
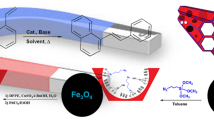
Abstract
A nitrogen ligand, i.e. 1,3-di-(o-aminophenoxy)-2-propyl propargyl ether (DPPE), has been synthesized and characterized. Magnetic mesoporous silica composite (MNP@SiO2-SBA) was obtained via embedding magnetite nano-particles (MNPs) between SBA-15 channels. DPPE palladium dichloride (MNP@SiO2-SBA-DPPE-Pd(II)) was then prepared via click chemistry and fully characterized. The Hiyama reaction conditions including solvent, amount of catalyst, base and temperature were optimized for the prepared catalyst. The activity and recyclability of supported magnetic Pd(II) catalyst were evaluated under the optimal reaction conditions. The catalyst was easily separated magnetically, reused in five runs sequentially, and no significant loss of activity was observed.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Doubtlessly, transition metal catalyzed reactions are very powerful and essential tools for carbon–carbon and carbon-heteroatom bonds formation. These methods provide high atom and step economy routes for constructing complex molecules under mild reaction conditions. Among various metals for catalyzing these reactions, palladium is more preferred and widely used due to enhanced activity and selectivity [1,2,3]. Some of these coupling reactions, e.g. Sonogashira, Stille, Negishi, Hiyama, Heck, Suzuki, and Kumada–Corriu reactions, have also found industrial applications [4]. However, these reactions have their own benefits and problems. For example, Suzuki coupling reaction suffers from hardly prepared and hardly purified arylboronic acids as starting materials. The Stille, Negishi, and Kumada–Corriu coupling reactions include using of toxic organotin, very reactive and less stable organozinc and organomagnesium reagents [5].
Among the Pd-involved organic preparations, the Hiyama cross-coupling reaction has a distinguished place. This reaction involve an organosilicon compound and a halogenated hydrocarbon which are easily prepared, accessible, low cost, and stable starting materials [6,7,8]. The main disadvantage of Hiyama coupling reaction is the less reactivity of C–Si bond toward electrophiles. This is due to the low polarizability of the C–Si bond, which decreases the nucleophilicity of the organosilicon compound. This problem can be overcome by using fluoride as a nucleophilic promoter. However, the fluoride ion may produce some difficulties with base‐sensitive protecting groups and acidic protons. The low reactivity of aryl chlorides, that are widely available commercially, is another shortcoming of the Hiyama coupling reaction [9].
In recent years, magnetite nanoparticles (MNPs) have attracted increased attention as a support for immobilization of catalysts [10,11,12]. They are nontoxic, highly dispersible, biocompatible, and they have a high surface-to-volume ratio. Furthermore, MNP-supported catalysts enjoy the benefit of easy and rapid separation from different reaction mixtures magnetically [13, 14]. It is highly recommended to coat MNPs before any treatment because they agglomerate intrinsically due to strong magnetic dipole–dipole interactions and high specific surface area [15, 16]. The most popular way to overcome this problem is using MNPs in the form of a composite with other metal oxides. Compositions of MNPs with different forms of mesoporous silica such as MCMs and SBAs have been widely investigated in recent years [17, 18]. Mesoporous silica provides high density of silanol groups inside the pores for further functionalization. There are plenty of reports on the incorporation of magnetic mesoporous for preparing magnetically retrievable supported catalyst in the literature [19,20,21].
In recent years, the preparation of a broad spectrum of supported Pd(II) complexes with different ligands as catalysts for coupling reactions has been reported. Improving the reaction yields and decreasing reactions time, processes costs, and byproducts are some the results of these investigations [22]. In the present study, synthesis, characterization, and grafting of 1,3-di-(o-aminophenoxy)-2-propyl propargyl ether (DPPE) on magnetic mesoporous silica via click reaction are explained. The supported ligand was then treated with a Pd(II) solution in EtOH to give a Pd(II)-supported magnetic catalyst (MNP@SiO2-SBA-DPPE-Pd(II)). The obtained recoverable magnetic catalyst is a quite effective catalyst in the Hiyama reaction and different derivatives were prepared.
2 Experimental
2.1 Materials
Fe3O4@SiO2-SBA magnetic composite [23] and 3-azidopropyltriethoxysilane [24] were prepared according to reported methods in the literature. 1,3-Di-(o-nitrophenoxy)-2-propanol (1) was synthesized via the method described by Zhang et al. [10] via the reaction of o-nitrophenol and 1,3-dichloro-2-propanol in hot DMF and in the presence of K2CO3. Laboratory grade solvents used in this work were dried according to reported procedures in the literature [25]. The remaining chemicals were obtained from Merck and used as received.
2.2 Synthesis of 1,3-Di-(o-nitrophenoxy)-2-propyl propargyl ether (3)
To a stirred solution of 1,3-di-(o-nitrophenoxy)-2-propanol 1 (0.830 g, 2.5 mmol) in dry DMF (5 mL) at 0 °C was added NaH (0.132 g, 3 mmol; 60% dispersion in mineral oil) in small portions under argon. The mixture was stirred at 0 °C for 1 h and then propargyl bromide (0.31 mL, 6 mmol) was added dropwise over 10 min. The reaction mixture was warmed to room temperature and stirred for additional 10 h. The reaction was quenched with MeOH (7 mL) and the white precipitated was filtered in vacuum, washed cold EtOH and dried to afford 0.630 g product [1]. (Yield: 66%, mp: 100–103 °C). FT-IR (KBr, cm–1): 3269 (m), 3061 (w), 2932 (w), 2869 (w), 1607 (m), 1521 (s), 1350 (s), 1280 (m), 1250 (m), 743 (m).
2.3 Synthesis of 1,3-Di-(o-aminophenoxy)-2-propyl propargyl ether (DPPE)
To a rapidly stirred suspension of powdered zinc (1.740 g, 26.6 mmol) in 4.25 mL of 37% aqueous ammonia was added a solution of 3 (1.000 g, 2.65 mmol) in 4.25 mL of THF. The mixture was refluxed for 6 h and then filtered. The filtrate was extracted with three 15 mL portions of ethyl acetate/water (50:50) mixture, and the combined extracts evaporated to dryness. The obtained residual orange oil chromatographed on 20 cm × 20 cm silica gel covered glass plates using 75:25 petroleum ether/acetone eluent to afford to give 0.510 g of orange oily product [26]. (Yield: 62.5%). FT-IR (KBr, cm–1): 3294 (s), 3048 (w), 2935 (w), 2869 (w), 1648 (s), 1526 (m), 1448 (w), 1310 (w), 1032 (m), 735 (m). 1H-NMR (250 MHz, CDCl3 δ): 6.85–7.00 (m, 2H); 6.70–6.80 (m, 2H); 4.47–4.50 (m, 2H); 4.40–4.46 (m, 1H); 4.23–4.32 (m, 4H); 3.90 (br, s, 4H); 2.54–2.56 (m, 1H). 13CNMR (62.5 MHz, CDCl3,δ): 141.1 (C–O, Ar.), 136.6 (C–N, Ar.), 122.0, 118.5, 115.4, 112.2, 79.8 (CH–O), 75.5 (≡C–CH2), 75.1 (≡C–H), 68.3 (O–CH2–CH), 57.9 (≡C–CH2). Mass: (m/z)+: 312.15 (M+, 84%).
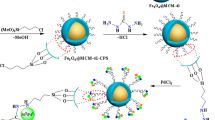
2.4 Synthesis of Azide Functionalized Magnetic Nanocomposite Fe3O4@SiO2-SBA-N3
A suspension of Fe3O4@SiO2-SBA (1.000 g) in 50 mL of toluene was irradiated in an ultrasonic bath for 10 min. To this suspension, 3-azidopropyltriethoxysilane (2.0 mL, 9.7 mmol) was added, and the mixture was stirred for 16 h at 80 °C under argon. The reaction mixture was then cooled, filtered and washed with toluene several times and then dried at 45 °C in a vacuum oven [11]. FT-IR (KBr, cm–1): 2935 (w), 2869 (w), 2104 (m), 1628 (m), 1526 (w), 1448 (w), 1088 (s), 735 (m).
2.5 Synthesis of MNP@SiO2-SBA-DPPE via Click Reaction
In a round-bottomed flask Fe3O4@SiO2-SBA-N3 (1.000 g) was dispersed in 20 mL of t-BuOH/water (1:1) containing CuSO4 (0.330 g, 1.32 mmol), sodium ascorbate (1.070 g, 5.4 mmol) and DPPE (1.200 g, 4.8 mmol). The mixture was irradiated in an ultrasonic bath for 15 min and stirred at 80 °C for 24 h under argon. The magnetic particles were then magnetically decanted, washed with EtOH and then dried in a vacuum oven at 80 °C [11]. FT-IR (KBr, cm−1): 3397 (br, s), 3067 (w), 2935 (w), 2869 (w), 2104 (m), 1628 (m), 1526 (w), 1448 (w), 1088 (s), 735 (m).
2.6 Synthesis of MNP@SiO2-SBA-DPPE-Pd(II)
DPPE functionalized magnetic mesoporous silica (1.000 g) was dispersed in a PdCl2 (0.03 g, 0.17 mmol) solution in EtOH (8 mL) in an ultrasonic bath for 15 min. The suspension was stirred at room temperature for 12 h. The catalyst was decanted magnetically, washed several times with EtOH and then dried in vacuum at 45 °C [25].
2.7 General Procedure for the Hiyama Reaction
In a 25 mL round-bottom flask, the magnetic catalyst (0.5 mol%) was dispersed in an ultrasonic bath in 1 mL of dry dimethylsulfoxide (DMSO) for 8 min. Aryl halide (0.2 mmol), triethoxyphenylsilane (0.2 mmol) and Et3N (0.4 mmol) were then added and the mixture was stirred at 120 °C. The reaction was followed by running TLC in an appropriated mixture of n-hexan/ethyl acetate as the eluent, and upon completion, the catalyst was removed and the residue was chromatographed on glass plates covered by silica gel.
3 Results and Discussion
3.1 Preparation and Characterization of the Catalyst
The new ligand (DPPE) has been prepared in two steps as shown in Scheme 1. In the first step, 1,3-di-(o-aminophenoxy)-2-propanol (1) was etherified with propargyl bromide 2 [1]. Selective reduction of nitro groups in the presence of ethynyl group by conventionl methods such as Pd/C/hydrazine; Pd/C/NaBH4; Fe/HCl; Sn/HCl, and Zn/HCl was unsuccessful. This was done by the specific method described by Kovar and Arnold for preparation of the ethynyl substuted o-diamines [14].

Synthesis of DPPE
Common spectroscopy methods were applied to confirm the structure of DPPE. More information are provided in the Supporting Information file.
Magnetite nanoparticles were prepared via co-precipitation method and then covered by a silica layer and then embedded into mesoporous silica (SBA-15) [27]. The as-prepared Fe3O4@SiO2-SBA particles were then silylated with 3-azidopropyltrimethoxysilane via post-synthesis grafting technique described by Malvi et al. [11]. The azide functionalized magnetic mesoporous silica was further functionalized with DPPE via the click methodology and then treated with PdCl2 solution in EtOH to afford supported Pd(II)-complex, Scheme 2.

Synthesis of Fe3O4@SiO2-SBA-DPPE-Pd(II)
The FT-IR spectra of DPPE and MNP@SiO2-SBA-DPPE are shown in Figures S-4a, b. For more discussion, please see the Supporting Information file.
Solid state 13C CP/MAS NMR spectrum of non-magnetic sample (SBA-DPPE), Fig. 1, exhibited three peaks of propyl chain carbon atoms at 8 (C1), 20 (C2) and 52 (C3) ppm [28]. The presence of peaks at 122 (C4) and 147 (C5) ppm are attributed to the two carbon atoms present in the triazole ring [29] and signals centered at 70 (C6–C8) ppm are due to the Csp3 directly linked to oxygen atoms. Peaks centered at the region 110–155 ppm are associated to the Csp2 atoms of the aniline group (C9–C14).

13C CP/MAS NMR spectrum of MNP@SiO2@SBA-DPPE
XPS measurements were performed les to obtain information about the structural features and the chemical environment. In the sample MNP@SiO2-SBA-DPPE-Pd(II), the XPS analysis was utilized to confirm the incorporation of the metal and evaluate the oxidation state of Pd in the catalyst. The high-resolution C1s spectra for MNP@SiO2-SBA-DPPE and MNP@SiO2-SBA-DPPE-Pd(II) are depicted in Fig. 2a and b. Both spectra are fitted with three components at 284.7, 286.1 and 287.8 eV, which are assigned to C–H/C–C/Car, C–N, and C–O, respectively [30]. As far as the N1s region (Fig. 3a and b), MNP@SiO2-SBA-DPPE shows a peak centered at 398.8 eV which is fitted with three components assigned to N–Harom (398.0 eV), N=N (399.2 eV) and N–C (401.0 eV), respectively. After Pd complexation, the N1s spectrum of MNP@SiO2-SBA-DPPE-Pd(II) was quite similar with three components centered at 398.2 (Pd-NHarom/N-Harom), 399.3 (N=N) and 401.0 (N–C) eV. The electronic state of palladium was further analyzed by XPS (Fig. 4a and b). The absence of palladium is clearly confirmed in the support MNP@SiO2-SBA-DPPE (Fig. 4a). In contrast, the Pd3d spectrum for MNP@SiO2-SBA-DPPE-Pd(II) exhibits two peaks centered at 337.8 and 343.1 eV that are associated to electron transitions of Pd3d5/2 and Pd3d3/2, respectively. These values of binding energy confirm that all species of Pd in the catalyst are present as Pd(II).

High-resolution XPS spectra of C1s region for MNP@SiO2@SBA-DPPE (a) and MNP@SiO2@SBA-DPPE-Pd(II) (b)

High-resolution XPS spectra of N1s region for MNP@SiO2@SBA-DPPE (a) and MNP@SiO2@SBA-DPPE-Pd(II) (b)

High-resolution XPS spectra of Pd3d region for MNP@SiO2@SBA-DPPE (a) and MNP@SiO2@SBA-DPPE-Pd(II) (b)
TGA was used to estimate the complex loading, Figure S-4. For this purpose, the weight loss (from ambient temperature to 590 °C) of MNP@SiO2-SBA (2.02%) was subtracted from the weight loss of the supported Pd(II) complex (35.18%). The obtained value (∆m = 33.16%) can be regarded as the loaded complex (33.16% or 331.6 mg g−1 of magnetic SBA). Atomic absorption spectroscopy revealed that the palladium loading is 0.10 mmol g−1 (1.10%wt).
Figure 5a and b display the low angle XRD patterns of MNP@SiO2-SBA and MNP@SiO2-SBA-DPPE-Pd(II), respectively. Pure SBA-15 exhibits an intense peak at 2θ = 0.84° corresponds XRD unit-cell parameter of 10.6 nm [31,32,33]. It is the characteristic of a well order hexagonal P6mm space group containing mesoporous phase. However, the interlayer spacing of MNP@SiO2-SBA is slightly smaller than pure SBA-15 and appeared at 2θ = 1.06° corresponds XRD unit-cell parameter of 8.3 nm, Fig. 5a. In the diffraction pattern of MNP@SiO2-SBA-DPPE-Pd(II), this peak is also displayed at 2θ = 1.06° with low intensity, Fig. 5b. It means that, the ordered mesoporous structure is retained. The formation of the Pd(II) complex inside the channels may be responsible for attenuation of the peak intensity. In the pattern of SBA-15, two additional peaks corresponding to (110) and (200) diffractions are also observed at 2θ = 1.58° and 1.82°. Because of disordering silica channels by impregnated magnetite nanoparticles, these peaks are not seen in the patterns of MNP@ SiO2-SBA and final catalyst [27, 34].

Low angle powder XRD patterns of a magnetic SBA and b the supported complex
Figure 6a and b shows the wide angle XRD patterns of magnetite and MNP@SiO2-SBA-DPPE-Pd(II), respectively. Both samples exhibit well-resolved diffraction peaks that can be indexed as (220), (311), (400), (422), (511) and (440) planes associated with cubic magnetite nanoparticles (JCPDS-ICDD Copyright 1938, file No. 01-1111) with the Fd-3 m space group [16]. However, due to confinement of MNPs between the channels of mesoporous silica, intensities of these characteristic diffraction peaks were attenuated in the pattern of supported catalyst, Fig. 6b.

Wide angle XRD patterns of a magnetite, b the supported Pd(II) complex
Figure 7a, b show N2 adsorption–desorption isotherms obtained by Brunauer–Emmett–Teller (BET) surface areas analyses. As shown, an H2 hysteresis loop of type-IV curve is observed for the MNP@SiO2-SBA [35]. The BET surface area and mean pore diameter of MNP@SiO2-SBA are 482.9 m2 g−1 and 7.63 nm, respectively. The sharp increase in P/P0 with the narrow size distribution (7.63 nm) highlights the formation of ordered mesoporous structure [19]. These values are less than the corresponding parameters reported for pure SBA-15 [32]. Comparison of the XRD unit cell parameters of magnetic SBA and pure SBA-15 also shows a similar trend, Table 1. The BET surface area and mean pore diameter in the supported catalyst are dropped to 83.8 m2 g−1 and 5.22 nm, respectively. Therefore, it can be concluded that the surface decoration by DPPE-Pd complex inside the pores of magnetic SBA is performed successfully without blocking the intrachannels space by MNPs. XRD pattern of the final catalyst also confirms that the ordered structure magnetic SBA is retained. Therefore, molecules can easily slip inside the channels for subsequent transformations (Table 2).

N2 Adsorption–desorption isotherms (a) and pore size distribution (b) of MNP@SiO2-SBA-DPPE-Pd(II)
The image obtained by TEM also confirmed ordered structure of magnetic SBA (Fig. 8). Highly ordered arrays of 1D mesoporous channels in which MNPs with 20 to 50 nm diameter are located between the channels of MNP@SiO2@SBA are seen in this image.

TEM images of magnetic SBA
Figure 9 shows the hysteresis loops of MNP, MNP@SiO2 and MNP@SiO2-SBA-DPPE-Pd(II) at room temperature and the results are tabulated in Table 3. Superparamagnetic behavior of the samples is shown by small remanence, hysteresis, coercivity values, and narrow width of the loop as seen in the inset of Fig. 9 [36, 37]. The saturation magnetizations of MNP, MNP@SiO2 and MNP@SiO2-SBA-DPPE-Pd(II) are 78.14, 34.52, and 1.56 emu g−1, respectively. Dropping of the maximal saturation magnetization of supported catalyst is due to (1) formation of a non-magnetic silica shell around MNPs; (2) incorporation of MNP@SiO2 into congested non-magnetic SBA channels; and (3) surface decoration of magnetic SBA by a non-magnetic Pd-complex. In spite of low Ms value, the catalyst could be separated magnetically by an external magnet, Figure S-5.

Magnetization curves for the MNP, MNP@SiO2 and MNP@SiO2-SBA-DPPE-Pd(II)
3.2 Hiyama Coupling Reaction
Iodobenzene and triethoxyphenylsilane were chosen as the model compounds for optimizing the Hiyama coupling reaction conditions. Reaction parameters such as the base, amount of the catalyst, solvent, amount of base, and reaction temperature were studied. The results are shown in Table 3 and Fig. 10a–e. The effect of the base was evaluated (Entries 1 to 7). The highest conversion is achieved with the Et3N base (Entry 1, 70.6% yield). The optimization of catalyst dosage shows that using the catalyst containing 0.5 mol% Pd(II) is the optimal value (Entry 1, 70.6% yield). Higher and lower amount of the catalyst loading gave poor results (Entries 8 to 9). According to solvent screening (Entries 10 to 16), the DMSO solvent is the best choice due to the highest yield (Entry 12, 88.5% yield). The amount of the base was also investigated. Lower or higher quantities of Et3N than that of 2.0 molar ratio diminish the reaction conversion (Entries 17 and 18). It should be noted that the reaction does not proceed properly at the temperatures lower or higher than 120 °C (Entries 19 to 21) and in the absence of the catalyst (Entry 22).

Optimization of the Hiyama reaction conditions: a base; b catalyst loading; and c solvent; d base mol ratio; and e reaction temperature. Reaction conditions: Iodobenzene (0.2 mmol), triethoxyphenylsilane (0.2 mmol), solvent (1 mL)
Various aryl halides were subjected to the optimized Hiyama reaction conditions with triethoxyphenylsilane, Table 4. As shown, aryl iodides and aryl bromides showed moderate to good conversions. Please see the Supporting Information for the NMR spectra. However, aryl bromides containing electron attracting substituents showed poor conversions and TOFs (Entries 8 and 9).
A possible mechanism for the Hiyama cross-coupling reaction catalyzed by MNP@SiO2-SBA-DPPE-Pd(II) can be proposed as Scheme 3. In the first step Pd(II) species leached in presence of base (Et3N) to giving [PdX4]2−. The leached palladium species are immediately re-deposited on the catalyst surface and further reacted with aryl halide on the catalyst surface to give desired coupling product. The leaching and re-deposition of Pd species are also been noted by Akira Sakon et al. [38] for Hiyama coupling reaction catalyzed by linear polystyrene-stabilized PdO nanoparticles.

Schematic illustration of the Hiyama reaction catalyzed by MNP@SiO2-SBA-DPPE-Pd(II)
Table 5 compares the catalytic activity of MNP@SiO2-SBA-DPPE-Pd(II) with a few reported catalysts in the literature. Comparing the TOFs reveals that the prepared catalyst shows better activity than some reported catalysts. The catalyst used in Entry 5 is also prepared in our group, but it obtains via a complicated process [3]. The catalyst reported in this study enjoys from enhanced activity and magnetic separation and easy work-up.
3.3 Recyclability of the Catalyst
Supported catalysts are usually prepared via complicated and expensive routes. Therefore, it is necessary to pay sufficient attention to retrieving of a used catalyst. The stability and re-usability of the magnetic catalyst were tested in the Hiyama coupling reactions of iodobenzene and triethoxyphenylsilane, Fig. 11. After each reaction, the catalyst was collected magnetically, washed with ethyl acetate, and then dried. As shown, the conversion reaches to 88.5% in the first run and then slightly dropped to 83.0% during 5 consecutive runs of the Hiyama reaction. Therefore, it can be concluded that the prepared supported catalyst is active and recyclable.

Recycling of MNP@SiO2-SBA-DPPE-Pd(II) for the Hiyama cross-coupling reaction of iodobenzene and triethoxyphenylsilane
3.4 The Catalyst Stability and Pd Leaching Study
The catalyst was retrieved and characterized by FT-IR, Figure S-6a and b. As seen, the spectrum of the used catalyst does not show any significant change compared to the fresh catalyst.
The leaching study was conducted by coupling reaction of equimolar ratio (0.2 mmol) of iodobenzene and phenyltriethoxy silane with 10 mg of the catalyst under optimal reaction conditions. The stability and heterogeneous nature of MNP@SiO2-SBA-DPPE-Pd(II) were studied by analyzing the aliquot of benzyl alcohol. The reaction liquor was analyzed by atomic absorption after separating the catalyst. The palladium content was found to be 0.0085 mg (leaching 8.0%). However, the remaining Pd(II) seems to be enough to catalyze the reaction, because the conversion drop are only 0.4% and 0.1% during the second and third runs (Fig. 11), respectively.
The XRD patterns of the fresh and used catalysts are shown in Figure S-7a and b. As seen, the signs related to Pd(0) particles are absent and no significant change is observed. Therefore, metal leaching may be responsible for the observed moderate catalyst deactivation rather than formation of Pd black aggregates.
Single electron microscope images (SEM) of the fresh and the used catalyst do not show any considerable change in the catalyst morphology, Fig. 12a and b. A uniform spherical morphology with particle size diameters greater than 100 nm is observed in both images. These particles seem to be aggregated possibly due to the very high hydrophilicity of the mesoporous silica shell and moisture adsorption.

SEM images of: a fresh MNP@SiO2-SBA-DPPE-Pd(II); and b used catalyst
4 Conclusion
In summary, the synthesis, characterization, and application of an efficient and retrievable magnetic catalyst are reported. The catalyst was obtained by supporting a Pd(II) complex onto magnetic mesoporous silica. It was fully characterized by conventional techniques. It was also shown that, the prepared supported complex could efficiently catalyze the Hiyama cross-coupling reaction, and excellent conversions are obtained. It was also shown that the catalyst could be retrieved magnetically and reused.
References
Nasrollahzadeh M, Issaabadi Z, Tohidi MM, Mohammad Sajadi S (2018) Chem Record 18:165
Rivas A, Pérez-Revenga V, Alvarez R, de Lera AR (2019) Chem A Eur J 25:14399
Nuri A, Mansoori Y, Bezaatpour A (2019) Appl Organomet Chem 33:e4904
Chen Y, Wang M, Zhang L, Liu Y, Han J (2017) RSC Adv 7:47104
Monfared A, Mohammadi R, Ahmadi S, Nikpassand M, Hosseinian A (2019) RSC Adv 9:3185
Komiyama T, Minami Y, Furuya Y, Hiyama T (2018) Angew Chem Int Ed 57:1987
González J, Schäfer P, Fletcher SP (2019) Organometallics 38:3991
Nuri A, Mansoori Y, Bezaatpour A, Shchukarev A, Mikkola J-P (2019) ChemistrySelect 4:1820
Karami K, Jamshidian N, Nikazma MM, Hervés P, Shahreza AR, Karami A (2018) Appl Organomet Chem 32:e3978
Kang T, Li F, Baik S, Shao W, Ling D, Hyeon T (2017) Biomaterials 136:98
Moon SH, Noh S-H, Lee J-H, Shin T-H, Lim Y, Cheon J (2017) Nano Lett 17:800
Tayeb Oskoie P, Mansoori Y (2018) J Part Sci Technol 4:1
Wei Y, Han B, Hu X, Lin Y, Wang X, Deng X (2012) Procedia Eng 27:632
Baig RN, Varma RS (2013) Chem Commun 49:752
Rutnakornpituk M, Puangsin N, Theamdee P, Rutnakornpituk B, Wichai U (2011) Polymer 52:987
Donescu D, Raditoiu V, Spataru CI, Somoghi R, Ghiurea M, Radovici C, Fierascu RC, Schinteie G, Leca A, Kuncser V (2012) Eur Polym J 48:1709
Li D, Yi R, Tian J, Li J, Yu B, Qi J (2017) Chem Commun 53:8902
Kheshti Z, Hassanajili S (2017) Int J Nanosci Nanotechnol 13:119
Huang S, Yang P, Cheng Z, Li C, Fan Y, Kong D, Lin J (2008) J Phys Chem C 112:7130
Tavman A, Ülküseven B, Agh-Atabay NM (2000) Transit Met Chem 25:324
Mousavi S, Mansoori Y, Nuri A, Koohi-Zargar B (2021) Catal Lett 151:1923
Polshettiwar V, Molnár Á (2007) Tetrahedron 63:6949
Wang S, Wang K, Dai C, Shi H, Li J (2015) Chem Eng J 262:897
Malvi B, Sarkar BR, Pati D, Mathew R, Ajithkumar TG, Sen Gupta S (2009) J Mater Chem 19:1409
Armarego WLF, Chai CLL (2003) Purification of laboratory chemicals, 5th edn. Butterworth-Heinemann, Burlington
Kovar RF, Arnold FE (1976) US Patent 3,975,444
Lai D-M, Deng L, Guo Q-X, Fu Y (2011) Energy Environ Sci 4:3552
Gao J, Zhang X, Xu S, Tan F, Li X, Zhang Y, Qu Z, Quan X, Liu J (2014) Chem Eur J 20:1957
Pathak A, Singh AP (2017) J Porous Mater 24:327
Graf N, Yegen E, Gross T, Lippitz A, Weigel W, Krakert S, Terfort A, Unger WES (2009) Surf Sci 603:2849
Kruk M, Jaroniec M, Ko CH, Ryoo R (2000) Chem Mater 12:1961
Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD (1998) Science 279:548
Mondal J, Sreejith S, Borah P, Zhao Y (2014) ACS Sustain Chem Eng 2:934
Khabazipour M, Shariati S, Safa F (2016) Synth React Inorg Metal-Org Nano-Met Chem 46:759
Sing KSW (1985) Pure Appl Chem 57:603
Choubey J, Bajpai AK (2010) J Mater Sci Mater Med 21:1573
Basavaraja C, Jo EA, Huh DS (2010) Mater Lett 64:762
Sakon A, Ii R, Hamasaka G, Uozumi Y, Shinagawa T, Shimomura O, Nomura R, Ohtaka A (2017) Organometallics 36:1618
Tetko IV, Tanchuk VY, Kasheva TN, Villa AE (2001) J Chem Inf Comput Sci 41:1488
Kriek E (1971) Chem Biol Interact 3:19
Ortgies DH, Barthelme A, Aly S, Desharnais B, Rioux S, Forgione P (2013) Synthesis 45:694
Burns AR, Wallace IM, Wildenhain J, Tyers M, Giaever G, Bader GD, Nislow C, Cutler SR, Roy PJ (2010) Nat Chem Biol 6:549
Zhang Y, Feng J, Li C-J (2008) J Am Chem Soc 130:2900
Pan C, Liu M, Zhao L, Wu H, Ding J, Cheng J (2008) Catal Commun 9:1685
Zhang L, Li P, Li H, Wang L (2012) Catal Sci Technol 2:1859
Keesara S, Parvathaneni S, Dussa G, Mandapati MR (2014) J Organomet Chem 765:31
Marset X, De Gea S, Guillena G, Ramón DJ (2018) ACS Sustain Chem Eng 6:5743
Acknowledgement
The financial support provided by Graduate Council of the University of Mohaghegh Ardabili (Grant No. 9960/13/D/96</) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no competing interests to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mousavi, S., Mansoori, Y., Nuri, A. et al. A Pd(II) Magnetically Retrievable Catalyst for Hiyama Reaction: Functionalization of Magnetic Mesoporous Silica via Click Reaction. Catal Lett 152, 3465–3478 (2022). https://doi.org/10.1007/s10562-021-03905-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03905-0