
Abstract
By impregnating 3 wt% Cr into the hydrothermally synthesized silicalite-1 with an average crystal size of 0.06–0.35 μm, the effects of the silanol-group density on the dispersion, structural and electronic characteristics of Cr(VI) oxides are studied for the oxidative dehydrogenation of propane with carbon dioxide (CO2-ODP). Characterization results reveal that the CrOx species are highly dispersed over silicalite-1 irrespective of the crystal sizes, and the highest CrOx dispersion is achieved over silicalite-1 with an average crystal size of 0.15 μm due to its highest density of the silanol nests. Moreover, the textural properties and the relative quantity of the Cr(VI) oxides with different structures are strongly affected by the crystal size of the zeolite. The catalyst by using 0.15 μm silicalite-1 as the support shows the most stable performance for CO2-ODP indexed by the C3H6 yield, which is much superior to the remaining catalysts. The correlation of the characterization data of the fresh and representative spent catalysts with the reaction results rigorously reveals that the catalytic stability of CrOx/silicalite-1 for CO2-ODP is mainly determined by the dual functions of the deposited coke, which is basically originated from the interactions between Cr(VI) oxides and the support. These understandings on the stability of CrOx/silicalite-1 raised essentially from the density of silanol nests over silicalite-1 are beneficial for developing more efficient Cr-based catalysts for the non-oxidative & oxidative dehydrogenation of short alkanes including CO2-ODP.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The propane dehydrogenation (PDH), which characterizes the endothermic and equilibrium-limited nature in thermodynamics, has been industrialized for the production of propene. In recent years, the rising market demand on propene and the additional propane supply from shale gas stimulate renewed interests on PDH from the industrial domain [1, 2]. Moreover, the key issue for the PDH reaction is the selective activation of C-H bonds in propane molecules, which is one of the most important unsolved issues for the catalysis science. Thus, great effort has been put from both industrial and academic fields on the design and development of more efficient PDH catalysts [3]. To date, however, the commercial PDH process is still limited to CrOx- or Pt-based catalysts, which require very frequent regenerations due to the quick deactivation. In contrast with PDH, the oxidative dehydrogenation of propane with CO2 as a mild oxidant (CO2-ODP) demonstrates the alleviated thermodynamics limitation and the effective conversion of the greenhouse gas of CO2 to valuable CO [4, 5]. Kinetically, the development of an efficient catalyst for the selective activation of C-H bonds accompanying the simultaneous activation of C = O bonds in CO2 molecules is favorable for advancing the heterogeneous catalysis science. Thus, different catalytic systems for CO2-ODP including CrOx, VOx, and supported Pt are extensively investigated [4], which may accelerate the practical application of CO2-ODP alternative to PDH as a more economically competitive and green process for producing propene.
As a widely used commercial catalyst for a number of oxidation reactions, the supported CrOx also shows a relatively higher activity and a reasonably high selectivity of propene for CO2-ODP [5]. However, due to the complex structure of the supported CrOx and the insufficient structural information of the supported Cr(VI) oxides during and after the catalytic reaction, the key factors determining the activity and selectivity of the supported CrOx catalysts are far from clear for the oxidation reactions involving hydrocarbons such as PDH and CO2-ODP [6], which impedes the rational design and development of the Cr-based catalysts with an improved activity and stability. Considering the toxic concern of Cr species on the environment and the lower theoretical surface density for the monolayer of isolated CrOx (0.6 Cr atoms per square nanometer), zeolites or mesoporous oxides such as silica with a high surface area and distinct structure are extensively investigated as supports of CrOx for CO2-ODP [5]. In this respect, we found that highly dispersed CrOx stabilized by the unique silanol nests over the silicalite-1 zeolite with the MFI topology (CrOx/silicalite-1) can promote both the activity and stability, and the catalyst with as small as of 3 wt% Cr shows the best performance for CO2-ODP [7]. More importantly, the isolated Cr(VI) oxides are concluded to be less active, but more selective to propene, and more stable that polymeric Cr(VI) oxides for CO2-ODP, which are essentially derived from the different strengths of adsorbed propane and the subsequent desorption behavior of the formed propene on the specific Cr(VI) oxides [7]. Following this result, the initial quick deactivation and subsequent promoting effects from coking during the CrOx/silicalite-1 catalyzed CO2-ODP reaction is proposed, in which the polymeric Cr(VI) oxides and isolated Cr(VI) oxides are the key factors, respectively [7]. Regarding the silanol nest, its density over zeolites is reported to be affected by crystal sizes related to the synthesis procedures, desilication, and metal loading [8,9,10]. Thus, the relative content of Cr(VI) oxides with different structures and the stability of the catalyst are expectably regulated by tuning the crystal size of the silicalite-1 zeolite provided that the textural and structural properties are retained.
In this work, the density of silanol groups over silicalite-1 was regulated via changing the average crystal size of the zeolite from 0.06 to 0.35 μm, and its impact on the dispersion, structural and electronic characteristics of Cr(VI) oxides over CrOx/silicalite-1 was investigated for CO2-ODP. We found that the relative quantity of the isolated Cr(VI) oxides and the polymeric Cr(VI) oxides with different polymerization degrees over CrOx/silicalite-1 was strongly dependent on the average crystal size of silicalite-1, and at least a twofold enhancement of the catalytic stability indexed by the C3H8 yield was achieved over the optimal catalyst of CrOx supported on silicalite-1 with an average crystal size of 0.15 μm. Importantly, the relative deactivation rate of the catalyst for CO2-ODP was rigorously revealed to be mainly determined by the dual functions of the deposited coke, the extents of which are dependent on the average crystal size of the specific silicalite-1.
2 Experimental
2.1 Catalyst Preparation
The detailed procedure for synthesizing silicalite-1 zeolites was reported in our previous work [7], and the synthesis parameters were adjusted to regulate the crystal size. Typically, the desired content of tetraethyl orthosilicate (TEOS, 98%) and tetrapropylammonium hydroxide solution (TPAOH, 25%) purchased from Alfa Aesar and Sinopharm Chemical Reagent Co., Ltd., respectively, were mixed with the molar ratio of 5SiO2/2TPAOH/75H2O in a Teflon container under stirring for 1 h, after which the hydrothermal synthesis was performed at 180 °C for one day within a Teflon-lined stainless-steel autoclave. After that the precipitate was sufficiently washed with the distilled water, dried in the oven, and calcined at 550 °C for 20 h, the silicalite-1 zeolite with the average crystal size of 0.35 μm was obtained. To prepare silicalite-1 with a smaller average crystal size, conditions for the gel formation, the hydrothermal synthesis, and the final calcination were adjusted. In the cases of silicalite-1 with the average crystal sizes of 0.15 and 0.22 μm, 25SiO2/9TPAOH/480H2O and 25SiO2/6TPAOH/480H2O were mixed for 4 h, respectively, while the conditions for the hydrothermal synthesis (170 °C for 72 h) and the calcination (550 °C for 3 h) were kept the same to those for synthesizing 0.35 μm silicalite-1. For synthesizing silicalite-1 with the smallest average crystal size of 0.06 μm, the gel with the composition of 4SiO2/1TPAOH/32H2O was mixed at 80 °C for 24 h. After this, it was hydrothermally synthesized at 170 °C for 24 h, the final calcination at 550 °C for 3 h led to the silicalite-1 with the average crystal size of 0.06 μm. For simplicity and consistence, the thus-prepared silicalite-1 zeolites were labeled as silicalite-1-y, where y represents the average crystal size of the zeolite, i.e., y = 0.06, 0.15, 0.22, and 0.35 μm, respectively.
CrOx/silicalite-1 catalysts were prepared by impregnating the desired content of the chromium (III) nitrate (Cr(NO3)3·9H2O, 99%, Sinopharm Chemical Reagent Co., Ltd.) aqueous solution on silicalite-1-y zeolites via the wet impregnation method. After drying at 110 °C in the oven for 12 h, the calcination was conducted at 550 °C in the furnace for 3 h, leading to the CrOx/silicalite-1-y catalysts. The nominal loading of Cr over all of catalysts was kept at 3 wt%.
2.2 Catalyst Characterizations
X-ray diffraction (XRD) patterns of the parent zeolites and the supported catalysts were obtained on a Bruker D8 Advance diffractometer. The scanning from 5 to 60° was performed at a rate of 6°·min−1. The relative crystallinity of the silicalite-1 zeolite was estimated with the fitted peak areas from 22 to 25° (2θ), and the 100% relative crystallinity of silicalite-1–0.06 is designated as the reference. Scanning electron microscopy (SEM) images of silicalite-1 zeolites were collected with a Hitachi FESEM SU8220 microscope. The actual Cr content in the samples was measured on an inductively coupled plasma optical emission spectrometer (ICP-OES, Prodigy 7).
The isotherms for the N2 adsorption and desorption of the zeolites and catalysts were measured at 77 K on an instrument of Micromeritics ASAP 2460. Before the N2 adsorption and desorption, the sample was degassed at 300 °C for 12 h under vacuum. The Brunauer–Emmett–Teller (BET) method was applied to determine the specific surface area of the catalysts, and the Barrett-Joyner-Halenda (BJH) model was employed to estimate the pore-size distributions. To compare the porous properties of the materials, the t-plot method was used to evaluate the surface area and volume of the micropores.
In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) experiments were conducted on a Thermo Scientific Nicolet iS50 spectrometer equipped with a diffuse reflectance cell (PIKE, DiffuseIR™). After the dehydration of the sample at 400 °C for 1 h, the spectrum was scanned from 3000 to 4000 cm−1 at 300 °C.
Temperature programmed reduction by hydrogen (H2-TPR) was performed using a Micromeritics Autochem 2920 instrument equipped with a thermal conductivity detector (TCD). Before the reduction process, the catalysts were fully pretreated at 400 °C under a flowing Ar. The H2-TPR profiles with the TCD signal were obtained by increasing the temperature from 100 to 800 °C under a flow of 10 vol.% H2/Ar.
In situ ultraviolet–visible diffuse reflectance spectroscopy (UV–Vis DRS) test was measured on the PerkinElmer spectrophotometer of Lambda 950. The dry helium was applied to dehydrate the sample at 400 °C in the DRS cell (Harrick, HVC-DR2), and then the spectrum of the dehydrated sample was scanned from 220 to 800 nm.
Ultraviolet Raman (UV-Raman) spectrum of the fresh catalyst in the region of 400–1300 cm−1 was collected on an instrument (HORIBA LabRAM HR Evolution) with an excitation source laser beam of 325 nm. In the cases of the spent catalysts, Raman shifts between 1200 to 1800 cm−1 were acquired with the excitation source laser beam of 532 nm.
The temperature programmed desorption of propane or propene (C3H8-TPD or C3H6-TPD) was measured on the same instrument with that of the H2-TPR experiments. After dehydrating at 400 °C in a flow of helium and saturating at 100 °C with C3H8 or C3H6, the TPD experiments under a flowing helium were carried out with increasing the temperature to 600 °C.
Thermogravimetric and differential scanning calorimetry (TG-DSC) was characterized for the spent catalysts on an instrument of NETZSCH STA 449F3. After dehydrating the sample at 100 °C for 1 h, TG-DSC was conducted until 800 °C in a flow of the air.
Temperature programmed oxidation of oxygen (O2-TPO) profiles were also measured on the instrument of Micromeritics Autochem 2920. After pretreating the used catalysts at 400 °C in a flow of argon for 30 min, the temperature was decreased to 100 °C. Then, 3 vol.% O2/Ar was switched. Finally, O2-TPO was performed from 100 to 800 °C, and the products (CO2 and CO) were monitored by the mass spectrometry (MS, HPR QIC-20).
2.3 Catalytic Reactions
The CO2-ODP reaction was tested in a quartz-tube reactor (i.d. 6 mm). 0.5 g of catalyst with particle sizes between 0.30 and 0.45 mm was packed into the isothermal zone of the reactor. Before starting the catalytic reaction, the fresh catalyst was heated to 550 °C in an Ar flow, and pretreated for 15 min. After this procedure, the feed gas (25 mL min−1) with the volumetric ratio of 20CO2/4C3H8/1Ar was well-mixed for 15 min, then immediately switched into the tube. The separated gaseous products were analyzed via an online GC-9560 instrument.
The conversions of C3H8 and CO2, C3H6 selectivity, and C3H6 yield were calculated using the following equations.
where, F is the flow rate of the gaseous products, which is calculated by using 4 vol.% Ar as the internal standard.
The relative deactivation rate of the catalyst for CO2-ODP was calculated from the C3H6 yield at the beginning (5 min, Y5) and end (305 min, Y305) of the reaction as shown in the following equation.
3 Results and Discussion
3.1 Physical Characteristics
As indicated from the XRD results (Figure S1a), all of the synthesized zeolites show the distinct diffractions of silicalite-1 at 2θ of 7.9, 8.8, 23.0, 23.9, and 24.5°, indicating the well crystallized MFI structure [10]. After loading Cr species, the MFI structure is still well preserved over the CrOx/silicalite-1 catalysts. Moreover, the absence of diffractions assigned to crystalline Cr2O3 indicates the high dispersion or amorphous nature of Cr species on silicalite-1. To give an estimate on the crystallinity of silicalite-1 with different average crystal sizes, the relative crystallinity was calculated by using silicalite-1–0.06 as a reference (100%). From the results given in Table 1, silicalite-1–0.15, silicalite-1–0.22, and silicalite-1–0.35 show quite similar relative crystallinities of about 85% (84–87%), which are slightly lower than that of silicalite-1–0.06 as the reference. This may be caused from the presence of some tiny crystals on the rim or surface of silicalite-1–0.15, silicalite-1–0.22, and silicalite-1–0.35, which can be clearly seen from the SEM images (Fig. 1). This explanation is supported by the further decreased but very comparable relative crystallinity of the zeolite for all of the CrOx/silicalite-1-y catalysts (65–72%, Table 1), the CrOx oxides of which are covered over the zeolite. From the crystal-size distributions (a-3 to d-3, Fig. 1), the average crystal size is determined to be ~ 0.06, ~ 0.15, ~ 0.22, and ~ 0.35 μm for silicalite-1–0.06, silicalite-1–0.15, silicalite-1–0.22, and silicalite-1–0.35, respectively.

SEM images with different magnitudes and the crystal-size distributions for silicalite-1–0.06 (a-1 to a-3), silicalite-1–0.15 (b-1 to b-3), silicalite-1–0.22 (c-1 to c-3), and silicalite-1–0.35 (d-1 to d-3)
According to the N2 adsorption/desorption patterns (Figures S1b1 to S1b4), both the silicalite-1-y and the catalysts show type I isotherms characterizing the microporous nature of the materials. Furthermore, an H4 hysteresis loop is observed at a P/P0 of about 0.85–0.99 especially for silicalite-1–0.06 and silicalite-1–0.15, revealing the presence of mesopores. This can be well interpreted as the agglomerates of silicalite-1 crystals as reported in references such as [11]. If the calculated pore-size distributions (Figure S1c) and textural data (Table 1) are examined, a more direct comparison can be made. Indeed, although all of the silicalite-1 zeolites show almost the same BET specific surface area (386–392 m2 g−1), the microporous surface area, the external surface area, and the pore volume are dependent on the average crystal size of silicalite-1. Specifically, the highest total pore volume and external surface area versus the smallest microporous surface area are observed for silicalite-1–0.06. This is consistent with the highest mesoporous volume of silicalite-1–0.06, which may be formed from the agglomerate of silicalite-1 with the smallest average crystal size of 0.06 μm. This explanation is supported from the continuously decreased mesoporous volume and the average pore size with increasing the average crystal size of silicalite-1 from 0.06 to 0.35 μm (Figure S1c and Table 1). In contrast, as a result of the largest average crystal size, silicalite-1–0.35 shows the smallest mesoporous volume and the average pore size. In the cases of silicalite-1–0.15 and silicalite-1–0.22, very comparable textural properties are observed (Table 1), which may be explained as the small difference in their average crystal sizes. As a result of the slightly higher mesoporous volume, larger total pore volume and the average pore size are obtained for silicalite-1–0.15.
When the catalysts are concerned, the loading of CrOx over silicalite-1–0.06 leads to a sharp decrease of the mesoporous volume from 0.39 to 0.06 cm3 g−1, which is consistent with the significant decrease of the average pore size. Accordingly, the total pore volume and the external surface area of CrOx/silicalite-1–0.06 are appreciably decreased. This can be well explained as that the loaded CrOx may mainly be located in the mesopores of the agglomerated silicalite-1–0.06, which is supported from the pore-size distributions (Figures S1c and S1d). In contrast, the most prominent observation for CrOx/silicalite-1–0.15 compared to silicalite-1–0.15 is the drastic decrease of the microporous surface area together with the microporous volume. However, the mesoporous volume is slightly increased from 0.18 cm3 g−1 for silicalite-1–0.15 to 0.21 cm3 g−1 for CrOx/silicalite-1–0.15. This indicates that the pore blocking in silicalite-1–0.15 occurs by the loaded CrOx species, which is supported by the narrower pore-size distribution and smaller peak pore size of CrOx/silicalite-1–0.15 than those of silicalite-1–0.15. When CrOx/silicalite-1–0.22 and CrOx/silicalite-1–0.35 are compared with the corresponding zeolite, the same changing trend of the calculated textural parameters to that of CrOx/silicalite-1–0.15 is ascertained although the extent is less significant.
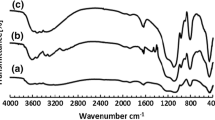
To understand the size effect of silicalite-1 on the anchoring of CrOx, silicalite-1-y was studied by in situ DRIFTS in the hydroxyl stretching region of 3000–4000 cm−1. From the spectra given in Fig. 2, two major absorptions are observed for all of silicalite-1-y samples. According to previous reports such as the reference [12], the sharp peak at ~ 3730 cm−1 and the broad absorption centered at about 3520 cm−1 can be assigned to the isolated terminal silanol and the silanol nest, respectively. The simple integration reveals that the peak intensity of silanol nests is increased in the sequence of silicalite-1–0.35 < silicalite-1–0.06 ≈ silicalite-1–0.15 < silicalite-1–0.22. However, silicalite-1–0.15 shows the highest intensity ratio of the silanol nest to the isolated terminal silanol, indicating the richest silanol nests. Considering the stronger interaction between CrOx and silanol nests than that between the isolated terminal silanol [12], the highest CrOx dispersion is expected over CrOx/silicalite-1–0.15, which is apparently supported from the calculated results of the pore-size distribution and pore volume. Thus, the amount of the silanol nest and the isolated terminal silanol is affected by the crystal size of silicalite-1, and the richest silanol nests are obtained over silicalite-1–0.15.

In situ DRIFTS spectra of silicalite-1-y zeolites
Thus, due to the varied extents of the agglomeration of silicalite-1 with different crystal sizes, the textural properties of both silicalite-1-y and CrOx/silicalite-1-y are altered. Moreover, the content of different silanol groups over silicalite-1 is regulated by changing its crystal size, and the highest CrOx dispersion over silicalite-1–0.15 is expectable as a result of the richest silanol nests over silicalite-1–0.15.
3.2 Size Effect of Silicalite-1 on the Structural Properties of CrOx
Because the number of Cr(VI) oxides in the Cr-based catalysts is recognized to be the crucial factor in determining the catalytic activity of CO2-ODP while the Cr(III) oxides in the forms of crystalline Cr2O3 show almost no activity [4], the reduction behavior of Cr species over the CrOx/silicalite-1-y catalysts was firstly probed by the H2-TPR method. As shown in Fig. 3, all of the samples show a prominent peak at about 430 °C and a small shoulder at around 280 °C, corresponding to the reduction of isolated and polymeric Cr(VI) oxides, respectively [7]. Thus, polymeric Cr(VI) oxides coexist with the isolated Cr(VI) oxide over all of the catalysts although the isolated Cr(VI) oxide is overwhelming among the Cr(VI) oxides irrespective of the catalysts. This result is further supported by the quantitative results for the calculated amount of the isolated and polymeric Cr(VI) oxides in Table 2. In the case of isolated Cr(VI) oxides, the reduction temperature is almost gradually increased with increasing the size of silicalite-1 from 0.06 to 0.35 μm, leading to the highest reduction temperature of ~ 432 °C for CrOx/silicalite-1–0.35. Moreover, very similar changing trend is observable for the reduction of polymeric Cr(VI) oxides. These results clarify the varied interactions of the CrOx oxides with the silicalite-1 support as well as different polymerization degrees of Cr(VI) oxides.

H2-TPR profiles of the CrOx/silicalite-1-y catalysts
To quantify the amount of Cr(VI) oxides in CrOx/silicalite-1-y catalysts, the H2 consumption based on the H2-TPR pattern is firstly calculated. As shown in Fig. 3, the CrOx/silicalite-1–0.15 catalyst consumes a clearly higher amount of H2 (0.57 mmol g−1) than the remaining catalysts showing very similar H2 consumptions (0.42–0.48 mmol g−1). Since that the amount of the reduced Cr(VI) oxides is directly proportional to the H2 consumption and the Cr content over all of the catalysts is essentially the same within analysis errors (2.72 ± 0.04 wt%, Table 2), this result suggests that the dispersion of Cr(VI) oxides over silicalite-1–0.15 is higher than those over the other silicalite-1 zeolites, the speculation of which in Sect. 3.1 is confirmed.
As revealed from our recent work [7], the differences in the activity, selectivity, and stability of CO2-ODP catalyzed by CrOx/silicalite-1 are closely related to the relative content of isolated and polymeric Cr(VI) oxides. Thus, to further study the effect of silicalite-1 size on the fraction of different structured Cr(VI) oxides, the obtained H2-TPR profiles were fitted into two Gaussian peaks, and the corrected amounts of isolated and polymeric Cr(VI) oxides are calculated from the content of consumed H2 with the assumption of the limited reduction of Cr(VI) to Cr(III) oxides during the H2-TPR test. From the calculated data in Table 2, the corrected amounts of isolated and polymeric Cr(VI) oxides over different CrOx/silicalite-1-y catalysts are dependent on the silicalite-1 size, the maximum of which are observed for the CrOx/silicalite-1–0.15 and CrOx/silicalite-1–0.22 catalysts, respectively. These results clearly indicate that the relative content of Cr(VI) oxides with different structures is indeed regulated by tuning the crystal size of the silicalite-1 zeolite.
From the results of in situ UV–Vis DRS under anhydrous conditions (Fig. 4a), two main peaks at ~ 240 and 350 nm attributed to isolated Cr(VI) oxides [13, 14] and one peak at around 460 nm characterizing the polymeric Cr(VI) oxides with different degrees of polymerization [15] are observed for all of the catalysts. Specifically, the peak intensity for the polymeric Cr(VI) oxides over CrOx/silicalite-1–0.22 is clearly higher than those over other catalysts, revealing the maximum content of the polymeric Cr(VI) oxides, consistent with the quantitative results from H2-TPR. Additionally, a weak peak at around 600 nm assigned to crystalline Cr2O3 is found for CrOx/silicalite-1-y except CrOx/silicalite-1–0.15. This suggests the higher dispersion of CrOx over silicalite-1–0.15, which is agreeable with the H2-TPR results.

In situ UV–Vis DRS (a) and UV-Raman spectra (b) of the CrOx/silicalite-1-y catalysts
To further reveal the crystal-size effect of silicalite-1 on the structures of Cr(VI) oxides, the UV-Raman test was conducted. According to the reference [16], the spectra given in Fig. 4b are deconvoluted into four peaks. The two distinctive bands at ~ 1070 and ~ 1150 cm−1 are corresponded to the transverse-optical asymmetric stretches and the longitudinal-optical silica network from silicalite-1, and the two intense bands at ~ 980 and ~ 1000 cm−1 are originated from the isolated dioxo and mono-oxo Cr(VI) oxides, respectively [16]. Moreover, the ratio of the intensity for the dioxo Cr(VI) oxide to that for the mono-oxo Cr(VI) oxide (Idioxo/Imono-oxo) is calculated as an estimate for the relative content of the isolated Cr(VI) oxides with different structures. From the results shown in Table 2, Idioxo/Imono-oxo is decreased in the order of CrOx/silicalite-1–0.22 (2.36) > CrOx/silicalite-1–0.15 (2.12) > CrOx/silicalite-1–0.06 (1.98) > CrOx/silicalite-1–0.35 (1.69). According to the references such as [14], the reduction of dioxo Cr(VI) oxide is easier than that of the mono-oxo Cr(VI) oxide. Thus, as a result of the lowest Idioxo/Imono-oxo of CrOx/silicalite-1–0.35, the highest reduction temperature for the isolated Cr(VI) oxide is observed in the H2-TPR results. These results indicate the important effect of the crystal size of silicalite-1 on the structure of the isolated CrOx because that both the Cr loading (2.72 ± 0.04 wt.%, Table 2) and the specific BET surface area of silicalite-1 (386–392 m2·g−1, Table 1) are almost identical.
Thus, the crystal size of silicalite-1 has a strong impact on the structure of CrOx over the catalysts. The dispersion of Cr(VI) oxides over silicalite-1–0.06, silicalite-1–0.22, and silicalite-1–0.35 is similar, which is lower than that over silicalite-1–0.15. Both polymeric Cr(VI) oxides and isolated Cr(VI) oxides are present over all of the catalysts although the isolated Cr(VI) oxide is the main component. In the case of the polymeric Cr(VI) oxide, its content over CrOx/silicalite-1–0.22 is the highest among the studied catalysts. When the isolated Cr(VI) oxides are concerned, the maximum concentration is obtained over CrOx/silicalite-1–0.15 and Idioxo/Imono-oxo is the lowest for CrOx/silicalite-1–0.35. In the case of crystalline Cr2O3, its contents over CrOx/silicalite-1–0.06, CrOx/silicalite-1–0.22 and CrOx/silicalite-1–0.35 are very limited while it is not detected over CrOx/silicalite-1–0.15.
3.3 Propane Adsorption and Propene Desorption Properties
As revealed from our recent work [7], the adsorption strength of propane and the subsequent desorption behavior of the formed propene on the specific Cr(VI) oxides are crucial in determining the initial activity and selectivity of the Cr-based catalysts for CO2-ODP. To study the crystal-size effect of silicalite-1 on the behaviors for the adsorption of propane and the desorption of propene on CrOx/silicalite-1-y catalysts, C3H8-TPD and C3H6-TPD experiments were conducted. Following the same method as illustrated in our recent work [7], the broad TPD profiles (Fig. 5) are deconvoluted into three peaks for the weak (peaks I and α), moderate (peaks II and β), and strong (peaks III and γ) adsorption of propane and propene, respectively. Moreover, the calculated amounts of the desorbed propane and propene are summarized in Tables S1 and S2. Irrespective of the catalysts, the amount of weakly adsorbed propane or propene is very similar, and is much lower than those of the medium or strongly adsorbed propane or propene. Moreover, a general trend for the amount of the medium or strongly adsorbed propane or propene over the catalysts is firstly increased and then decreased with increasing the crystal size of silicalite-1 from 0.06 to 0.35 μm, leading to the highest amount of the medium or strongly adsorbed propane or propene over CrOx/silicalite-1–0.22. Since that the polymeric Cr(VI) oxide can adsorb propane or propene more strongly than the isolated Cr(VI) oxide over CrOx/silicalite-1 catalysts [7], and the changing manner of propane or propene adsorption is similar to the variation for the relative content of the different Cr(VI) oxides. Thus, the crystal size of silicalite-1 has a clear effect on the desorption properties of propane and propene as a result of the impacted structural properties of CrOx over silicalite-1-y.

Profiles of C3H8-TPD (a) and C3H6-TPD (b) over CrOx/silicalite-1-y catalysts
3.4 Catalytic Performance
The time-on-stream (TOS) results of CO2-ODP over CrOx/silicalite-1-y at 550 °C are given in Fig. 6. The initial conversions of C3H8 (Fig. 6a) and CO2 (Fig. 6b) at a TOS of 5 min are concurrently increased in the order of CrOx/silicalite-1–0.35 ≈ CrOx/silicalite-1–0.15 < CrOx/silicalite-1–0.06 < CrOx/silicalite-1–0.22. In contrast, the initial C3H6 selectivity at a TOS of 5 min is increased in the sequence of CrOx/silicalite-1–0.22 < CrOx/silicalite-1–0.15 < CrOx/silicalite-1–0.06 < CrOx/silicalite-1–0.35 (Fig. 6c). As a result, an increased order of CrOx/silicalite-1–0.15 < CrOx/silicalite-1–0.35 ≈ CrOx/silicalite-0.06 ≈ CrOx/silicalite-1–0.22 is observed for the initial C3H6 yield (Fig. 6d). These results indicate that both the catalytic activity and the initial C3H6 selectivity are slightly affected by the crystal size of silicalite-1. Noteworthy, with increasing the crystal size of silicalite-1 from 0.06 to 0.35 µm, the changing pattern of the initial catalytic activity coincides with that for the amount of the medium or strongly adsorbed C3H8 while the varying sequences for the C3H6 selectivity and the amount of the medium or strongly adsorbed C3H6 are in an inverse order (Sect. 3.3, Tables S1 and S2). Thus, the impact of the crystal size of silicalite-1 on the initial catalytic activity and the C3H6 selectivity for CO2-ODP can be directly explained based on the desorption of C3H8 and C3H6 over the CrOx/silicalite-1 catalysts, which are essentially induced from the varied contents of CrOx with different structures.

The time-on-stream conversions of C3H8 (a) and CO2 (b), the C3H6 selectivity (c), and the C3H6 yield (d) over the CrOx/silicalite-1-y catalysts for CO2-ODP under the conditions of 0.5 g catalyst, T = 550ºC, CO2: C3H8: Ar = 20: 4:1, a total flowrate = 25 mL·min−1 (The experimental results within 5% errors are reproducible from the parallel experiments for three times)
When the catalytic stability is concerned, the conversions of C3H8 and CO2 are quickly decreased within the initial period of about 120 min, and are almost levelled off until the end of the reaction at a TOS of 305 min, the extent of which is closely dependent on the specific catalyst. In contrast, the C3H6 selectivity over all of the catalysts is rapidly increased until a TOS of around 120 min, and is kept almost constant until the end of the test. These results agree well with our proposed two-stage reaction mechanism for CO2-ODP over CrOx/silicalite-1 catalysts with the increase of TOS, i.e., the fast deactivation at the initial period and the subsequently stable stage [7]. As a result, a gradually decreased C3H6 yield with increasing TOS is obtained, the extent of which is obviously dependent on the catalyst. To make a quantitative estimation on the catalytic stability, the relative deactivation rate is calculated from the C3H6 yields at TOS of 5 and 305 min. As shown in Table 2, the stability is increased in the order of CrOx/silicalite-1–0.35 (21.8%) < CrOx/silicalite-1–0.06 (18.8%) < CrOx/silicalite-1–0.22 (12.9%) < CrOx/silicalite-0.15 (6.6%). Thus, the impact of the crystal size of silicalite-1 on the catalytic stability of CrOx/silicalite-1 for CO2-ODP is significant, and this will be discussed in the next section together with the characterization results of the representative spent catalysts.
3.5 Origin of the Enhanced Catalytic Stability
As a common fact, coking is inevitable during CO2-ODP irrespective of the catalyst. Moreover, our previous results indicate that the coke deposited on the polymeric Cr(VI) oxides originated from the toughly adsorbed propene over CrOx/silicalite-1 is beneficial for the catalytic stability [7]. Thus, the spent CrOx/silicalite-1–0.35 catalyst with the worst stability and the used CrOx/silicalite-1–0.15 showing the best stability were studied as representatives by TG-DSC, and the results are given in Fig. 7 and Table S3. Considering different performances of the catalysts for CO2-ODP, the coke deposition rate, which is defined as the amount (g) of the coke deposited over the catalyst by producing 1 mol of C3H6, is also calculated. As revealed from Fig. 7, the changing patterns of the weight and thermal flow with increasing the temperatures are very similar for the two spent catalysts. However, the peak temperature for the thermal flow of the spent CrOx/silicalite-1–0.15 (~ 360 °C) is slightly higher than that of the used CrOx/silicalite-1–0.35 (~ 353 °C), which may suggest a slightly higher graphitization extent of the deposited coke over CrOx/silicalite-1–0.15. If the calculated data in Table S3 are compared, both the amount of the coke and the coke deposition rate are higher over the more stable catalyst of CrOx/silicalite-1–0.15, which agrees with its higher content of polymeric Cr(VI) oxides (Table 2) and strongly adsorbed propene (Table S2). This observation is consistent with our previous findings [7], indicating a promotional effect of the deposited coke on the stability of CrOx/silicalite-1-y for CO2-ODP.

TG-DSC curves of CrOx/silicalite-1 after CO2-ODP for 305 min under the conditions of 0.5 g catalyst, T = 550 °C, CO2: C3H8: Ar = 20:4:1, and a total flowrate of 25 mL·min−1
As a matter of fact, the stability of a solid catalyst is mainly affected by the catalyst structures. Thus, the XRD characterization was applied to find out the possible impact of coking on the structural properties of silicalite-1 and CrOx. For the convenient comparison, the XRD patterns of both the fresh and used catalysts of CrOx/silicalite-1–0.35 and CrOx/silicalite-1–0.15 are combined in Fig. 8. Irrespective of the used catalysts, there are still no XRD diffractions assigned to CrOx species including crystalline Cr2O3. This indicates that CrOx species over the spent catalysts are similar to those over the fresh catalysts, i.e., highly dispersed or amorphous states. Thus, the impact of coking on the structural properties of CrOx species is very limited if it has. Moreover, the main XRD diffractions of silicalite-1 for the used CrOx/silicalite-1–0.35 coincides with those of the corresponding fresh catalyst (Fig. 8a and the enlargements). Contrary to this, the intensity for the main XRD peaks of silicalite-1 over the used CrOx/silicalite-1–0.15 is slightly decreased in comparison with that over the fresh catalyst (Fig. 8b and the enlargements). According to the references [17, 18], the weakened XRD diffractions are originated from the coke deposited in the zeolite channels. As discussed in Sect. 3.1, the blocking of the silicalite-1–0.15 micropores by CrOx is revealed from the N2 adsorption/desorption results. Thus, the decreased intensity of XRD peaks for silicalite-1–0.15 over the used catalyst can be reasonably attributed to the deposited coke over CrOx in the silicalite-1–0.15 channels. In contrary, the blocking of silicalite-1–0.35 channels by CrOx is much less obvious than that of the silicalite-1–0.15, leading to the almost overlapped XRD diffractions of the zeolites in the fresh and spent CrOx/silicalite-1–0.35 catalysts. These results suggest that the distribution of the deposited coke over the catalysts is varied depending on the average size of the silicalite-1 support, which should be one of the reasons for the different stability of CrOx/silicalite-1 catalysts. In contrast, the contribution from the structural changes of the CrOx species and the zeolite to the deactivation of the CrOx/silicalite-1 catalysts for CO2-ODP can be neglected.

Comparative XRD patterns for the fresh and spent catalysts of CrOx/silicalite-1–0.35 (a) and CrOx/silicalite-1–0.15 (b)
To get more information on the deposited coke, the used CrOx/silicalite-1–0.15 and CrOx/silicalite-1–0.35 were further characterized with different techniques. As indicated from the O2-TPO results (Fig. 9), CO2 is the main product for the burning of the deposited coke irrespective of the catalysts, indicating the easy combustion of the coke with a relatively low graphitization degree. However, the peak temperature for releasing CO2 over the spent CrOx/silicalite-1–0.35 (366 °C) is slightly higher than that over the spent CrOx/silicalite-1–0.15 (356 °C), which is contradictory to the TG-DSC result (Fig. 7), i.e., a lower DSC peak temperature for the spent CrOx/silicalite-1–0.35 (353 °C) than that for the spent CrOx/silicalite-1–0.15 (360 °C). Since that the differences of the peak temperatures in both TG-DSC and O2-TPO are very limited for the two spent catalysts (7–10 °C), the more sensitive Raman spectra are recorded (Figure S2). Following the reported method [19], the spectrum is fitted into G, D1, and D3 bands corresponding to the ideal graphitic, disordered, and amorphous carbon, respectively. Moreover, the intensity ratio of the D1 band to G band (ID1/IG) characterizing the graphitization extent of carbonaceous species is calculated. As shown in Table S3, ID1/IG of the spent CrOx/silicalite-1–0.15 (0.28) is clearly lower than that of the spent CrOx/silicalite-1–0.35 (0.47), revealing the slightly higher graphitization degree of the coke. Thus, the graphitization degree of the coke over the spent CrOx/silicalite-1–0.15 is slightly higher than that over the spent CrOx/silicalite-1–0.35 although the graphitization extent of the coke over both of the catalysts is relatively low.

The changing of the MS signals for CO2 and CO with increasing the temperature during O2-TPO of the used CrOx/silicalite-1–0.15 and CrOx/silicalite-1–0.35 catalysts
However, a clear CO2 peak centered at a much higher temperature of about 520 °C is observed for the used CrOx/silicalite-1–0.15 catalyst while it is only a tail in the case of the used CrOx/silicalite-1–0.35 (Fig. 9). Since that a higher peak temperature of O2-TPO is directly related to a higher graphitization extent of the coke, these results and discussion suggest that the cokes with a higher and lower graphitization degrees are simultaneously present over the spent catalysts. Moreover, the amount of the coke with a higher graphitization degree over the used CrOx/silicalite-1–0.15 is more significant than that over the used CrOx/silicalite-1–0.35. Indeed, the temperatures for the oxidation of the coke over the active phases including CrOx species are found to be clearly lower than those over the supports [20, 21], and are generally explained as the lower graphitization degree of the coke over the active phase than that over the support [22, 23]. Following the explanation [20,21,22,23], the distributions of the deposited coke over the spent CrOx/silicalite-1 catalysts can be proposed, i.e., the majority of the coke with a lower graphitization extent is present on the surface of CrOx species while a small part of the higher graphitized coke is located on the silicalite-1 support. Moreover, the graphitization degree of the coke deposited over CrOx is very similar irrespective of the silicalite-1 supports. As a result of the much intense peak at around 520 °C for the spent CrOx/silicalite-1–0.15 (Fig. 9), the higher graphitized coke on the silicalite-1–0.15 support is more significant than that on the silicalite-1–0.35 support.
To explore the origin of the higher graphitized coke, the C3H6-TPD experiment was applied for the silicalite-1–0.15 support. In comparison with the C3H6-TPD pattern of the fresh CrOx/silicalite-1–0.15 catalyst, the silicalite-1–0.15 shows only a greatly sharper and much stronger peak for the desorption of the very weakly adsorbed C3H6 (Figure S3), suggesting that coke cannot be directly formed on the silicalite-1 support during CO2-ODP. Considering the high dispersion & the amorphous nature of the CrOx species, the higher graphitized coke may be located on the periphery of the CrOx species over the CrOx/silicalite-1 catalyst, which can be directly contacted with the zeolite. This explanation is supported by the more obvious blocking of silicalite-1–0.15 channels by CrOx than that of the silicalite-1–0.35. Thus, as a result of the small size of the silicalite-1 channels (~ 0.5 nm), the coke deposited over the CrOx species in blocking the silicalite-1 channels can be directly contacted with the silicalite-1 channels, leading to a higher amount of the coke with a higher graphitization extent over the used CrOx/silicalite-0.15.
As revealed from our previous work [7], the isolated Cr(VI) oxides and polymeric Cr(VI) oxides with a lower polymerization degree are the key factor in determining the catalytic activity and propene selectivity at the later stage of the CO2-ODP because the majority of the polymeric Cr(VI) oxides with a higher polymerization degree are gradually covered by the deposited coke in the initial period of reaction. Moreover, the stability of the catalyst is quantified with the C3H6 yield, which is closely related to the activation of C3H8 and the desorption of C3H6. Thus, to reveal the effect induced from the deposited coke, C3H8-TPD and C3H6-TPD of the spent catalysts were studied, and the results are given in Fig. 10 and Tables S4, S5. In comparison with the fresh catalysts, both the spent CrOx/silicalite-1–0.15 and the used CrOx/silicalite-1–0.35 show a clearly lower amount of the desorbed C3H8, which is mainly contributed from the significant decrease of the strongly adsorbed C3H8 (Fig. 10a and Table S4). Moreover, the peak temperatures for the desorption of the medium and strongly adsorbed C3H8 over the spent catalysts are obviously lower than those over the fresh catalysts (Table S4). These results indicate the negative effect of the deposited coke on the C3H8 conversion, which is agreeable with the catalytic results (Fig. 6a). Since that the peak temperatures for the desorption of the medium or strongly adsorbed C3H8 and the amount of the desorbed C3H8 over the spent CrOx/silicalite-1–0.15 are higher than those over the spent CrOx/silicalite-1–0.35, higher TOS C3H8 conversions are achieved over CrOx/silicalite-1–0.15 (Fig. 6a). The analysis of the C3H6-TPD results in Fig. 10b and Table S5 reveals that the deposited coke over CrOx/silicalite-1 catalysts favors the C3H6 desorption, leading to the higher C3H6 selectivity over CrOx/silicalite-1–0.35 than that over CrOx/silicalite-1–0.15 (Fig. 6c). These observations coincide with our previous findings [7], indicating that the coke are mainly deposited on the polymeric Cr(VI) oxides with a higher degree of polymerization, and less coke on isolated Cr(VI) oxides and polymeric Cr(VI) oxides with a lower polymerization degree over CrOx/silicalite-1 catalysts triggers more exposed active sites for CO2-ODP, characterizing the slightly decreased C3H8 conversion and enhanced C3H6 selectivity at the end of the reaction to the initial catalytic performance.

The profiles for C3H8-TPD (a) and C3H6-TPD (b) of the used CrOx/silicalite-1–0.15 and CrOx/silicalite-1–0.35 catalysts
Based on these facts and the discussion, the mechanism for the enhanced stability of the CrOx/silicalite-1 for CO2-ODP is summarized as follows. The coke deposition mainly originated from the strongly adsorbed propene plays a promotional effect on the stability of CrOx/silicalite-1 catalyzed CO2-ODP. Moreover, the coke, which preferably deposits over the more active but less selective polymeric Cr(VI) oxides, plays dual functions of (1) decreasing the C3H8 conversion and (2) increasing the C3H6 selectivity, the extents of which are dependent on the amount and the location of the coke induced from the structural properties of the CrOx/silicalite-1 catalyst. Although the coke deposition over CrOx/silicalite-1–0.15 is slightly significant than that over CrOx/silicalite-1–0.35, the amount of the coke with a higher graphitization extent directly contacted with the zeolite is also higher over CrOx/silicalite-1–0.15, leading to an alleviated decrease of the TOS C3H8 conversion but a slightly enhanced increase of the TOS C3H6 selectivity. As a result, CrOx/silicalite-1–0.15 shows a much higher stability indexed by the C3H6 yield than CrOx/silicalite-1–0.35. In comparison with CrOx/silicalite-1–0.15, the effects of the coke with a higher graphitization extent over CrOx/silicalite-1–0.22 are expectedly diminished due to the less obvious blocking of the silicalite-1–0.22 channels by CrOx species. Thus, CrOx/silicalite-1–0.22 shows a lower catalytic stability than CrOx/silicalite-1–0.15 for CO2-ODP although it has the highest content of polymeric Cr(VI) oxides.
As shown in Fig. 6d, the TOS deactivation of the most stable CrOx/silicalite-1–0.15 catalyst for CO2-ODP is still observable. To reveal the structural stability of the catalyst, the regeneration experiments after CO2-ODP for 3 h were performed at 550 °C in the air atmosphere by burning the deposited coke over the catalyst for 0.5 h. As seen from Fig. 11, the C3H6 yield can be fully restored even after 3 reaction-regeneration cycles. This indicates that the deactivation of CrOx/silicalite-1–0.15 is reversible and the structure of the catalyst is stable. Thus, the prolonged lifetime of CrOx/silicalite-1–0.15 for CO2-ODP is reasonably expected.

Reaction-regeneration cycles of CrOx/silicalite-1–0.15 for CO2-ODP under the conditions of 0.5 g catalyst, T = 550 °C, CO2:C3H8:Ar = 20:4:1, and a total flowrate of 25 mL min−1
4 Conclusions
In summary, well crystallized silicalite-1 with an average crystal size of 0.06, 0.15, 0.22, and 0.35 μm was successfully synthesized by the hydrothermal method, and silicalite-1–0.06 exhibits a slightly higher crystallinity than the remaining silicalite-1 zeolites, which have very similar crystallinities. After loading CrOx, crystallinities of silicalite-1 are comparable for all of the catalysts. In the cases of the textures, the porous properties of silicalite-1 and CrOx/silicalite-1 are sensitive to the average crystal sizes of the zeolite, which is originated from the size-dependent agglomeration of the zeolite. Indeed, the density of different silanol groups is adjusted via regulating the average crystal size of silicalite-1, and the most abundant silanol nests are obtained over silicalite-1–0.15. As a result, the highest CrOx dispersion is achieved over silicalite-1–0.15, which is agreeable with the non-detected crystalline Cr2O3. Irrespective of the catalysts, both isolated and polymerized Cr(VI) oxides are present although the relative content of the polymeric Cr(VI) oxide and Idioxo/Imono-oxo are the highest over CrOx/silicalite-1–0.22. From the CO2-ODP results, the initial catalytic activity is slightly increased in the order of CrOx/silicalite-1–0.15 ≈ CrOx/silicalite-1–0.35 < CrOx/silicalite-0.06 < CrOx/silicalite-1–0.22, which is reasonably explained as the size impact of the silicalite-1 on the CrOx structures induced from varied contents of silanol groups over the zeolite. In contrast, a different changing pattern of CrOx/silicalite-1–0.35 < CrOx/silicalite-1–0.06 < CrOx/silicalite-1–0.22 < CrOx/silicalite-0.15 is observed for the catalytic stability. Together with the characterization results of the representative spent catalysts, this is mainly attributed to the dual functions of the deposited coke over the catalysts, the amount and the location of which are essentially induced from the effects of the silanol groups on CrOx structures. Thus, a more active and stable CrOx/silicalite-1 catalyst for CO2-ODP is expectable if silicalite-1 with a further improved content of silanol nests can be synthesized via carefully regulating its crystal size.
References
Li XY, Pei CL, Gong JL (2021) Chem 7:1755
Chen S, Chang X, Sun GD, Zhang TT, Xu YY, Wang Y, Pei CL, Gong JL (2021) Chem Soc Rev 50:3315
Li CY, Wang GW (2021) Chem Soc Rev 50:4359
Jiang X, Sharma L, Fung V, Park SJ, Jones CW, Sumpter BG, Baltrusaitis J, Wu ZL (2021) ACS Catal 11:2182
Li GM, Liu C, Cui XJ, Yang YH, Shi F (2021) Green Chem 23:689
Otroshchenko T, Jiang G, Kondratenko VA, Rodemerck U, Kondratenko EV (2021) Chem Soc Rev 50:473
Wang J, Song YH, Liu ZT, Liu ZW (2021) Appl Catal B: Environ 297:120400
Cheng YH, Miao CX, Hua WM, Yue YH, Gao Z (2017) Appl Catal A: Gen 532:111
Barbera K, Bonino F, Bordiga S, Janssens TVW, Beato P (2011) J Catal 280:196
Grand J, Talapaneni SN, Vicente A, Fernandez C, Dib E, Aleksandrov HA, Vayssilov GN, Retoux R, Boullay P, Gilson JP, Valtchev V, Mintova S (2017) Nat Mater 16:1010
Groen JC, Peffer LAA, Pérez-Ramı́rez J (2003) Microporous Mesoporous Mater 60:1
Cheng YH, Lei TQ, Miao CX, Hua WM, Yue YH, Gao Z (2018) Catal Lett 148:1375
Weckhuysen BM, Wachs IE, Schoonheydt RA (1996) Chem Rev 96:3327
Chakrabarti A, Wachs IE (2014) Catal Lett 145:985
Baek J, Yun HJ, Yun D, Choi Y, Yi J (2012) ACS Catal 2:1893
Lee EL, Wachs IE (2008) J Phys Chem C 112:6487
Rojo Gama D, Nielsen M, Wragg DS, Dyballa M, Holzinger J, Falsig H, Lundegaard LF, Beato P, Brogaard RY, Lillerud KP, Olsbye U, Svelle S (2017) ACS Catal 7:8235
Zhang Y, Wu SD, Xu X, Jiang HQ (2020) Catal Sci Technol 10:835
Sattler JJ, Beale AM, Weckhuysen BM (2013) Phys Chem Chem Phys 15:12095
Xia K, Lang WZ, Li PP, Yan X, Guo YJ (2016) J Catal 338:104
Jiang F, Zeng L, Li SR, Liu G, Wang SP, Gong JL (2014) ACS Catal 5:438
Larsson M, Hultén M, Blekkan EA, Andersson B (1996) J Catal 164:44
Mimura N, Okamoto M, Yamashita H, Oyama ST, Murata K (2006) J Phys Chem B 110:21764
Acknowledgements
The financial supports from the National Natural Science Foundation of China (21636006) and the Fundamental Research Funds for the Central Universities (GK201901001 and 2017CBZ002) are highly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10562_2022_4012_MOESM1_ESM.docx
Supplementary file1 (DOCX 351 kb) The additional results including XRD, N2 adsorption/desorption isotherms, Raman spectra of the representative spent catalysts, and C3H8/C3H6-TPD are available free of charge
Rights and permissions
About this article

Cite this article
Wang, J., Song, YH., Yuan, EH. et al. Elucidating the Support-Size Effect on the Catalytic Stability of CrOx/Silicalite-1 for Oxidative Dehydrogenation of Propane with CO2. Catal Lett 153, 790–804 (2023). https://doi.org/10.1007/s10562-022-04012-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-022-04012-4