
Abstract
The hyper-cross-linked porous poly(2-naphthol) fabricated by the Friedel–Crafts alkylation of 2-naphthol has been functionalized with sulfonic acid to obtain a solid acid catalyst. The catalyst is applied for the protection of phenol, alcohols, thiols, amines and aldehydes with acetic anhydride at room temperature. The catalytic protection using the new solid acid is featured by achieving high yield at neat condition, needing no aqueous work-up and/or chromatographic separation, and showing excellent recycling efficiency, suggesting the potential of this sulfonated porous polymers as a new protection protocol in a wide range of sustainable chemical reactions.
Graphical Abstract

An efficient and eco-friendly method has been developed for the protection of phenols, alcohols, thiols, amines and aldehydes with acetic anhydride in presence of sulfonic-acid-functionalized hyper-cross-linked poly(2-naphthol) as a solid acid catalyst.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The protection of functional group is essential throughout the course of different organic transformations in multi-step synthesis of organic compounds, particularly within the preparation of poly functional molecules like nucleosides, carbohydrates, and natural products [1]. Environmentally clean protection protocol has emerged as part of immense interest in the view of economic and environmental aspects. Even though different reagents are adopted for the protection of functional groups such as phenol and alcohol [2], thiols [3], amines [4], and aldehydes [5, 6], acetic anhydride is most widely used, because it is cheap and easily available, and easy to deprotect [6, 7]. In general the protection reactions were performed in the presence of either acid [8] or base catalysts [9]. However, most of the reports to date possess fundamental drawbacks including employing harsh conditions with long reaction time, using harmful organic solvents, and need tedious work-up procedures [10, 11].
The use of heterogeneous catalyst and environmentally friendly reaction conditions can be a candidate to circumvent of drastic methods and hazardous solvents. Heterogeneous polymer functionalized materials (PFMs) have been efficiently working due to their functional availability and efficient workup procedures [12,13,14]. Suitably crosslinked PFMs are featured by their high surface areas, unique properties, and a broad range of potential uses in disciplines including catalysis, sensing, energy storage and catalysis support applications [15,16,17,18]. Moreover, the PFMs catalysts show high dispersion and reactivity with a high degree of chemical stability [19].
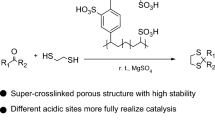
As a means of developing green and eco-friendly protection protocol of functional groups a crosslinked FPM has been fabricated by the alkylation of 2-naphthol, followed by self-crosslinking (Sch 1). The crosslinked poly(2-naphthol) (p2NPh–OH) was subjected to sulfonic acid functionalization and employed the sulfonic acid-functionalized poly(2-naphthol) (p2NPh–OSO3H) for solvent-free acylation of phenol, alcohols, thiols, amines, and aldehydes. The p2NPh–OSO3H catalyst is featured by high efficiency for targeted protection and recyclability.

Fabrication of sulfonic acid functionalized poly(2-naphthol) and its applications for the protection of phenols, alcohols, thiols, amines and aldehydes
2 Experimental
2.1 General
2-naphthol (2NPh–OH), and formaldehyde dimethyl acetal (FDA; 98%) obtained from TCI, chlorosulfonic acid and various phenols, alcohols, thiols, amines, and aldehydes obtained from Merck, 1,2-dichloroethane (DCE) from Daejung Chem. (Seoul, Korea), anhydrous FeCl3 (98%) from Acros were used as received. 1H and 13C NMR (400 MHz and 100 MHz) spectra were checked by using Varian INOVA 400 NMR spectrometer at 25 °C. The chemical shift values are quoted relative to Me4Si. Fourier transform infrared (FT-IR) spectra were recorded at 25 °C, by Shimadzu IR Prestige 21 spectrometer using KBr disk in the range between 4500 to 500 cm−1. The x-ray diffraction (XRD) study was performed by using an automatic Philips powder diffractometer with nickel-filtered Cu Kα radiation. The diffraction pattern was collected the 2θ range in between 0 and 80° in steps of 0.02 and counting times of 2 s per step. The microstructures of the samples were carryout using an S-3000 scanning electron microscope (SEM; Hitachi, Japan), and thermogravimetric analysis (TGA) was carried out using a TGA N-1000 (Scinco, Seoul, and Republic of Korea). X-ray photoelectron spectroscopy (XPS) analysis was performed in a Theta Probe AR-XPS system with a monochromatic Al Kα x-ray source (1486.6 eV). The Brunauer–Emmett–Teller (BET) and density functional theory (DFT) methods (Nova 3200e system, Quantachrome Instruments, USA) were used to examine the BET specific surface area and pore size distribution of the samples.
2.2 Synthesis of Porous p2NPh–OH and Sulfonation of p2NPh–OH
The FDA (8 mmol) was added to a solution containing 2-napthol (4 mmol) in 15 mL of DCE in a 50 mL flask, and the mixture was stirred for 20–30 min. After that anhydrous FeCl3 (8 mmol) was then added, and the reaction was continued for 18 h at 70 °C. After cooling, the reaction was quenched with MeOH and the polymer was washed with deionized water, acetone, and MeOH. The solid was additional refined by Soxhlet extraction with methanol for 24 h, then dried beneath vacuum at 70 °C for 12 h. The polymer was denoted as p2NPh–OH.
For the sulfonation of p2NPh–OH, 0.3 g of p2NPh–OH in 10 mL of CH2Cl2 at 0 °C was added drop-by-drop to a solution of chlorosulfonic acid (2.5 mL) in CH2Cl2 (10 mL) and the resultant mixture was stirred for 2 h at 25 °C. The black solid filtered and rinsed with CH2Cl2 and dried to afford p2NPh–OSO3H. The concentration of acid sites of the p2NPh–OSO3H catalysts was measured by a reverse titration with HCl (0.337 N). 10 mL of NaOH (0.130 N) was added to 0.1 g of catalyst and stirred for 30 min at room temperature. The resultant mixture was filtered and washed with deionized water. The additional amount of NaOH in the filtrate was titrated with HCl in the presence of phenolphthalein as an indicator. The concentration of acid sites was 1.14 mmol g−1.
2.3 General Procedure for Acylation Reaction
To a 10 mL reaction flask phenol/alcohol/thiol/amine/aldehyde (1 mmol), acetic anhydride (1 mmol) and p2NPh–OSO3H (10 mg) as a catalyst was added and the resulting mixture stirred at 25 °C. The progress of the reaction was checked using thin layer chromatography (TLC). After completion, ethyl acetate (EA) was added and the catalyst was separated from the reaction mixture by straightforward filtration. The separated catalyst was cleaned with EA (10 mL) and then dried in an oven for 2 h and reused for further reaction. The reaction mixture was evaporated under reduced pressure to get the products which were characterized by 1H and 13C NMR spectroscopy. All of the obtained products are renowned within the literature.
2.4 Molecular Simulations of Hypercrosslinked Poly(2-naphthol)
Molecular models for the crosslinked polymer networks were generated using the BIOVIA Materials Studio 5.0 software package (Dassault Systemes, BIOVIA Corp., San Diego, CA) with the polymer consistent force field (COMPASS II) [20]. Molecular simulations were performed with the Forcite module, using a time step of 1 fs, the Nosé-Hoover thermostat with a Q ratio of 0.01, and the Andersen barostat with a time constant of 1 ps. Monomers are packed into a periodic cell using amorphous cell module in Materials Studio. The enclosed script implemented for a crosslinking simulation using Forcite was adapted to join monomer units via a condensation polymerization based on a set of predetermined connectivity rule with the removal of simple molecules. Packing monomers and defining their reactive atoms and crosslinking sites, the crosslinked structure can be generated with any degree of crosslinking. This script is composed of three steps: (a) initial packing of the simulation box (using amorphous cell), (b) dynamics for initial equilibration: i.e. NVT dynamics run followed by NPT run, (c) a cross-linking procedure until the target degree of cross-linking is reached, and (d) dynamics for a final energy minimization. Default values were used to run the script. Close contact exclusion rules were applied during the condensation polymerization. Close contacts were deleted if (i) they are not between predetermined reactive atoms, (ii) they would exceed the conversion target, and (iii) intra-topology, meaning they would result in more than one concurrent crosslinks to an atom.
3 Results and Discussion
Post-functionalization of the hypercrosslinked p2NPh–OH with chlorosulfonic acid was monitored using FT-IR spectroscopy. The FT-IR spectra of p2NPh–OH, p2NPh–OSO3H are shown in Fig. 1. Both p2NPh–OH and p2NPh–OSO3H show –OH stretching frequencies at 3448 and 3456 cm−1, respectively, and the asymmetric and symmetric stretching bands of p2NPh–OSO3H sample at 1227 and 1058 cm−1, correspondingly, and the S–O stretching band at 775 cm−1, confirming the existence of the –SO3H cluster. Figure 1b shows the XRD patterns of the sample before and after sulfonic acid functionalization. The p2NPh–OH sample shows a very broad peak centered at 2θ = 18.5° and the p2NPh–OSO3H sample at 2θ = 23.5°. Both samples are basically amorphous and the shift of the center is related with the functionalization of mainly peripheral –OH groups in p2NPh–OH to bulky –OSO3H groups.

a FT-IR spectra, b XRD patterns, and c TGA thermograms of p2NPh–OSO3H
The TGA curves of p2NPh–OH and p2NPh–OSO3H, are shown in Fig. 1c. The TGA curve of the sulfonated materials show the first weight losses 6.5% at 100 °C, which correspond to the loss of water molecules present in the pore surface, followed by the loss of adsorbed water molecules having a non-covalent interaction with the free sulfonic acid groups. The further, weight losses of 55.4% in the temperature range 130–280 °C could be attributed to the loss of free sulfonic acid groups followed by the cleavage of the organic framework beyond 530 °C. The high thermal stability of the porous polymers could be attributed to the hyper crosslinking and the rigid polymeric networks in the molecular skeletons of p2NPh–OSO3H, (Fig. 1c). The SEM images of p2NPh–OH and p2NPh–OSO3H in Fig. 2a–d shows a mixed tubular and sphere morphology, and, after sulfonation, the tubular and sphere morphology of the polymers was retained.

SEM images of p2NPh–OH (a, b) and p2NPh–OSO3H (c, d)
Nitrogen adsorption, desorption isotherms and the textural properties of p2NPh–OH and p2NPh–OSO3H are shown in Fig. 3. The BET surface area of p2NPh–OH, 408 m2g−1, decreases significantly once sulfonic acid functionalization to 180 m2g−1. The large sulfonic acid groups are grafted onto the porous surface of the p2NPh–OH, resulting in overcrowd partial pores in p2NPh–OH. Additionally, the decrease in surface area after the functionalization of the p2NPh–OH is due to the raise in the mass of the fabric with regard to the load of weight percent of the polymer. Furthermore, the p2NPh–OSO3H shows significantly lower mesoporous volume than that of p2NPh–OH. These various changes within the textural properties of p2NPh–OSO3H indicate the productive functionalization of p2NPh–OH. The pore size distribution of p2NPh–OH and p2NPh–OSO3H was determined by DFT, indicating foremost pore widths of p2NPh–OH and p2NPh–OSO3H centered at about 9.2 and 8.2 Å (Fig. 3b), respectively. These results clearly suggest that these polymers contain micropores as well.

a Nitrogen adsorption/desorption isotherms and b pore size distribution of p2NPh–OH and p2NPh–OSO3H
For the atomic simulations [21] of p2NPh–OH the amorphous cells were produced with 200 seed molecules, 1,6-alkylated 2-naphthol, as shown in Fig. 4. The crosslinks were formed between atoms labelled R1 and R2 in 1,6-bis(methoxymethyl)naphthalen-2-ol monomer. For the crosslinking close contacts are searched between R1 and R2 within threshold (here < 4Å). A spring connects R1 and R2 and gradually stiffer by harmonic restraint. Then, the spring between R1 and R2 is replaced by bond. In order to fit stoichiometry methoxy group on R1 and hydrogen atom on R2 is removed as methanol molecule. After the new bond formation and the removal of methanol, atom types, charges and charge groups in the amorphous cell are recalculated and then repeat the same processes to achieve the target conversion. The amorphous cells for the 200-unit cluster with solvent accessible surfaces (blue) according to the degree of crosslinking (X) are shown in Fig. 4b. The simulation box size is 38.50 Å at X = 11.00% and 35.62 Å at X = 100%. At low degree of crosslinking, both samples are essentially nonporous, since the flexible and mobile nature of growing polymer chains prevents pores from forming. As the degree of crosslinking increases to a specific degree expected micropores are formed.

a 1,6-bis(methoxymethyl)naphthalen-2-ol monomer used for the synthesis of hypercrosslinked poly(2-naphthol) networks showing polymerization procedures and b snapshots of molecular simulation boxes of p2NPh–OH taken at various degrees of crosslinking (X), showing the solvent-accessible surface areas (blue areas). For amorphous cells construction, 200 units monomer were used. Dark blue areas represent the micropore surface and grey balls and sticks represent newly formed crosslink sites
Figure 4b displays the pore volumes as defined by the solvent-accessible surface [22] at different degrees of crosslinking. To compare experimental BET surface area with geometric one the solvent-accessible surface area is commonly used. Even though the geometric and BET surface areas are fundamentally different and should be compared with care, geometric surface areas obtained for p2NPh–OH using an N2-size probe (dN2 = 3.681 Å) in Materials Studio. The surface area is found to record the maximum value, 503.88 m2g−1, at X = 68.25%. Strict trend of the variation of surface area according to the degree of crosslinking is not observed; however, the surface area decreases as the X value increases. Finally the surface area becomes 200.28 m2g−1 at X = 100%. Considering the experimental BET surface area of p2NPh–OH is 408 m2g−1, the calculated solvent-accessible surface areas show reasonable agreement with the experimental BET value as measured by N2 sorption. The origination of discrepancies between BET surface area and geometrical one have been well described in the literature [23].
The p2NPh–OSO3H, is characterized by XPS. As shown in Fig. 5a, peaks corresponding to oxygen, carbon, and sulfur are a unit clearly determined, confirming the presence of sulfonic acid groups in p2NPh–OSO3H. The S(2p) peak of p2NPh–OSO3H corresponds to S(2p3/2) [24, 25], indicating the presence of SO3H within the p2NPh–OSO3H. The reduced binding energy of the S(2p) peak is determined for p2NPh–OSO3H (Fig. 5d), suggesting a rise within the electron density on the sulfur atoms.

XPS spectrum of p2NPh–OSO3H (a) and its expanded spectra (b–d) corresponding to C(1 s), O(1 s) and S(2p), respectively
The catalytic activity of p2NPh–OSO3H was investigated for the acylation of phenols, alcohols, thiols, amines, and aldehydes with acetic anhydride (Sch 1). To pre-optimize the reaction conditions, a model reaction using phenol with acetic anhydride was conducted at various conditions in the presence of p2NPh–OSO3H catalyst. Various protic (ethanol, methanol, and water) and aprotic (CH2Cl2, CHCl3, and toluene) solvents were firstly used for the acylation, resulting in insufficient yields (50–75%) even after 3 h of reaction (see entries 1–6 in Table 1). The acylation of phenol was then conducted under solvent free conditions. Entry 7 in Table 1 clearly shows the acylation of phenol gives much better yield (97%) in much shorter reaction time (15 min) under neat condition. The decrease of catalyst amount from 10 to 5 mg results in the decrease of yield (89%) and the increase of catalyst amount to 15 mg showed no further increase of yield. As expected, no products were observed in the absence of catalyst even after a prolonged reaction time (entry 10 in Table 1).
Having the optimized conditions, we further examined for the acylation of varied phenols and benzylic alcohols. As shown in Table 2, all phenols and benzylic alcohols were under gone smoothly to the resultant acetates in sensible yields. The benzylic alcohols bearing electron-releasing groups resulted in better yields with shorter reaction times than aromatic substrates bearing electron-withdrawing groups (entries 3 and 4). The acylations of a range of thiols, amines and aldehydes were also attempted to confirm the potential of the solid acid catalyst. The acylations of phenols and thiols were proceeded slowly, comparing to amines and aldehydes. Specifically high activities were achieved for the acylations of varied substituted aromatic amines and substituted aldehydes. No chemo-selectivity was observed for compounds with two different functional groups like –SH and –OH in a molecule (entry 8, Table 2). However, the p2NPh–OSO3H catalyst showed very good substrate selectivity between the phenol and benzyl alcohol: i.e. it gave predominantly acylated product of benzylic alcohol. In the case of ethane-1,2-dithiol and salicylaldehydes the corresponding diacetates and triacetates were formed.
Based on the obtained results, the following reaction mechanism might be proposed for the acylation of phenols, alcohols, thiols, amines in the presence of p2NPh–OSO3H (Sch 2). The mechanism involves the protonation of the carbonyl group of acetic anhydride by p2NPh–OSO3H catalyst. Then the nucleophilic addition of various substrates on the electron deficient carbonyl carbon results in the corresponding acylated products.

Proposed mechanism for the p2NPh–OSO3H-catalyzed acylation of phenols, alcohols, thiols, and amines
For the comparison of the catalyst performance, the acylation of benzyl alcohol with acetic anhydride was summarized in Table 3. The polydopamine sulfamic acid-functionalized magnetic Fe3O4 nanoparticles (Fe3O4@PDA–SO3H) showed high activity at neat condition [26]. However, it needs tedious multi-step procedures to prepare the catalyst, which makes commercially troublesome. In situ generated Ph3P(OAc)2 acted as reasonable organo-catalyst for the protection of alcohols and thiols at mild conditions [27]. Harmful polar solvent like acetonitrile was used to achieve 87% yield in 120 min for the acylation of benzyl alcohol. Sodiumacetate trihydrate (NaOAc·3H2O) was also used for the acylation of benzyl alcohol, needing laborious workup procedure [28]. Bromodimethylsulfonium bromide (Me2S+Br Br−) was used as a catalyst for the protection of phenols, alcohols, thiols, amines and aldehydes [29]. For the acylation of 1 mmol of benzyl alcohol large amount of catalyst (111 mg) and 2 mmol of acetic anhydride were used to achieve 90% yield in 25 min.
The reusability of p2NPh–OSO3H was also examined for the preparation of acylation of phenol, from the reaction of phenol with acetic anhydride. The catalyst was easily recovered by adding EA to the reaction mixture; the insoluble p2NPh–OSO3H was separated by simple filtration, washed twice with EA (15 mL), and finally dried under vacuum. The catalyst displayed good reusability after 10 runs (Fig. 6).

Effect of p2NPh–OSO3H recycling on the yield of the compound 1
4 Conclusions
Hyper-cross-linked and porous poly(2-naphthol) was prepared by using Friedel–Crafts alkylation-induced polymerization of 2-naphthol and was acid-functionalized to get solid acid p2NPh–OSO3H catalyst (1.14 mmol –OSO3H g−1). The catalyst showed excellent activities for the acylation of phenols, alcohols, thiols, amines, and aldehydes at eco-friendly reaction conditions. The catalyst also showed perfect chemo-selectivity for the acylation of phenol and alcohol. In addition the catalyst retained its activity up to 10 consecutive recycle reactions. The straightforward preparation method of the porous solid acid catalyst, neat and mild reaction conditions, high yields of acetylated products, versatility to variety of organic compounds, facile workup, and recyclability of the catalyst must promote this method as one of the candidates for the protection of phenols, alcohols, thiols, amines, and aldehydes in organic chemistry.
References
Greene TW, Wuts PGM (1999) In protective groups in organic synthesis. Wiley, New York
Sefkow M, Kaatz H (1999) Tetrahedron Lett 40:6561–6562
Arjona O, Medel R, Rojas J, Costa AM, Vilarrasa J (2003) Tetrahedron Lett 44:6369–6373
Kalla RMN, Lim J, Bae J, Kim I (2017) Tetrahedron Lett 58:1595–1599
Kalla RMN, Park H, Hoang TTK, Kim I (2014) Tetrahedron Lett 55:5373–5376
Kalla RMN, Kim MR, Kim YN, Kim I (2016) New J Chem 40:687–693
Gangadhar KN, Vijay M, Prasad RNB, Devi BLAP (2013) Green Sus Chem 3:122–128
Khaligh NG (2012) J Mol Catal A 363–364:90–100
Vedejs E, Diver ST (1993) J Am Chem Soc 115:3358–3359
Tamaddon F, Amrollahi MA, Sharafat L (2005) Tetrahedron Lett 46:7841–7844
Kumar P, Pandey RK, Bodas MS, Dagade SP, Dongare MK, Ramaswamy AV (2002) J Mol Catal A 181:207–213
Maly KE (2009) J Mater Chem 19:1781–1787
Sun Q, Dai Z, Meng X, Xiao F-S (2015) Chem Soc Rev 44:6018–6034
Kalla RMN, Varyambath A, Kim MR, Kim I (2017) Appl Catal A 538:9–18
Ding S-Y, Dong M, Wang Y-W, Chen Y-T, Wang H-Z, Su C-Y, Wang W (2016) J Am Chem Soc 138:3031–3037
Zhou Y-B, Wang Y-Q, Ning LC, Ding ZC, Wang W-L, Ding CK, Li R-H, Chen J-J, Lu X, Ding Y-J, Zhan Z-P (2017) J Am Chem Soc 139:3966–3969
Liu T-T, Liang J, Huang Y-B, Cao R (2016) Chem Commun 52:13288–13291
Xu H, Gao J, Jiang DL (2015) Nat Chem 7:905–912
Zhang Y, Xiong Y, Ge J, Lin R, Chen C, Peng Q, Wang D, Li Y (2018) Chem Commun 54:2796–2799
Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG (1995) J Chem Phys 103:6951–6958
Trewin A, Willock DJ, Cooper AI (2008) J Phys Chem C 112:20549–20559
Duren T, Millange F, Fe´rey G, Walton KS, Snurr RQ (2007) J Phys Chem C 111:15350–15356
Hart KE, Abbott LJ, Colina CM (2013) Mol Simul 39:397–404
Russo PA, Antunes MM, Neves P, Wiper PV, Fazio E, Neri F, Barreca F, Mafra L, Pillinger M, Pinna N, Valente AA (2014) J Mater Chem A 2:11813–11824
Ho TTM, Bremmell KE, Krasowska M, Stringer DN, Thierry B, Beattie DA (2015) Soft Matter 11:2110–2124
Veisi H, Taheri S, Hemmati S (2016) Green Chem 18:6337–6348
Iranpoor N, Firouzabadi H, Davan EE (2013) Tetrahedron Lett 54:1813–1816
Mojtahedi MM, Samadian S (2013) J Chem 2013:1–7
Khan AT, Islam S, Majee A, Chattopadhyay T, Ghosh S (2005) J Mol Catal A 239:158–165
Acknowledgements
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea (2018R1D1A1A09081809). The authors are also grateful to the BK21 PLUS Program for partial financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kalla, R.M.N., Reddy, S.S. & Kim, I. Acylation of Phenols, Alcohols, Thiols, Amines and Aldehydes Using Sulfonic Acid Functionalized Hyper-Cross-Linked Poly(2-naphthol) as a Solid Acid Catalyst. Catal Lett 149, 2696–2705 (2019). https://doi.org/10.1007/s10562-019-02811-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-02811-w