Fe3O4@C crystalline colloidal arrays. Sens Actuators 236:399–407
Wang Y, Wang Z, Wu K, Wu J, Meng G, Liu Z (2017) Synthesis of cellulose-based double-network hydrogels demonstrating high strength, self-healing, and antibacterial properties. Carbohydr Polym 168:112–120
Yang J, Han CR, Duan JF, Ma MG, Zhang XM, Xu F, Sun RC (2013) Synthesis and characterization of mechanically flexible and tough cellulose nanocrystals–polyacrylamide nanocomposite hydrogels. Cellulose 20:227–237
Yin L, Fei L, Cui F, Tang C, Yin C (2007) Superporous hydrogels containing poly(acrylic acid-acrylamide)/-carboxymethyl chitosan interpenetrating polymer networks. Biomaterials 28:1258–1266
Yue Y, Han J, Han G, French AD, Qi Y, Wu Q (2016) Cellulose nanofibers reinforced sodium alginate-polyvinyl alcohol hydrogels: core–shell structure formation and property characterization. Carbohydr Polym 147:155–164
Zhang J, Rong J, Wendi LI, Lin Z, Zhang X (2011) Preparation and characterization of bacterial cellulose/polyacrylamide hydrogel. Acta Polym Sin 6:602–607
Zhao D, Huang J, Zhong Y, Li K, Zhang L, Cai J (2016) High-strength and high toughness double-cross-linked cellulose hydrogels: a new strategy using sequential chemical and physical cross-linking. Adv Funct Mater 26:6279–6287
Zhao J, Zheng K, Nan J, Tang C, Chen Y, Hu Y (2017) Synthesis and characterization of lignosulfonate-graft-poly (acrylic acid)/hydroxyethyl cellulose semi-interpenetrating hydrogels. React Funct Polym 115:28–35
Zhou C, Wu Q (2011) A novel polyacrylamide nanocomposite hydrogel reinforced with natural chitosan nanofibers. Colloids Surf B 84:155–162
Zhou J, Chang C, Zhang R, Zhang L (2007) Hydrogels prepared from unsubstituted cellulose in NaOH/urea aqueous solution. Macromol Biosci 7:804–809
Zhou C, Wu Q, Yue Y, Zhang Q (2011) Application of rod-shaped cellulose nanocrystals in polyacrylamide hydrogels. J Colloid Interface Sci 353:116–123
Zhou Q, Xu D, Zhi-Nian LZ (2013) Synthesis and pH sensitivity of polyacrylamide hydrogel. J Southwest Univ Natl Nat Sci Ed 39:695–698
Acknowledgments
We acknowledge the generous financial support of the Special Scientific Research Fund for Public Service Sectors of Forestry (No. 201504603), the National Natural Science Foundation of China (Nos. 21402027, 31370560), the Natural Science Foundation of Fujian Province (No. 2015J05046), Science Foundation for Distinguished Young Scholars of Fujian Agriculture and Forestry University (No. xjq201503), Scientific Research Foundation of Graduate School of Fujian Agriculture and Forestry University (No. 324-1122yb041). B. Lu also thanks the support of the outstanding youth scientific research personnel training plan of colleges and universities in Fujian Province. G. Lin thanks the support of Program for New Century Excellent Talents in Fujian Province University, the outstanding youth scientific research personnel training plan of colleges and universities in Fujian Province.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Lin, F., Lu, X., Wang, Z. et al. In situ polymerization approach to cellulose–polyacrylamide interpenetrating network hydrogel with high strength and pH-responsive properties. Cellulose 26, 1825–1839 (2019). https://doi.org/10.1007/s10570-018-2171-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2171-y