
Abstract
The S100 protein family genes play a crucial role in multiple stages of tumorigenesis and progression. Most of S100 genes are located at chromosome locus 1q21, which is a region frequently rearranged in cancers. Here, we examined the expression of the S100 family genes in paired pancreatic ductal adenocarcinoma (PDAC) samples and further validated the expression of S100A16 by immunohistochemistry staining. We found that S100A16 is significantly upregulated in clinical PDAC samples. However, its roles in PDAC are still unclear. We next demonstrated that S100A16 promotes PDAC cell proliferation, migration, invasion, and metastasis both in vitro and in vivo. Knockdown of S100A16 induces PDAC cell cycle arrest in the G2/M phase and apoptosis. Furthermore, we also demonstrated that S100A16 promotes PDAC cell proliferation, migration, and invasion via AKT and ERK1/2 signaling in a fibroblast growth factor 19 (FGF19)-dependent manner. Taken together, our results reveal that S100A16 is overexpressed in PDAC and promotes PDAC progression through FGF19-mediated AKT and ERK1/2 signaling, suggesting that S100A16 may be a promising therapeutic target for PDAC.
Graphical abstract

S100A16 was upregulated in PDAC and associated with prognosis of PDAC patients.
S100A16 regulates apoptosis and the cell cycle of pancreatic cancer cells.
S100A16 promotes the progression of pancreatic cancer by AKT-ERK1/2 signaling.
S100A16 may be a promising therapeutic target for PDAC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a devastating disease, pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal causes of cancer-related disease. Due to its aggressive feature, most of PDAC patients are diagnosed at advanced disease stages, with a 5-year survival rate of less than 8% (Manji et al. 2017; Fang et al. 2017). Previous studies reported that many risk factors contribute to PDAC, such as smoking, alcohol drinking, adiposity, diabetes, pancreatitis, and some genetic factors (Iodice et al. 2008; Tramacere et al. 2010; Pang et al. 2019; Maisonneuve and Lowenfels 2015; Kirkegard et al. 2018). Though these traditional factors and some biomarkers helped us in understanding risk prediction and early diagnosis of PDAC, there are few treatment options for PDAC patients (Hessmann et al. 2017). Surgical resection and chemotherapeutics remain the mainstay of treatment for PDAC. Nevertheless, only approximately 10–20% of PDAC patients were diagnosed in the early stages and suitable for surgical resection. Moreover, these patients undergoing surgical resection have a very high rate of recurrence and only survive between 24 and 30 months (Riquelme et al. 2019; Gillen et al. 2010; Nevala-Plagemann et al. 2019). For patients diagnosed at advanced disease stages, because of metastasis presentation, chemotherapeutics are the mainstay option. However, chemotherapeutics resistance, low response rate, and toxic effect lead to only modestly extending survival (Halbrook and Lyssiotis 2017; Ding et al. 2019). Molecular-targeted therapy is used in multiple kinds of tumors while having a little positive impact on PDAC patients so far (Kumarasamy et al. 2019). On this occasion, more strenuous efforts are needed to understand the molecular pathology of PDAC and develop novel therapeutic strategies.
The S100 protein family is composed of small acidic proteins and plays a crucial role in vertebrate evolution. In response to calcium ions, S100 proteins regulate many processes of cellular biology by interacting with distinct proteins (Sturchler et al. 2006; Marenholz and Heizmann 2004). We examined the expression of the S100 genes in paired PDAC samples, and S100A16 was one of the most overexpressed genes in tumors compared to matched normal tissues. As a new member of the S100 proteins, S100A16 is highly conserved in mammals (Babini et al. 2011). Recently, S100A16 was reported as a prognostic marker in various kinds of cancer, such as lung adenocarcinoma, colorectal cancer, and prostate cancer. Overexpression of S100A16 in tumors associated with the growth and EMT of cancer cells (Chen and Liang 2018; Sun et al. 2018; Zhu et al. 2016). However, the potential functions and mechanisms of S100A16 in PDAC are still unclear. In this study, we demonstrate that S100A16 is highly expressed in PDAC and correlates with poor prognosis. Knockdown or knockout of S100A16 significantly attenuated the progression of PDAC via downregulating AKT and ERK1/2 pathways in an FGF19-dependent manner. S100A16 may provide a potential therapeutic target for PDAC.
Materials and methods
Gene expression profiling analysis
The expression of S100 family genes was obtained from TCGA, and the differential expression of S100A16 was verified by two different datasets GSE16515 and GSE15471 from the GEO database. The survival curve was obtained from the online database, GEPIA.
Plasmid and lentivirus
The lentiviral vector pLKO.1 TRC cloning vector (Addgene plasmid 10878) was used for constructing shRNA for S100A16. The restriction enzymes EcoR1 and Agel were used for cloning the sequence of S100A16 into the pLKO.1 empty vector. Two S100A16 shRNAs sequences used here were 5′-CAGCCTGGTCAAGAACAAGAT-3′ and 5'-CAGAACCTGGATGCCAATCAT-3'. For further silencing S100A16 in SW1990 and PANC1, S100A16-targeted small interfering RNAs (siRNAs) were ordered from GenePharma Company (Shanghai, China), and the ExFect®2000 Transfection Reagent was bought from Vazyme Company. The siRNAs for S100A16 (siS100A16-1, sense 5′-CCAAUCAUGAUGGGCGCAUCAGCUU-3′, antisense 5-AAGCUGAUGCGCCCAUCAUGAUUGG-3; siS100A16-2, sense 5′-CAGTCATTGTCCTGGTGGAAA-3′, antisense 5′-TTTCCACCAGGACAATGACTG-3′; scrambled siRNA (siNC), sense 5′-UUCUCCGAACGUGUCACGUTT-3′, antisense 5′-ACGUGACACGUUCGGAGAATT-3′) were synthesized. To construct cell lines stably overexpressing S100A16, the pLenti-CMV-GFP-Puro empty vector was purchased from Addgene. The coding area of S100A16 was amplified from SW1990 and then connected to the carrier. The cells were screened with puromycin (2 μg/mL) for 7 days, and the stable strain was obtained. The pLenti-CRISPR V2 vector (Addgene) was used to construct stable cell lines. The S100A16 has been knocked out. The sequences are as follows: sense 5′-CACCGGGAGAAGGCAGTCATTGTCC-3′, antisense 5′-CCCTCTTCCGTCAGTAACAGGCAAA-3′.
Quantitative real-time PCR
Total RNA was isolated from cells with TRIzol reagent (Tiangen), purified by trichloromethane. Then 500–1000 ng RNA was stably transcribed into cDNA using a RNA reverse transcription kit purchased from Abm. The expression of S100A16 was detected by a BIORAD Real-Time PCR system. The specific primers used here were as follows: S100A16 5′-TGGAGAGGAGGCAGACTGAG-3′ and 5′-CCACCAGGACAATGACTGC-3′; GAPDH 5′-ACCCAGAAGACTGTGGATGG-3′ and 5′-TTCAGCTCAGGGATGACCTT-3′.
Xenograft models
Four- to six-week-old BALB/c-nude male mice were purchased from Shanghai Vital River Laboratory Animal Technology Co., Ltd. Then, the mice were injected subcutaneously with 5 × 106 Aspc1 cells. To track the process of tumor formation, the size of the xenograft was measured once a week, and the tumor volume was calculated using the formula volume length × width 2/2. After 4 weeks, the tumor was surgically stripped off and weighed. Parts of the tumors were fixed by 4% paraformaldehyde overnight and immunohistochemistry assay was conducted. The remaining tissue was lysed for protein extraction for the western blot experiment.
Tail vein injection and metastasis models
For tail vein injection assay, the immunodeficient mice were also ordered from Vitalriver (Shanghai, China) and raised in a specific pathogen-free (SPF) Laboratory Animal Center at Nanjing Medical University. Two hundred microliters Aspc1 cell suspensions containing 2 × 106 cells were injected through the tail vein. After the mice were euthanized, the tumors were photographed and saved for subsequent experiments.
Statistical analysis
All the results represent three independent experiments, and all data are mean ± s.e.m. Statistical differences were calculated using the Student’s t test, and P < 0.05 was considered significant.
Results
S100A16 expression is significantly elevated in PDAC and correlates with poor prognosis
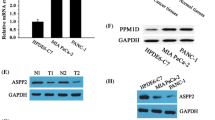
To investigate the potential role of the S100 family genes in PDAC development, we examined the expression of the S100 genes in paired PDAC samples, and S100A16 was one of the most overexpressed genes in tumors compared to matched normal tissues (Fig. 1a). Based on both The Cancer Genome Atlas (TCGA) and The Human Protein Atlas (HPA) datasets, the expression of S100A16 was shown significantly higher in most of PDAC tumor tissues but not/low expression in non-tumor pancreas tissues (Fig. S1). We next examined S100A16 expression in PDAC tissues. Immunohistochemical (IHC) staining of S100A16 in PDAC tissues and adjacent non-tumor pancreatic tissues revealed that S100A16 expression was elevated in PDAC tissues (Fig. 1b). Moreover, S100A16 protein expression level in PDAC cell lines was apparently higher than in the normal pancreatic cells (HPNE cells) (Fig. 1c). Consistently, S100A16 expression was also shown to be significantly upregulated in tumor samples compared to normal tissues in other PDAC datasets (GSE16515 and GSE15471, P < 0.0001) as well as the TCGA datasets. Patients with high S100A16 expression were associated with poor overall and disease-free survival compared to patients with low S100A16 expression (Fig. 1e). Taken together, these data verify that S100A16 expression was upregulated in PDAC and associated with the prognosis of PDAC patients.

S100A16 is highly expressed in PDAC tissues and pancreatic cancer cell lines. a The expression of S100 family genes in 60 pairs of PDAC tissue samples. b Representative hematoxylin and eosin and IHC staining for S100A16 in PDAC and adjacent noncancerous tissue (scale bar, 50 μm). The expression of S100A16 is higher in tumors than in adjacent tissues. c The expression of S100A16 is higher in pancreatic cancer cells than normal pancreatic cells. GAPDH was used as a control. d S100A16 is overexpressed in PDAC compared to noncancerous tissues in both GEO and TCGA databases (P < 0.05). e The overexpression of S100A16 in PDAC is significantly associated with poor overall and disease-free survival in the TCGA dataset based on the GEPIA2 analysis
S100A16 promotes growth and proliferation of PDAC cells
The upregulation of S100A16 in PDAC prompted us to investigate its potential functions in PDAC progression. Thus, the effects of S100A16 on the growth of PDAC cells were evaluated. As shown in Fig. 2a–c and Fig. S2a, b, knockdown of S100A16 expression by siRNA or shRNA significantly attenuated the proliferation of SW1990 or Aspc1 cells by performing trypan blue staining and CCK-8 assays. Similarly, in Fig. S2e, f, knockdown of S100A16 expression by siRNA effectively reduced the cell proliferation rate of KPC cells by CCK-8 assays. For better exploring of the biological functions of S100A16, we generated an S100A16-knockout cell line using the CRISPR/Cas9 system (Fig. 2d). Trypan blue staining and CCK-8 assays also revealed that knockout of S100A16 suppressed the growth of SW1990 cells (Fig. 2e, f). Consistent with these, stable overexpression of S100A16 in SW1990 and PANC1 cells by lentivirus dramatically promoted the growth of PDAC cells (Fig. 2g–i). Furthermore, a colony-forming assay was performed. As shown in Fig. 2j–l, knockout of S100A16 inhibited the ability of cell clone formation, while overexpression of S100A16 promoted this process. Taken together, our results demonstrate that S100A16 promotes the growth of PDAC cells in vitro.

S100A16 promotes growth and proliferation of PDAC cells. a Western blot analysis for S100A16 expression in the knocked down cell lines using two siRNAs. b, c The cell growth and proliferation capacity of S100A16 were tested by the trypan blue staining and CCK-8 assay. The knockdown of S100A16 reduced cell growth and proliferation of pancreatic cancer cells. d CRISPR/Cas9-mediated knockout of S100A16 in SW1990 cell line was detected by western blot. e, f Using the trypan blue staining and CCK-8 assay, S100A16 knockout in SW1990 cells significantly decreased the proliferation rate compared to S100A16+/+ cells. g Western blot assay shows the overexpression level of S100A16 in SW1990 and PANC1 cell lines. h, i S100A16 overexpression groups have a higher growth rate in SW1990 and PANC1 cell lines. j The colony formation ability was significantly reduced in S100A16 knocked out SW1990 cells. k, l The colony formation experiment was conducted in PANC1 and SW1990 cells which are overexpressed S100A16. (All data are mean ± s.e.m. **P < 0.01; ***P < 0.001)
S100A16 promotes migration and invasion of PDAC cells
Metastasis is one of the major obstacles to improve the outcome of PDAC patients. Thus, we want to evaluate whether S100A16 influences the process of PDAC metastasis. As shown in Fig. 3a, knockdown of S100A16 by siRNA significantly inhibited the migration and invasion of PANC1 cells. Knockout of S100A16 also obviously inhibited the migration of SW1990 cells, while re-expression of S100A16 in S100A16-knockout SW1990 cells restored the migration ability (Fig. 3b). EMT is a crucial step for cancer cell metastasis. To understand the potential mechanism of S100A16 on PDAC cell metastasis, expression levels of EMT markers, E-cadherin and vimentin were measured. As shown in Fig. 3c, the knockout of S100A16 led to the increased protein level of E-cadherin, while re-expression of S100A16 decreased E-cadherin expression. On the contrary, knockout of S100A16 led to the reduced protein level of vimentin, while re-expression of S100A16 restored vimentin expression. Besides, wound healing assay also revealed that knockout of S100A16 significantly inhibited PDAC cell migration (Fig. S2d). The knockdown of S100A16 by shRNA has a similar phenomenon in different PDAC cells (Fig. 3d and Fig. S2c). These results suggested that S100A16 promoted the migration and invasion of PDAC cells. To confirm that, we further overexpressed S100A16 in SW1990 and PANC1 cells and found that the upregulation of S100A16 dramatically promoted the migration and invasion of PDAC cells (Fig. 3e, f). Consistent with these, S100A16 suppressed the expression of E-cadherin while increased vimentin expression (Fig. 3g). Collectively, our data suggested that S100A16 facilitates migration and invasion of PDAC cells and possess their oncogenic phenotypes.

The influence of S100A16 on migration and invasion of PDAC cells. a The migration and invasion capacity of the cells decreased significantly in S100A16 silenced PANC1 cell lines. b Knockout of S100A16 can reduce the migration ability of SW1990 cells, and overexpression of S100A16 on this basis can restore the migration capacity. c EMT-related markers in SW1990 cells were detected by western blot. d The migration and invasion ability of the cells is slower in S100A16 knocked down PANC1 cell line. e, f Overexpression of S100A16 increased the migration and invasion capacity of PANC1 and SW1990 cell lines. g EMT-related markers in SW1990 cells with overexpression of S100A16 were detected by western blot. (All scale bars, 200 μm. **P < 0.01; ***P < 0.001)
Knockdown of S100A16 induces PDAC cell apoptosis and cell cycle arrest
To investigate the impact of S100A16 on cell survival, apoptosis was assessed by Annexin V/propidium iodide (PI) staining. As shown in Fig. 4a, b, knockdown of S100A16 in Aspc1 by shRNA increased the percentage of Annexin V+ cells, which suggested promoting cancer cell apoptosis. A similar phenomenon was found in PANC1 cells (Fig. 4c). In SW1990 cells, cell cycle analysis showed that knockout of S100A16 increased the percentage of cells in the G2/M phase, whereas reduced the rate of cells in the G1 phase (Fig. 4d). Taken together, these results demonstrate that S100A16 suppressed PDAC cells apoptosis and knockdown of S100A16 induced PDAC cell cycle arrest in the G2/M phase.

The knockdown of S100A16 affects the apoptosis and cell cycle of human PDAC cells. a The western blotting data confirmed the knockdown effects of shS100A16 in both Aspc1 and SW1990 cells. b, c Knockdown of S100A16 can increase the Annexin V+ apoptosis cell percentage of Aspc1 (b) and PANC1 (c) cell lines. d Cell cycle assay was carried out in SW1990 cell, and the cell cycle was blocked in the G2 phase in the S100A16 knockdown cells(KO-S100A16), Data are mean ± s.e.m. *P < 0.05; **P < 0.01;***P < 0.001
The functions of S100A16 in PDAC progression are partially through FGF19-mediated AKT and ERK signaling pathways
PI3K/Akt, MEK/ERK and Wnt/β-catenin signaling pathways are critical for PDAC progression. To elucidate the mechanism of S100A16 mediated PDAC progression, phosphorylated AKT (p-ATK), phosphorylated ERK1/2 (p-ERK1/2), and β-catenin protein levels were evaluated by Western Blot. As shown in Fig. 5a, overexpression of S100A16 in SW1990 and PANC1 cells dramatically promoted the phosphorylation of ATK and ERK1/2, while having little influence on the phosphorylation of JNK and expression levels of β-catenin. That is implying a dominant role for AKT and ERK1/2 signaling pathways in S100A16-mediated PDAC progression. To verify whether S100A16 promotes PDAC progression through modulation of the AKT and ERK signaling pathways, AKT and ERK1/2 were blocked by MK2206 and U0126, respectively. As shown in Fig. 5b–d, S100A16-mediated migration was significantly blocked by ATK inhibitor MK2206 in PANC1 cells. ERK1/2 inhibitor U0126 also eliminates S100A16-mediated migration in PANC1 and SW1990 cells. Moreover, knockout of S100A16 in SW1990 cells significantly inhibited the phosphorylation of ATK and ERK1/2, while re-expression of S100A16 in S100A16-knockout SW1990 cells restored these processes (Fig. 5e). Consistent with these, S100A16-mediated proliferation could be reduced by MK2206 and U0126 (Fig. 5f, g). Taken together, our results demonstrate that S100A16 promotes PDAC progression partially through AKT and ERK signaling pathways.

The functions of S100A16 in PDAC progression are partially through AKT and ERK signaling pathways. a The effects of S100A16 overexpression of the expression of different signaling pathways. Overexpression of S100A16 increases the phosphorylation level of AKT and ERK. b, c PANC1 cells transfected with Plenti-con and plenti-S100A16 were treated with DMSO and MK2206 inhibitor for AKT (b) or U0126 inhibitor for ERK (c), then the cell migration experiment was conducted. d SW1990 cells transfected with Plenti-con and plenti-S100A16 were treated with DMSO and U0126 inhibitor for ERK, then the cell migration experiment was conducted. e Knockout of S100A16 in the SW1990 cell line declines the phosphorylation level of AKT and ERK, but the re-expression of S100A16 restores the phosphorylation level of AKT and ERK. f, g SW1990 cells transfected with Plenti-con and plenti-S100A16 were treated with DMSO and MK2206 (f) and U0126 (g) in the SW1990 cell line then CCK-8 assay was carried out to test the proliferation ability. (Data are mean ± s.e.m. **P<0.01; ***P < 0.001;ns, not significant.)
To further explore how S100A16 regulates AKT and ERK signaling pathways, RNA-seq analysis was performed after S100A16 knockout in SW1990 cells to analyze the genes whose expression was regulated by S100A16. As shown in Fig. 6a, we found that the expression of FGF19 was dramatically decreased after S100A16 knockout. Since FGF19 was previously reported to interact with FGFR4 and then activates AKT and ERK signaling pathways (Liu et al. 2020), we further validated that S100A16 positively regulates the expression of FGF19 by QPCR and western blot assays (Fig. 6b, c, e, f). And when we knocked down FGF19 by siRNA in Aspc1 cells, the proliferation ability of cells was suppressed (Fig. 6g). We have proven that overexpressed of S100A16 obviously promoted the migration ability of SW1990 cells, while knocked down of FGF19 in S100A16-overexpressed SW1990 cells limited the migration ability (Fig. 6h). Moreover, when we knockdown FGF19 in PDAC cells, we also observed that the phosphorylation level of AKT and ERK was significantly inhibited (Fig. 6d). FGFR4 is one of the receptors of FGF19, and we found that the expression of both FGF19 and FGFR4 were overexpressed in PDAC cells and tumor tissues (Fig.S3a–c and Fig. 6i). Moreover, overexpression of FGF19 was significantly correlated with the expression of S100A16 and also associated with poor overall survival of PDAC patients (Fig. S3d, e). To further explore the function of FGF19 in pancreatic cancer, Aspc1 and SW1990 cells were starved and added FGF19 cell growth factor, and then the protein level of P-ERK and P-AKT was detected at different time points. As shown in Fig. 7a, b, Western Blot assays showed that as time and concentration increased, the protein level of P-ERK and P-AKT increased gradually. Furthermore, when we increased the concentration of FGF19 cell growth factor, trypan blue staining was used to detect the proliferation ability of Aspc1 cells on the fourth day, and it was found that there was a dose-dependent relationship between the number of cell proliferation and FGF19 cell growth factors (Fig. 7c). Different doses of FGF19 cell growth factor was added to S100A16-knockout Aspc1 cells; the proliferation ability of cells increased gradually (Fig. 7d). Similarly, a Transwell assay was used to test the migration ability of Aspc1 cells added with different doses of the FGF19 growth factor. As shown in Fig. 7e, with an increased dose of the FGF19 growth factor, the migration ability of Aspc1 cells has also been enhanced. Knockout of S100A16 obviously inhibited the migration of SW1990 cells, while added FGF19 growth factor in S100A16-knockout SW1990 cells restored the migration ability (Fig. 7f). Taken together, these results demonstrate that S100A16 can modulate AKT and ERK signaling pathways partially by regulating the expression of FGF19 in PDAC cells.

The result of RNA sequencing and validation shows that FGF19 obviously reduced in S100A16 knocked out cells. a The represent heatmap of dysregulated genes in RNA sequencing. b, c qPCR assays are carried out to detect the mRNA level of FGF19 in SW1990 and Aspc1 cells. d The knockdown of FGF19 significantly inhibits the phosphorylation level of AKT and ERK in SW1990 cells. e FGF19 is effectively decreased in S100A16 knocked out the SW1990 cell line. f FGF19 is significantly increased in S100A16 overexpressed PANC1 cell lines. g CCK8 assay was used to test the proliferation ability of FGF19 knocked down Aspc1 cells. h After overexpression of S100A16 in Aspc1 cells, a migration experiment was conducted to test the transferability of FGF19 knocked down Aspc1 cells. i The expression of FGF19 and FGFR4 in pancreatic cancer cells and normal pancreatic cells were detected by western blot. Data are shown as mean ±SEM. ***p<0.001

The influence of FGF19 cell growth factor on proliferation and migration of PDAC cells. a Western blot assay was used to detect the protein level of P-ERK and P-AKT in Aspc1 cells added a different dose of FGF19 cell growth factor. b Western blot assay was used to detect the protein level of P-ERK and P-AKT in SW1990 cells added a different dose of FGF19 cell growth factor. c CCK8 assay was used to test the proliferation ability of Aspc1 cells added different concentrations of FGF19 cell growth factor. d CCK8 assay was used to test the proliferation ability of S100A16-knockout Aspc1 cells. e The migration ability of Aspc1 cells was increased after adding the FGF19 cell growth factor. f Adding the FGF19 growth factor in S100A16-knockout SW1990 cells restored the migration ability. Data are shown as mean ±SEM. *p<0.05; **p<0.01; ***p<0.001
S100A16 promotes the growth and metastasis of PDAC in vivo
To explore the influence of S100A16 on the growth of PDAC in vivo, a subcutaneous xenograft tumor model was performed to study the tumorigenicity of S100A16-knockdown Aspc1 cells. As shown in Fig. 8a, b, knockdown of S100A16 in Aspc1 cells significantly inhibits tumor growth in vivo. To confirm that, we also generated an S100a16-knockdown stable KPC cell line and injected into wild-type C57BL/6 mice subcutaneously. As shown in Fig. 8c, compared with control tumors, knockdown of S100a16 in KPC cells inhibited tumor growth. TUNEL staining assay also revealed that knockdown of S100a16 in KPC promotes apoptosis in vivo (Fig. 8d). IHC staining of KPC xenograft tumors also observed that knockdown of S100A16 resulted in the reduction of p-ATK and p-ERK1/2 (Fig. 7e). To further investigated whether S100A16 was necessary for mediating the effect of metastasis in PDAC, a mouse tail vein injection assay was performed. As shown in Fig. 8f, g, knockdown of S100A16 in Aspc1 cells dramatically reduced lung metastasis. Taken together, these results demonstrate that S100A16 promotes the growth and metastasis of PDAC in vivo, which reveals that S100A16 might be a potential target for PDAC (Fig. 8).

S100A16 silencing suppresses tumor growth and metastasis of PDAC cells in vivo. a, b Representative images of Aspc1 subcutaneous tumor-bearing nude mice. Pictures of tumors stripped from nude mice and show that S100A16 knockdown decreases the tumor size. Knockdown of S100A16 decreases the tumor weight in nude mice injected with Aspc1 cells. Tumor volumes were measured once a week for a month. c Representative images of KPC subcutaneous tumor-bearing B6 mice. S100A16 Knockdown decreases the tumor weight in B6 mice, injected KPC cells. d The slices of the tumor are stained with TUNEL and DAPI then photographed. e Representative IHC staining for p-ERK1/2, p-AKT, and S100A16 in KPC tumor paraffin sections. f Lung metastasis was detected according to the number of metastatic nodules apparent on the lung surface of nude mice. g The representative images of H&E staining confirmed the lung metastasis inhibition in the shS100A16 group mice. (Data are mean ± s.e.m.*P<0.05; **P < 0.01; ***P < 0.001)
Discussion
Though enormous efforts have been made by researchers, the treatment outcome of PDAC is discouraging because of its high metastatic nature and poor prognosis (Law et al. 2019). There is an urgent requirement for novel therapeutic targets. As a unique calcium-binding protein, S100A16 was reported to be abnormally overexpressed in many kinds of cancer (Tomiyama et al. 2018). However, its potential roles in PDAC are still unclear. Thus, in this study, we aimed to investigate the function of S100A16 in PDAC. We found that S100A16 was highly expressed in PDAC compared to normal tissue, and its high expression was correlated to poor survival. High expression of S100A16 promoted PDAC cell growth, migration, and invasion while knockdown of S100A16-induced PDAC cell apoptosis and cell cycle arrest. These demonstrated the oncogenic roles of S100A16 in PDAC.
To explore the potential mechanism of S100A16-mediated PDAC progression, we first tested the activation status of some classical oncogenic signaling pathways by western blot. PI3K/AKT and MEK/ERK pathways are crucial for the development of human cancers. Previous studies also reported that PI3K/AKT and MEK/ERK pathways are constitutively active in PDAC. The PI3K/AKT pathway inhibits apoptosis and promotes cell proliferation when activated by various growth factors (Yamamoto et al. 2004; Luo et al. 2003). In parallel to the PI3K/AKT pathway, the MEK/ERK pathway activation triggered by RAS or RAF in PDAC contributes to the regulation of cell proliferation, differentiation, apoptosis, and epithelial-mesenchymal transition (EMT) (Garcia et al. 2014; Hu et al. 2018). We found that overexpression of S100A16 dramatically promoted the phosphorylation of AKT and ERK1/2. PI3K/AKT and MEK/ERK pathways were dysregulated in many human cancers (Riquelme et al. 2016; Cao et al. 2019; Jokinen and Koivunen 2015). The previous study also reported that S100A16 promoted human prostate cancer progression via AKT and ERK signaling pathways (Zhu et al. 2016). Thus, an RNA-seq was performed after S100A16 knockout in PDAC cells. Our results revealed that knockout of S100A16 dramatically reduced FGF19 expression, which could interact with FGFR4 and subsequently activate AKT and ERK signaling pathways (Gao et al. 2019). Aroosha Raja has found that the process of liver cancer can be regulated by FGF19 through AKT and ERK pathways (Raja et al. 2019). And Lixia Gao had proved that the protein levels of p-ERK1/2 and p-AKT were decreased when they knocked out FGF19, and the protein level can be restored after re-expression of FGF19 (Gao et al. 2019). Thus, our data demonstrated that S100A16 could regulate AKT and ERK signaling pathways by modulating the expression of FGF19.
AKT and ERK signaling pathways are critical oncogenic cascades for cancer cell growth, survival, motility, and invasion (Dhillon et al. 2007; Testa and Bellacosa 2001). As the first step for metastasis, EMT is also regulated by AKT and ERK signaling pathways (Yoo et al. 2011; Wang et al. 2018). In this study, we found that knockout of S100A16 significantly decreased the expression of vimentin while increasing the expression of E-cadherin. On the contrary, overexpression of S100A16 increased the expression of vimentin and reduced the expression of E-cadherin, which confirmed that S100A16 promotes the process of EMT. Besides, to ensure whether S100A16 induces PDAC progression via AKT and ERK signaling pathways, we used AKT and ERK inhibitors to block these two pathways. Our results revealed that S100A16-mediated migration and proliferation were blocked by these inhibitors, which suggested that S100A16 promotes PDAC progression in an AKT and ERK signaling pathway–dependent manner.
Conclusion
Our study revealed that S100A16 is abnormally overexpressed in PDAC, which in turn induces the expression of FGF19 and subsequently activates AKT and ERK signaling pathways. This ultimately promotes PDAC cell proliferation, migration and invasion. S100A16 may provide a potential candidate therapeutic target for PDAC.
References
Babini E, Bertini I, Borsi V, Calderone V, Hu X, Luchinat C, et al. Structural characterization of human S100A16, a low-affinity calcium binder. J Biol Inorg Chem: JBIC: a Publ Soc Biol Inorg Chem. 2011;16(2):243–56. https://doi.org/10.1007/s00775-010-0721-3.
Cao Z, Liao Q, Su M, Huang K, Jin J, Cao D. AKT and ERK dual inhibitors: the way forward? Cancer Lett. 2019;459:30–40. https://doi.org/10.1016/j.canlet.2019.05.025.
Chen LL, Liang C. Aberrant S100A16 expression might be an independent prognostic indicator of unfavorable survival in non-small cell lung adenocarcinoma. PloS ONE. 2018;13(5):e0197402. https://doi.org/10.1371/journal.pone.0197402.
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. https://doi.org/10.1038/sj.onc.1210421.
Ding L, Madamsetty VS, Kiers S, Alekhina O, Ugolkov A, Dube J et al. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR-mediated DNA damage response. Clin Cancer Res. 2019;25(21):6452–62. https://doi.org/10.1158/1078-0432.CCR-19-0799.
Fang C, Guo X, Lv X, Yin R, Lv X, Wang F, et al. Dysbindin promotes progression of pancreatic ductal adenocarcinoma via direct activation of PI3K. J Mol Cell Biol. 2017;9(6):504–15. https://doi.org/10.1093/jmcb/mjx043.
Gao L, Lang L, Zhao X, Shay C, Shull AY, Teng Y. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma. Oncogene. 2019;38(13):2394–404. https://doi.org/10.1038/s41388-018-0591-7.
Garcia MN, Grasso D, Lopez-Millan MB, Hamidi T, Loncle C, Tomasini R, et al. IER3 supports KRASG12D-dependent pancreatic cancer development by sustaining ERK1/2 phosphorylation. J Clin Invest. 2014;124(11):4709–22. https://doi.org/10.1172/jci76037.
Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7(4):e1000267. https://doi.org/10.1371/journal.pmed.1000267.
Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31(1):5–19. https://doi.org/10.1016/j.ccell.2016.12.006.
Hessmann E, Johnsen SA, Siveke JT, Ellenrieder V. Epigenetic treatment of pancreatic cancer: is there a therapeutic perspective on the horizon? Gut. 2017;66(1):168–79. https://doi.org/10.1136/gutjnl-2016-312539.
Hu J, Li L, Chen H, Zhang G, Liu H, Kong R, et al. MiR-361-3p regulates ERK1/2-induced EMT via DUSP2 mRNA degradation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9(8):807. https://doi.org/10.1038/s41419-018-0839-8.
Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbeck's Arch Surg. 2008;393(4):535–45. https://doi.org/10.1007/s00423-007-0266-2.
Jokinen E, Koivunen JP. MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol. 2015;7(3):170–80. https://doi.org/10.1177/1758834015571111.
Kirkegard J, Cronin-Fenton D, Heide-Jorgensen U, Mortensen FV. Acute pancreatitis and pancreatic cancer risk: a nationwide matched-cohort study in Denmark. Gastroenterology. 2018;154(6):1729–36. https://doi.org/10.1053/j.gastro.2018.02.011.
Kumarasamy V, Ruiz A, Nambiar R, Witkiewicz AK, Knudsen ES. Chemotherapy impacts on the cellular response to CDK4/6 inhibition: distinct mechanisms of interaction and efficacy in models of pancreatic cancer. Oncogene. 2019;39:1831–45. https://doi.org/10.1038/s41388-019-1102-1.
Law HC, Lagundzin D, Clement EJ, Qiao F, Wagner ZS, Krieger KL et al. The proteomic landscape of pancreatic ductal adenocarcinoma liver metastases identifies molecular subtypes and associations with clinical response. Clin Cancer Res. 2020;26(5):1065-76. https://doi.org/10.1158/1078-0432.CCR-19-1496
Liu Y, Cao M, Cai Y, Li X, Zhao C, Cui R. Dissecting the role of the FGF19-FGFR4 signaling pathway in cancer development and progression. Front Cell Dev Biol. 2020;8:95.
Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–62. https://doi.org/10.1016/s1535-6108(03)00248-4.
Maisonneuve P, Lowenfels AB. Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int J Epidemiol. 2015;44(1):186–98. https://doi.org/10.1093/ije/dyu240.
Manji GA, Olive KP, Saenger YM, Oberstein P. Current and emerging therapies in metastatic pancreatic cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. 2017;23(7):1670–8. https://doi.org/10.1158/1078-0432.ccr-16-2319.
Marenholz I, Heizmann CW. S100A16, a ubiquitously expressed EF-hand protein which is up-regulated in tumors. Biochem Biophys Res Commun. 2004;313(2):237–44. https://doi.org/10.1016/j.bbrc.2003.11.115.
Nevala-Plagemann C, Hidalgo M, Garrido-Laguna I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat Rev Clin Oncol. 2019;17:108–23. https://doi.org/10.1038/s41571-019-0281-6.
Pang Y, Holmes MV, Chen Z, Kartsonaki C. A review of lifestyle, metabolic risk factors, and blood-based biomarkers for early diagnosis of pancreatic ductal adenocarcinoma. J Gastroenterol Hepatol. 2019;34(2):330–45. https://doi.org/10.1111/jgh.14576.
Raja A, Park I, Haq F, Ahn SM. FGF19-FGFR4 signaling in hepatocellular carcinoma. Cells. 2019;8(6):536. https://doi.org/10.3390/cells8060536.
Riquelme E, Behrens C, Lin HY, Simon G, Papadimitrakopoulou V, Izzo J, et al. Modulation of EZH2 expression by MEK-ERK or PI3K-AKT signaling in lung cancer is dictated by different KRAS oncogene mutations. Cancer Res. 2016;76(3):675–85. https://doi.org/10.1158/0008-5472.can-15-1141.
Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178(4):795–806.e12. https://doi.org/10.1016/j.cell.2019.07.008.
Sturchler E, Cox JA, Durussel I, Weibel M, Heizmann CW. S100A16, a novel calcium-binding protein of the EF-hand superfamily. J Biol Chem. 2006;281(50):38905–17. https://doi.org/10.1074/jbc.M605798200.
Sun X, Wang T, Zhang C, Ning K, Guan ZR, Chen SX et al. S100A16 is a prognostic marker for colorectal cancer. 2018;117(2):275–83. https://doi.org/10.1002/jso.24822.
Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(20):10983–5. https://doi.org/10.1073/pnas.211430998.
Tomiyama N, Ikeda R, Nishizawa Y, Masuda S, Tajitsu Y, Takeda Y. S100A16 up-regulates Oct4 and Nanog expression in cancer stem-like cells of Yumoto human cervical carcinoma cells. Oncol Lett. 2018;15(6):9929–33. https://doi.org/10.3892/ol.2018.8568.
Tramacere I, Scotti L, Jenab M, Bagnardi V, Bellocco R, Rota M, et al. Alcohol drinking and pancreatic cancer risk: a meta-analysis of the dose-risk relation. Int J Cancer. 2010;126(6):1474–86. https://doi.org/10.1002/ijc.24936.
Wang K, Ji W, Yu Y, Li Z, Niu X, Xia W. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial-mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene. 2018;37(39):5340–54. https://doi.org/10.1038/s41388-018-0311-3.
Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res: Off J Am Assoc Cancer Res. 2004;10(8):2846–50. https://doi.org/10.1158/1078-0432.ccr-02-1441.
Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71(22):7061–70. https://doi.org/10.1158/0008-5472.can-11-1338.
Zhu W, Xue Y, Liang C, Zhang R, Zhang Z, Li H, et al. S100A16 promotes cell proliferation and metastasis via AKT and ERK cell signaling pathways in human prostate cancer. Tumour Biol: J Int Soc Oncodev Biol Med. 2016;37(9):12241–50. https://doi.org/10.1007/s13277-016-5096-9.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81902803, 82072739), Overseas Young Talents Project of China, Zhejiang Provincial Natural Science Foundation (No.LY21H160057) and Science and Technology Bureau of Wenzhou (Y20190069), “Innovative and Entrepreneurial Team” (No. (2018)2015), Science and Technology Grant (BE2019758), and the Natural Science Foundation (BK20190657) of Jiangsu Province, Southeast University-Nanjing Medical University Cooperative Research Project (2242018K3DN33), and Funding of “Peak” Training Program for ScientificResearch of Yijishan Hospital, Wannan Medical College(No.GF2019G19), as well as by China Postdoctoral Science Foundation (2019M651897) and Science and Technology Development Fund of Nanjing Medical University (NMUB2018015).
Author information
Authors and Affiliations
Contributions
DF and HX conceived and designed the experiments. DF, CZ, PX, YL, CH, AA, XM, and ZY performed the experiment and data analysis/bioinformatics analysis. YL, QS, GZ, and LL contributed clinical materials and some funding support. HX contributed to the coordination of the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Ethical approval was obtained from the medical/animal ethical committee of the Nanjing Medical University (China).
Consent to participate
Informed consent was obtained where applicable.
Consent for publication
All authors have read and agreed to the published version of the manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 1282 kb)
Rights and permissions
About this article

Cite this article
Fang, D., Zhang, C., Xu, P. et al. S100A16 promotes metastasis and progression of pancreatic cancer through FGF19-mediated AKT and ERK1/2 pathways. Cell Biol Toxicol 37, 555–571 (2021). https://doi.org/10.1007/s10565-020-09574-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-020-09574-w