
Abstract
Near infrared (NIR) spectroscopy was tested as a rapid monitor of cellulose features by analyzing structurally distinguishable cellulose from different organisms ranging from Monera to Plantae as well as Animalia. The optimal spectral region was first identified using intra-crystalline deuteration, and then statistically analyzed based on second derivative spectra by principal component analysis. The score plots clearly distributed the samples according to crystalline structure such as relative crystallinity and allomorphism. These characteristics and the corresponding loading factors provided key NIR absorption criteria for identifying structural properties, especially in bands at 6527 and 6383 cm−1, which correspond to Iα and Iβ, respectively. In addition, calibration models were created for relative cellulose crystallinity using partial least square regression and for allomorph ratios using simple absorption band shifts at 6476–6446 cm−1. NIR spectra of cellulose from various organisms combined with multivariate analysis can be used as a database for simple and rapid assessment of unknown cellulose materials.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The plant cell wall has a central role in the mechanical function of the plant body, and its microstructure has evolved to adapt to environmental conditions (Sarkar et al. 2009). Therefore, survey of cell wall structure is a good approach to understand biological diversity as well as the progress of evolution. Cellulose is the main component of the plant cell wall, and is composed of β-1,4-homoglucan chains that are highly extended and packed to form microfibrils. It is synthesized by various organisms, including terrestrial plans, algae, bacteria, and tunicates (Tsekos 1999). However, the crystalline structure varies among biological organisms in terms of crystalline size, allomorphs, and uniplanar orientation. Therefore, the comparison of microfibril structures provides useful information for a better understanding of how the evolutionary process occurred.
Native cellulose has been characterized by different structural analyses such as X-ray diffraction, electron diffraction, nuclear magnetic resonance (NMR), and Fourier transform infrared spectroscopy (FTIR). Cross polarization/magic angle spinning (CP/MAS) 13C-NMR spectra first demonstrated that cellulose microfibrils are composed of two crystalline units, namely Iα and Iβ (Attala and VanderHart 1984), which electron diffraction determined that correspond to unit cells as one chain triclinic and two chains monoclinic, respectively (Sugiyama et al. 1991a). Synchrotron X-ray and neutron diffraction further clarified the precise atomic coordinates and hydrogen bond formation (Nishiyama et al. 2002, 2003). The ratio of these allomorphs is different depending on the source of cellulose, and can be detected by NMR (VanderHart and Attala 1984) and some diffraction technique (Wada et al. 1995). FTIR is also a powerful tool to classify allomorphs, as demonstrated by infrared (IR) spectra combined with electron diffraction using crystalline phase transition of Iα to Iβ (Sugiyama et al. 1991b). The assignment of IR bands characteristic of the corresponding crystalline units has been widely applied to monitor the crystalline phase during chemical hydrolysis (Wada and Okano 2001), enzymatic degradation (Hayashi et al. 1998), and development of the chordate tunicate Oikopleura dioica (Nakashima et al. 2011).
Near infrared (NIR) spectroscopy is similar to IR spectroscopy in that the absorption of the electromagnetic wave is detected from chemical bond vibrations. However, in NIR spectroscopy, measurement is based on overtone and combinational vibration of functional groups, and NIR bands are less intense compared to those of IR. Therefore, NIR absorption is not saturated even in high crystalline material or wet samples, which allows monitoring of sugar content in fruit juice (Rodriguez-Saona et al. 2001) or biomass digestion in ethanol production (Horikawa et al. 2015). Cellulose structural properties have also been reported to successfully estimate relative crystallinities (Inagaki et al. 2010) and degree of polymerization (Horikawa et al. 2016). However, these reports are limited to cellulose from terrestrial plants, and few studies have explored the structural diversity of cellulose using NIR spectral analysis.
In the present work, I investigated structurally distinguishable cellulose from approximately 20 species of Monera, Plantae, and Animalia. NIR spectra were combined with multivariate analysis to evaluate correlation between sources and crystalline structure. First, I determined the NIR region corresponding to the crystalline structure using intra-crystalline deuteration. Second, principal component analysis (PCA) was carried out to study source distribution based on cellulose suprastructure. Third, I developed calibration models to predict cellulose crystalline features from NIR spectroscopy.
Experimental
Cellulose samples
Bacterial cellulose was obtained from Acetobacter aceti (Aj-12368) as described by Nge and Sugiyama (2007). Bacterial cells were statically cultured in Hestrin–Schramm medium at 27 °C for 2 weeks and then a thick gelatinous membrane (BC pellicle) was squeezed to collect the cells. The cell suspension was transferred to fresh medium and cultured statically at 27 °C for 9 days. The BC pellicles obtained were boiled in distilled water for 1 h, followed by 0.4% NaOH at 80 °C for 2 h, and then washed with distilled water several times.
Cell wall samples of Glaucocystis nostochinearum, Oocystis apiculate, Valonia macrophysa, Cladophora spp., Boergesenia forbesii, Gelidium sesquipedale, and Erythrocladia subintegra were boiled in 1% NaOH for 8–10 h followed by treatment in 0.05 N HCl at room temperature. The samples were then subjected to bleaching treatment in 0.3% NaClO2 at pH 4.9 in acetate buffer at 70 °C for 3 h. The samples obtained were thoroughly rinsed with distilled water. The G. sesquipedale samples were boiled in water to remove agarose before the chemical treatment mentioned above.
Cellulose was obtained from Halocynthia papillosa as described by Sugiyama et al. (1991b). Halocynthia roretzi and Ciona intestinalis were treated with 5% KOH at room temperature overnight and then washed with distilled water. The samples were incubated in a bleaching solution at 70 °C for 2 h, which was repeated several times. After becoming colorless, cellulose samples were washed with distilled water.
Micrasterias crux-melitensis samples were boiled in 1% NaOH for 8 h, washed with distilled water, and then treated at 70 °C for 2 h in a bleaching solution. The bleaching treatment was followed by thorough washing and overnight dispersion into 5% KOH. The bleaching and KOH treatments were repeated several times before washing with water as described by Koyama et al. (1997).
Chara australis and Egeria densa were purchased from Kagaku Club (Shiga, Japan). To purify cellulose, the specimens were treated with NaClO2 solution at pH 4–5 at 70 °C, which is known as the Wise method (Wise et al. 1946). After repeating the process three times, the samples were boiled in 5% NaOH for 2 h and then washed with distilled water.
Cryptmeria japonica, Eucalyptus globulus, and sugarcane bagasse were subjected to cellulose extraction as described by Horikawa et al. (2013). After two-step milling using Orient mill VM-16 (Seishin, Tokyo, Japan) followed by Bantam mill AP-BL (Hosokawa Micron, Osaka, Japan), the products were subjected to the Wise method for lignin removal. The treatment was repeated and then samples were boiled in 5% NaOH for several hours. The cellulose samples obtained were washed with distilled water.
Cellulose was obtained from ramie as described by Sugiyama et al. (1991a).
Dewaxed cotton cellulose was purchased from Sogo Laboratory Glass Works (Kyoto, Japan).
Avicel PH-101, which is the commercial name of microcrystalline cellulose, was purchased from Asahi Kasei (Tokyo, Japan).
Nanocellulose was obtained from the purified cellulose from C. japonica. A cellulose suspension of approximately 0.8% was prepared and then passed through a grinder (Masuko, Saitama, Japan) at 1500 rpm as previously described (Abe et al. 2007).
Suspension-cultured cells induced from Nicotiana tabacum, known as BY-2 cells, were maintained in modified LS-medium (Nagata et al. 1981). Seven-day cultured cells were collected and subjected to the Wise method three times to remove proteins and lignin. The holocellulose obtained was treated with boiling 5% NaOH for 2 h and washed in distilled water for cellulose purification.
All purified cellulose samples were freeze-dried and molded to make 3 tablets from each source for measurement deviation. The tablets were made by collecting 10–40 mg of dehydrated cellulose and then hand-pressing it with a special die set (Kyoto Pastec, Kyoto, Japan) following a previously published protocol (Horikawa et al. 2011).
Intra-crystalline deuteration
Halocynthia roretzi cellulose was annealed in 0.1 N NaOD for 1 h at 210 °C, during which intra-crystalline deuteration reached completion (Nishiyama et al. 1999). The deuterated samples were rinsed in distilled water several times.
IR spectroscopy
IR spectra were obtained using a PerkinElmer Frontier system equipped with microscopic accessory (PerkinElmer, Waltham, MA, USA). Cellulose suspension was placed on the BaF2 window (13 mm diameter × 2 mm thickness) and dried completely. The spectra were recorded in the range of 4000–700 cm−1 using a low noise detector (HgCdTe) that was cooled to − 196 °C with a spectral resolution of 4 cm−1 and acquisition of 128 scans.
NIR spectroscopy
NIR spectra were recorded under air-dry conditions using a PerkinElmer Spectrum 100 N system equipped with integrated sphere diffuse reflectance accessory (PerkinElmer, Waltham, MA, USA). Cellulose tablets were put directly on the sampling window where it was irradiated from below. The spectra were obtained in the range of 10,000–4000 cm−1 employing a triglycine sulfate detector with a spectral resolution of 16 cm−1 and acquisition of 32 scans.
Multivariate analysis
PCA and partial least square (PLS) regression analysis, which correspond to multivariate analyses, were performed using commercial software (Unscrambler v.10.3; CAMO Software, Woodbridge, NJ). To obtain more structural information, the original NIR spectral data with 2 cm−1 of interval, which were interpolated using spectral analysis software, were subjected to multiplicative scatter correction (MSC) and then converted with the Savitzky–Golay second derivative (1964) using a fifth-order polynomial for smoothing before multivariate analysis. Both algorithms for PCA and PLS regression analysis were executed on NIR second derivative spectra. PC loading was obtained from the model built for score plots. PLS regression model was created using optimal factors determined by cross-validation (leave-one-out): one sample was excluded from the model, and then its characteristics were predicted by making a model without the sample. The coefficient of determination for calibration (R 2 c ) and the root mean square error of calibration (RMSEC) were applied for evaluation of calibration performance. The models created were assessed using the coefficient of determination of prediction (R 2 p ) and the root mean square error of prediction (RMSEP). A greater value of relative predictive determinant (RPD) reflects a more accurate fitting of data by calibration, and an RPD higher than 2.5 is considered satisfactory for screening (Williams and Sobering 1993).
Results and discussion
Optimal NIR region for cellulose crystal analysis suggested by intra-crystalline deuteration
When a cellulose sample is annealed in 0.1 N NaOD in D2O, all OH bonds involved in microfibril formation are replaced by OD bonds (Fig. 1a i, ii). After cool down, the deuterated cellulose is washed with water, where surface OD bonds are accessible and revert to OH bonds whereas OD bonds in the crystalline core remain unaltered (Fig. 1a iii) (Horikawa and Sugiyama 2008). This behavior was detected by IR spectroscopy as shown in the cellulose spectra of H. roretzi before and after intra-crystalline deuteration (Fig. 1b). Intra-crystalline deuteration was reflected in a series of shaper bands around 3400 cm−1, corresponding to OH stretching vibrations, shifting to 2400 cm−1, where OD stretching vibrations are detected. However, broad and small IR absorption regions were observed around 3600 cm−1, indicating that surface OD bonds reverted to OH bonds due to water access. This technique was applied to NIR spectra to determine OH stretching regions involved in cellulose crystal formation. Similar behavior can be seen in NIR spectra in which part of the overtones related to OH stretching vibrations in the range of 7000–6000 cm−1 disappeared and new bands appeared in 5200 cm−1 (Fig. 1c). A region with small absorption around 6900 cm−1 was observed after intra-crystalline deuteration, and should correspond to surface OH bonds by rehydration. In addition, combinational vibrations around 4800 cm−1 vanished after intra-crystalline deuteration, and these correspond to OH vibration during formation of the cellulose crystal. However, combinational vibrations include stretching and bending mode, which makes it difficult to assign the bands in light of the crystalline cellulose structure. NIR bands in the range of 6600–6200 cm−1 were, therefore, used for the following multivariate analysis.

a Schematic models of H. roretzi cellulose microfibrils (i) before and (ii) after intra-crystalline deuteration, and then (iii) washed with water where only surface OD bonds are replaced by OH bonds. b IR and c NIR spectra of H. roretzi cellulose before (black line) and after (red line) intra-crystalline deuteration. IR and NIR spectra after intra-crystalline deuteration correspond to model (iii) shown in a. (Color figure online)
Determination of structural variety of cellulose from various sources using PCA
To better understand the structural relationships among cellulose produced by various organisms, PCA score plots were computed based on NIR spectra of the relevant region. Original spectra were transformed to second derivatives before multivariate analysis to extract minor spectral features such as shoulder bands. Figure 2 exhibits score plots in PC1 (76%) and PC2 (19%) loading vectors, in which samples were clearly dispersed although some terrestrial plants and algae were clustered. Marine algae, tunicates, and bacterial cellulose localized in the left area, whereas higher plants, Gelidium, and Chara localized in right area. It is generally recognized that marine organisms generate large cellulose microfibrils, whereas higher plants produce small microfibrils of approximately 3–4 nm. Therefore, the PC1 vector might correspond to cellulose crystallinity. To verify this hypothesis, Fig. 3a shows the PC1 loading vector calculated with second derivative spectra of Boergesenia and Egeria (Fig. 3b). PC1 vector had three positive regions around 6527, 6454, and 6291 cm−1, all of which coincided with the negative bands observed in the second derivative spectra of Boergesenia. Additionally, almost no or small negative band was observed at 6300 cm−1 in Egeria cellulose. The comparison of spectra obtained from different cellulose crystallinity samples further supported this hypothesis.

Principal component analysis plot of the first and second principal components of second derivative NIR spectra in the range of 6600–6200 cm−1 of cellulose from different organisms. (Color figure online)

a PC 1 loading vector for PCA in the range of 6600–6200 cm−1 and b second derivative spectra of Egeria (red line) and Boergesenia (blue line), which synthesize high and low crystalline cellulose microfibrils, respectively. (Color figure online)
In the PCA score plots, terrestrial plants were ordered in left to right along PC 1 vector as follows: cotton, ramie, avicel, soft and hard wood, nanocellulose, grass, and cultured cells. Avicel can be prepared by removal of amorphous regions by chemical hydrolysis in microfibrils composing wood pulp, which shows a higher relative crystallinity. Grass is mainly constituted of parenchyma cells with the primary cell wall, and cultured cells also have only the thinner primary cell wall without lignification. Therefore, it is generally accepted that their cellulose crystallinities are lower than those of wood, which has a thick and lignified secondary cell wall. Interestingly, nanocellulose prepared from soft wood cellulose by grinder was clearly positioned to the right compared to its source. This distribution indicates that mechanical treatment decreases crystallinity, which is consistent with previously published reports (Hideno et al. 2016). To evaluate cellulose crystallinity, I performed X-ray diffractometry and estimated the full-width at half-maximum (FWHM) of the peak (200) presented in Table 1. FWHM is sometimes applied as an index of crystallinity and the values increase with lower crystalline cellulose. The FWHM values for terrestrial plants were mostly in accordance with their arrangement along the PC1 direction, suggesting that NIR spectroscopy successfully detected the molecular arrangements that form microfibrils.
In the PC2 direction, score plots show Glaucocystis, Acetobacter, Oocystis, Cladophora, Valonia, Boergesenia, and tunicates in order from top to bottom. This suggests that PC2 loading arranged cellulose samples based on allomorphs as shown in Table 1. Figure 4a shows PC2 loading vector with Glaucocystis and H. papillosa, each representative of cellulose Iα and Iβ, respectively (Fig. 4b). The bands in the PC2 loading vector at 6526 and 6446 cm−1 were negatively correlated, whereas shoulder bands at 6484 and 6383 cm−1 were positively correlated, with second derivative spectra. This can be interpreted as the former 2 bands being characteristic of Iα and the latter bands of Iβ, which coincided with negative bands in the second derivative spectra of Glaucocystis and H. papillosa, respectively. Fackler and Schwanninger (2010) have tentatively assigned bands at 6270 and 6340 cm−1 to Iα and Iβ, respectively, although these bands are different from those observed in this work. The characteristic bands in this study were determined using Iα-rich and Iβ-dominant cellulose samples, which is an improvement on previously published information.

a PC 2 loading vector for PCA in the range of 6600–6200 cm−1 and b second derivative spectra of Glaucocystis and H. papillosa, which synthesize Iα-rich and Iβ-dominant cellulose microfibrils, respectively. (Color figure online)
Interestingly, terrestrial plants seemed to localize in score plots as an extension of Ciona and Hallocynthia samples, which consist mainly of Iβ crystal units. Though it is difficult to determine the allomorph type in lower crystalline cellulose microfibrils, Wada et al. (1997) showed by synchrotron X-ray diffraction analysis that wooden cellulose microfibrils are Iβ type. Algae such as Micrasterias, Chara, and Glidium are reported to be of type Iβ (Kim et al. 1996; Koyama et al. 1997), and they localized close to terrestrial plants. In contrast, Erythrocladia cellulose also has lower crystallinity and consists of Iα units (Koyama et al. 1997), and localized outside the line from tunicate to terrestrial plants. Therefore, score plots built using NIR spectra obtained from various sources further indicate that terrestrial plants synthesize cellulose microfibrils comprised of monoclinic units.
Calibration model for relative cellulose crystallinity
Based on the score plots in the PCA described above, I created calibration models for cellulose crystallinity by employing PLS regression. Statistics of the FWHM values is summarized in Table 2, where a wide variance is shown in both the calibration and prediction sets for randomly selected samples. To obtain a better model, second derivative spectra in the rage of 6600–6200 cm−1 as well as other regions were obtained as follows: (1) 10,000–7500 cm−1, in which the second and third overtones were present; (2) 7500–6000 cm−1, where mainly OH overtone vibrations were detected; (3) 6000–5500 cm−1, where the CH and aromatic framework vibrations were detected; and (4) 5500–4000 cm−1, which corresponded to several combinational vibrations. Next, the optimum number of PLS factors to create the calibration model was determined to reduce the sum of squared residuals using the cross-validation algorithm. Although the sum of the residuals decreased as the number of PLS factors increased without any inflection point, too many numbers lead to overfitting, which results in unreliable models. Therefore, the number of PLS factors used in this study was fewer than 6.
These selected regions, including the full-length NIR range of 10,000–4000 cm−1, generated the PLS calibration models summarized in Table 3. The range of 6600–6200 cm−1 using 4 of PLS factor numbers produced a best model with an RMSEP of 0.273°, Rp 2 of 0.895, and RPD of 3.17 (Fig. 5). When using 5 PLS factors, regression performance became worse, with an RMSEP of 0.274°, Rp 2 of 0.860, and RPD of 2.74. The reduction in regression quality might be due to an overfitting problem. Table 3 also shows the regression performance of full length and other selected regions. With the exception of the model obtained for the NIR spectra ranging from 7500 to 6000 cm−1 and 5500–4000 cm−1, PLS regression calibrations showed poor performance, as all Rp 2 were < 0.790 and all RMSEP were > 0.387°. Combinational vibrations in the range of 5500–4000 cm−1 gave an acceptable calibration model with an RMSEP of 0.377°, Rp 2 of 0.800, and RPD of 2.29. Considering this achievement, the range of combinational vibrations also involved crystalline structural information, as the bands at 4800 cm−1 indicated in the intra-crystalline deuteration. The results in this study demonstrated that a broader range is not requisite to improve the model performance, and accordingly, the logical selection of the absorption range is an important process to create a more reliable regression model as a determination by the intra-crystalline deuteration.

Relationships between the full-width at half-maximum (FWHM) of peak at (200) (°) calculated by X-ray diffractometry and predicted by near infrared spectroscopy in the range of 6600–6200 cm−1. Black and red plots correspond to calibration and prediction data sets, respectively. (Color figure online)
Rambo and Ferreira (2015) recently showed that near infrared spectroscopy combined with multivariate analysis allowed the evaluation of cellulose crystallinity of banana residues using almost the full-length NIR region. Inagaki et al. (2010) have successfully reported a calibration model for cellulose crystallinity using several specific NIR absorption regions assigned to amorphous, semi-crystalline, or crystalline structure in the range of 7200–6100 cm−1. However, both published reports are limited to cellulose samples from terrestrial plants. This study demonstrates that short ranges such as 6600–6200 cm−1 allowed to estimate cellulose relative crystallinity from different organisms ranging from bacteria to higher plants as well as tunicates in Animalia.
Calibration model to determine the ratio of cellulose crystalline allomorph
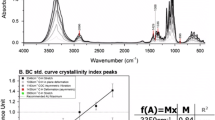
I also designed a standard curve to determine the crystalline ratio of allomorphs in cellulose. The available allomorph data set is insufficient, as only six samples with NMR and FTIR data were reported (Imai and Sugiyama 1998; Imai et al. 1999). I initially surveyed the bands of second derivative spectra in the range of 6600–6200 cm−1, which are summarized in Table 4. Bands around 6527 or 6383 cm−1 were characteristic of Iα and Iβ, respectively, and both were independent of allomorph ratios in the various samples. In contrast, the band observed around 6450 cm−1 shifted position depending on the allomorph ratio. The cellulose Iα fraction was therefore plotted against the corresponding band position in each cellulose sample, clearly indicating a linear correlation (Fig. 6). The estimated cellulose Iα ratio is calculated as shown below,
where W6476 – 6446 is the wavenumber of the band position present in the range of 6476–6446 cm−1 in the second derivative spectra. Nakashima et al. (2011) have described a calibration model for the Iα fraction, which was created using IR band shifts at 3345 cm−1 and assigned to O3–H···O5. The authors describe a moving range in IR spectroscopy as narrow as 3350–3340 cm−1, whereas in this study I describe a three times wider range of 6476–6446 cm−1. The observed wide range of band shifts is promising to easily determine the cellulose crystalline allomorph faction, and allows a rapid evaluation of unknown cellulose samples even in small amounts. IR or NIR band shift is a result of changes in atomic distances. Therefore, a band shift can be observed, for example, when the measurement temperature is changed (Altaner et al., 2014). Given this background, a change in allomorph ratio should not result in a band shift but in a change in absorbance of specific bands. Therefore, specific bands of Iα and Iβ are considered to be originally close to one another, and merging of these bands will appear as a shift with changed allomorph ratio.

Relationships between allomorph ratio and near infrared band shift ranging from 6476 to 6446 cm−1. Cellulose Iα fractions of standard samples are applied as shown in Table 1
The other cellulose samples were characterized in terms of crystalline allomorphism using the calibration curve created in this study (Table 5). Bacterial cellulose had a highly Iα type crystalline structure, which is consistent with a previous report (Attala and VanderHart 1984). The two samples from Rhodophyta (Erythrocladia and Gelidium) were rich in Iα and Iβ, which is in agreement with a published report by Koyama et al. (1997). The authors have discussed that the Iα domain in the primitive red algae Erythrocladia (Bangiophyceae) may be replaced by Iβ in a higher one such as Gelidium (Florideophyceae). In the case of Charophyta, it is well-known that Micrasterias produces flatter microfibrils (Kim et al. 1996), whereas Chara synthesizes smaller ones (Koyama et al. 1997). However, both crystalline types were reported to be of the Iβ type (Koyama et al. 1997), which is similar to the results estimated by the standard curve in this study. For higher plants, the calibration model based on NIR spectra demonstrated that their microfibrils are fully composed of Iβ allomorph, which is in agreement with previous reports (Wada et al. 1997) and with the discussion of PCA score plots mentioned above. In addition, the calibration curve indicated that Ciona microfibrils are predominantly composed of Iβ type crystalline units. Given that tunicates are generally recognized to synthesize Iβ type cellulose microfibrils, the results obtained for Ciona are as expected.
In this study, using structurally distinguishable cellulose samples, I have designed a distribution map in terms of crystalline features. Though NIR spectra lack sharper bands compared to IR spectroscopy, the combination with multivariate analysis and monitoring of band shifts provided calibration models, which allowed the quantitative estimate of cellulose structural properties. The score plots and calibration models obtained will be useful for understanding the structural characteristics of unclear cellulose samples such as chemically treated or artificially synthesized cellulose microfibrils.
Conclusions
I have demonstrated that the short NIR range of 6600–6200 cm−1, suggested by intra-crystalline deuteration, involves structural information for relative cellulose crystallinity and allomorphism. PCA of this region revealed the crystalline structure characteristics in a linear arrangement from tunicates to cultured cells, which supports the hypothesis that cellulose microfibrils synthesized by trees are composed of Iβ units. Furthermore, PLS regression allowed the quantitative evaluation of cellulose relative crystallinity in a wide range of organisms, from marine algae to cultured cells. Finally, I successfully designed a standard curve to estimate the ratio of Iα and Iβ by a simple detection of NIR band shifts. Further investigation of spectral bands combined with its measurement may allow the detection of other crystalline features such as the uniplanar orientation or shape of the microfibril cross section. NIR spectroscopy therefore has the potential for accurate and quantitative assessment of unknown cellulose samples, and provides a better understanding of the relationships among cellulose-synthesizing organisms in terms of crystalline structure.
References
Abe K, Iwamoto S, Yano H (2007) Obtaining cellulose nanofibers with a uniform width of 15 nm from wood. Biomacromolecules 8:3276–3278
Altaner CM, Horikawa Y, Sugiyama J, Javis MC (2014) Cellulose Iβ investigated by IR-spectroscopy at low temperatures. Cellulose 21(5):3171–3179
Atalla RH, Vanderhart DL (1984) Native cellulose—a composite of two distinct crystalline forms. Science 223:283–285
Belton PS, Tanner SF, Cartier N, Chanzy H (1989) High-resolution solid-state 13C nuclear magnetic-resonance spectroscopy of tunicin, an animal cellulose. Macromolecules 22:1615–1617
Fackler K, Schwanninger M (2010) Polysaccharide degradation and lignin modification during brown rot of spruce wood: a polarised fourier transform near infrared study. J Near Infrared Spectrosc 18:403–416
Hayashi N, Sugiyama J, Okano T, Ishihara M (1998) Selective degradation of the cellulose Iα component in Cladophora cellulose with Trichoderma viride cellulase. Carbohydr Res 305:109–116
Hideno A, Abe K, Uchimura H, Yano H (2016) Preparation by combined enzymatic and mechanical treatment and characterization of nanofibrillated cotton fibers. Cellulose 23:3639–3651
Horikawa Y, Sugiyama J (2008) Accessibility and size of Valonia cellulose microfibril studied by combined deuteration/rehydrogenation and FTIR technique. Cellulose 15:419–424
Horikawa Y, Imai T, Takada R, Watanabe T, Takabe K, Kobayashi Y, Sugiyama J (2011) Near-infrared chemometric approach to exhaustive analysis of rice straw pretreated for bioethanol conversion. Appl Biochem Biotech 164:194–203
Horikawa Y, Konakahara N, Imai T, Kentaro Abe, Kobayashi Y, Sugiyama J (2013) The structural changes in crystalline cellulose and effects on enzymatic digestibility. Polym Degrad Stabil 98:2351–2356
Horikawa Y, Imai M, Kanai K, Imai T, Watanabe T, Takabe K, Kobayashi Y, Sugiyama J (2015) Line monitoring by near-infrared chemometric technique for potential ethanol production from hydrothermally treated Eucalyptus globulus. Biochem Eng J 97:65–72
Horikawa Y, Imai T, Abe K, Sakakibara K, Tsujii Y, Mihashi A, Kobayashi Y, Sugiyama J (2016) Assessment of endoglucanase activity by analyzing the degree of cellulose polymerization and high-throughput analysis by near-infrared spectroscopy. Cellulose 23:1565–1572
Imai T, Sugiyama J (1998) Nanodomains of Iα and Iβ cellulose in algal microfibrils. Macromolecules 31:6275–6279
Imai T, Sugiyama J, Itoh T, Horii F (1999) Almost pure Iα cellulose in the cell wall of Glaucocystis. J Struct Biol 127:248–257
Inagaki T, Siesler HW, Mitsui K, Tsuchikawa S (2010) Difference of the crystal structure of cellulose in wood after hydrothermal and aging degradation: a NIR spectroscopy and XRD study. Biomacromolecules 11:2300–2305
Kim N-H, Hearth W, Vuong R, Chanzy H (1996) The cellulose system in the cell wall of Micrasterias. J Struct Biol 117:195–203
Koyama M, Sugiyama J, Itoh T (1997) Systematic survey on crystalline features of algal celluloses. Cellulose 4:147–160
Larsson PT, Westermark U, Iversen T (1995) Determination of the cellulose Iα allomorph content in a tunicate cellulose by CP/MAS 13C-NMR spectroscopy. Carbohydr Res 278:339–343
Nagata T, Okada K, Takebe I, Matsui C (1981) Delivery of tobacco mosaic-virus RNA into plant-protoplasts mediated by reverse-phase evaporation vesicles (Liposomes). Mol Gen Genet 184:161–165
Nakashima K, Nishino A, Horikawa Y, Hirose E, Sugiyama J, Satoh N (2011) The crystalline phase of cellulose changes under developmental control in a marine chordate. Cell Mol Life Sci 68:1623–1631
Nge TT, Sugiyama J (2007) Surface functional group dependent apatite formation on bacterial cellulose microfibrils network in a simulated body fluid. J Biomed Mater Res A 81A:124–134
Nishiyama Y, Isogai A, Okano T, Muller M, Chanzy H (1999) Intracrystalline deuteration of native cellulase. Macromolecules 32:2078–2081
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Nishiyama Y, Sugiyama J, Chanzy H, Langan P (2003) Crystal structure and hydrogen bonding system in cellulose Iα, from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 125:14300–14306
Rambo MKD, Ferreria MMC (2015) Determination of cellulose crystallinity of banana residues using near infrared spectroscopy and multivariate analysis. J Braz Chem Soc 26:1491–1499
Rodriguez-Saona LE, Fry FS, McLaughlin MA, Calvey EM (2001) Rapid analysis of sugars in fruit juices by FT-NIR spectroscopy. Carbohydr Res 336:63–74
Sarkar P, Bosneaga E, Auer M (2009) Plant cell walls throughout evolution: towards a molecular understanding of their design principles. J Exp Bot 60:3615–3635
Savitzky A, Golay MJE (1964) Smoothing+differentiation of data by simplified least squares procedures. Anal Chem 36:1627–1639
Sugiyama J, Vuong R, Chanzy H (1991a) Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 24:4168–4175
Sugiyama J, Persson J, Chanzy H (1991b) Combined infrared and electron-diffraction study of the polymorphism of native celluloses. Macromolecules 24:2461–2466
Tsekos I (1999) The sites of cellulose synthesis in algae: diversity and evolution of cellulose-synthesizing enzyme complexes. J Phycol 35:635–655
VanderHart DL, Atalla RH (1984) Studies of microstructure in native celluloses using solid-state 13C NMR. Macromolecules 17:1465–1472
Wada M, Okano T (2001) Localization of Iα and Iβ phases in algal cellulose revealed by acid treatments. Cellulose 8:183–188
Wada M, Sugiyama J, Okano T (1995) Two crystalline phase (Iα/Iβ) system of native celluloses in relation to plant phylogenesis. Mokuzai Gakkaishi 41:186–192
Wada M, Okano T, Sugiyama J (1997) Synchroton-radiated X-ray and neutron diffraction study of native cellulose. Cellulose 4:221–232
Williams PC, Sobering DC (1993) Comparison of commercial near infrared transmittance and reflectance instruments for analysis of whole grains and seeds. J Near Infrared Spectrosc 1:25–32
Wise L, Murphy M, D’Addieco A (1946) Chlorite holocellulose, its fractionation and beating on summative wood analysis and studies on the hemicelluloses. Pap Trade J 122:35–43
Acknowledgments
I would like to express my grateful appreciation to Dr. J. Sugiyama of Kyoto University for the cellulose samples. I also thank Dr. S. Mizuno-Tazuru and Dr. K. Abe of Kyoto University for their technical support. This study was supported by the Japan Society for the Promotion of Science (JSPS) (KAKENHI Grant Numbers 15K18723, 17H03840, and 17K19283).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Horikawa, Y. Assessment of cellulose structural variety from different origins using near infrared spectroscopy. Cellulose 24, 5313–5325 (2017). https://doi.org/10.1007/s10570-017-1518-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1518-0