
Abstract
Two series of Fe/Cu/K catalysts were prepared by co-precipitation (CP-xK) and by co-precipitation accompanied with hydrothermal treatment (HY-xK), respectively. The catalysts were investigated by N2 adsorption, X-ray diffraction (XRD), Mössbauer effect spectroscopy (MES), scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM), H2 temperature-programmed reduction (H2-TPR) and thermogravimetric analysis. N2 adsorption revealed that HY-xK catalysts displayed relatively low surface area, but generated additional macropore. XRD and SEM showed that HY-xK catalysts were well grown crystallite, while the CP-xK catalysts were amorphous. In addition, hydrothermal treatment remarkably influenced the growth orientation of hematite nanocrystals, resulting in the preferential exposure of the (110) plane. HRTEM also indicated that HY-1.32K was dominated by Fe2O3 nanocrystals with (110) plane, and the CP-1.38K primary particle mainly exposed (104) plane. The H2-TPR profiles for the two series of catalysts were similar, though the merged and smooth peak of HY-xK catalysts possibly suggested that the component elements were more uniform. MES and XRD indicated that the catalyst after hydrothermal treatment can be easily reduced into active carbide phases in Fischer–Tropsch synthesis (FTS) reaction. In FTS reaction, the HY-xK catalysts showed higher activity and better stability than the CP-xK catalysts given comparable K content.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Fischer–Tropsch synthesis (FTS), a well-known process to convert syngas into liquid fuels and chemicals, has been attracting great interest due to petroleum resource shrinking in the past decades [1]. Iron-based catalysts are preferred for FTS reaction due to their low cost, high activity and excellent water–gas-shift reactivity, which match the syngas from coal gasification with low H2/CO ratio [2]. Remarkably, three principal challenges come for iron-based FTS catalyst: activity, selectivity and stability [3]. At present, modification of iron-based catalyst by introducing promoters or supports, or change of the preparation methods is being explored to achieve the goal.
Among the chemical promoters, Cu can enhance the hematite reducibility. When copper oxide is reduced to metallic Cu, the crystallites provide H2 dissociation sites, which spillover to iron oxides and promote its reduction at lower temperatures [4, 5]. The well-known electronic agent K in the iron-based catalysts contributes to improving the dissociative adsorption of CO [6], strengthening the bond of Fe–C by increasing the electron density on Fe while weakening Fe–H and C–O bonds; [6–8] favoring the formation of olefins and longer-chain hydrocarbon molecules [8] and the suppression of CH4 formation [9, 10]; facilitating the water–gas-shift activity [11, 12] and the carburization of surface Fe as well [13]. However, excessive K loading will break the balance between the CO dissociation and the carbon hydrogenation, leading to the deactivation of catalysts for an excessive carbon deposition [14]. So, the appropriate amount of K promoter is essential for iron catalysts.
The method of catalyst preparation also plays an important role in the physical property and catalytic performance of the catalysts [15]. Nowadays, the iron-based catalyst has been prepared by the co-precipitation [11, 16], fusion [9, 17], sol–gel [18], microemulsions [19] and so on [20–22]. It is known that during the preparation of typical iron-based FTS catalyst, the precipitate is boiled and maintains for several minutes [11, 23]. Kölbel et al. [23] reported that brief boiling of the precipitate facilitates the filtration and titrimetric standardization of the precipitant against the iron solution. In essence, this operation is a hydrothermal treatment at room pressure. In this paper, hydrothermal treatment is operating at high temperature and pressure. However, few reports refer to the iron-based catalysts prepared by co-precipitation accompanied with hydrothermal treatment. The FTS reaction is a structure sensitive reaction [24]. It is well known that hydrothermal treatment operating at elevated temperature and pressure [25] allows the synthesis material with particular morphology [26, 27]. In addition, various structures may be formed such as morphology, dispersion, phases, if different conditions of hydrothermal method are applied. These differences may affect the FTS activity, selectivity and stability of catalysts. Therefore, the effect of hydrothermal treatment on the structure of iron catalysts needs to be investigated in more detail.
In our previous studies, the CoFe nanocrystals were obtained by the hydrothermal treatment and thermal decomposition of the iron-doped cobalt carbonate hydroxide hydrate intermediate on an iron substrate, and were tested in fixed-bed reactor. From the results of characterization study demonstrate that, CoFe nanowires catalyst and nanosheets catalyst exposed distinguished crystal planes and showed different FTS performance [28].
The purpose of the present study is to investigate the catalytic performance and the changes in structure and crystal plane of precipitated iron-based catalysts which involves hydrothermal treatment, instead of being untreated to be clear of its effect. Since K promoter influences the performance greatly, Fe/Cu/K catalysts of varying K content are designed.
2 Experimental
2.1 Catalyst Preparation
The HY-xK catalysts were prepared by co-precipitation accompanied with hydrothermal treatment. In a typical synthesis, 10 mmol of Fe(NO3)3, 0.53 mmol of Cu(NO3)2 and 40 mmol of (NH2)2 CO were dissolved in 100 mL of deionized water under stirring and then transferred into a Teflon-lined stainless steel autoclave, sealed and maintained at 110 °C for 8 h. After cooling down to the room temperature, the obtained solid product was recovered by filtration and washed with deionized water. The potassium promoter was added in a calculated amount to the precursor by impregnation with aqueous solution of K2CO3 to give the desired K content. The final products were further dried in air at 100 °C for 10 h and then calcined at 350 °C for 6 h.
For comparison, the CP-xK catalysts were prepared by the co-precipitation method. A solution containing both 10 mmol of Fe(NO3)3 and 0.53 mmol of Cu(NO3)2 and a separate solution of NH4OH (0.5 mol L−1) were used in the precipitation processes. Under stirring, the ammonium hydroxide solution with a certain flow rate was added into the salt solution to achieve a pH value of 8.8–9.0. The precipitate was filtered, washed completely with deionized water. The potassium promoter was added in a calculated amount to the precursor by impregnation with aqueous solution of K2CO3 to give the desired K content. The final products were further dried in air at 100 °C for 10 h and then calcined at 350 °C for 6 h.
The series catalysts prepared by co-precipitation accompanied with hydrothermal treatment are denoted as HY-xK and the series catalysts prepared by co-precipitation method are denoted as CP-xK; in both cases x denotes the wt% of K2O.
2.2 Catalyst Characterization
The textural properties of catalysts were determined via N2 adsorption at −195.8 °C with a TriStar 3000 Gas Absorption Analyzer (Micromeritics Instrument Co., USA) and surface area was calculated using the BET method. The pore diameter was calculated by applying the Barret–Joyner–Halenda method (BJH) to the adsorption branches of the N2 isotherms. The samples were degassed at 200 °C and 6.7 Pa for 2 h prior to the measurement.
The actual composition of the catalysts was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). To prepare the solution for elemental analysis, 2 mL of concentrated nitric acid was used to dissolve 40 mg of catalyst sample, followed by adding 2 mL of 30 wt% H2O2; the solution was then diluted to 1,000 mL with deionized water.
X-ray diffraction (XRD) measurements were carried out with a D/max-RA X-ray diffractometer (Rigaku, Japan), equipped with Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 150 mA. The measurements were made at room temperature in the 2θ range of 10–80 °C with a scanning rate of 2 or 4 min−1.
The Mössbauer effect spectroscopy (MES) of catalysts was obtained on an MR-351 constant-acceleration Mössbauer spectrometer (FAST, Germany) at room temperature, using 25 mCi 57Co in a Pd matrix. The spectrometer was operated in a symmetric constant- acceleration mode.
The morphology of catalysts was investigated by scanning electron microscopy (SEM) (JSM-6701F, Japan). The catalysts were treated by dessication and spray-gold.
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images of the catalysts were obtained by using a JEM 2010 microscope operating at 200 kV. The catalysts were dispersed in ethanol and mounted on a carbon foil supported on a copper grid.
H2 temperature-programmed reduction (H2-TPR) was carried out using a dynamic analyzer (Micromeritics, Model 2920). About 30 mg of catalyst was treated in 5 % H2/95 % Ar (v/v) (flow rate 50 mL min−1), and the reduction temperature was increased from room temperature to 1,000 °C at a heating rate of 10 °C min−1. The hydrogen consumption was calibrated using the H2-TPR of CuO (Aldrich, 99.99+ %) as the standard sample under the same conditions.
Thermogravimetric analysis (TGA) was conducted on a thermogravimetric analyzer of Setsys Evolution TGA 16/18 in nitrogen atmosphere from ambient temperature to 650 °C with a heating rate of 10 °C min−1.
2.3 Reactor System
The FTS performance of the catalysts was tested in a stainless steel fixed-bed reactor with inner diameter of 12 mm loaded with 2 mL of catalyst.
The syngas (H2/CO = 2) was passed through a series of purification traps to remove trace amounts of iron carbonyls, water, and other impurities. The flow rate of syngas was controlled by mass flow controller (Brooks, model 5850E). The exit stream passed through a hot trap (130 °C), a back-pressure regulator and a cold trap (0 °C) to collect liquid products. A wet gas flow meter was used to monitor the flow rate of tail gas.
The catalyst was reduced in situ in syngas at 290 °C, 0.5 MPa and 1,000 h−1 for 20 h. After reduction, steady-state reaction conditions were set as 220–290 °C, 1.5 MPa, H2/CO = 2 and 2,000 h−1.
The tail gas was analyzed online by gas chromatography (GC) (models 6890N and 4890D, Agilent) equipped with a 16-port sampling valve and two sampling loops. In one sampling loop, the hydrocarbons were analyzed with an Al2O3 capillary column (HP-PLOT, 30 m × 0.53 mm) and a flame ionization detector (FID). In the other loop, H2, N2, CO, CO2, and CH4 were analyzed with a Porapak Q stainless steel packed column (2 m × 3 mm), a PLOT Q packed column (HP-PLOT, 25 m × 0.53 mm), a molecular sieve 5A packed column (HP-PLOT, 30 m × 0.53 mm), and a TCD. The products in oil phase were analyzed off-line by GC (model 6890N, Agilent) with a quartz capillary column (DB-1, 60 m × 0.25 mm) and a FID. The products in wax were analyzed offline using a gas chromatograph (6890N, Agilent, USA) with a stainless steel capillary column [UA+-(HT), 30 m × 0.53 mm] and a FID. Oxygenates in water were analyzed off-line using GC (model 6890N, Agilent) with a quartz capillary column (J&W DB-WAX, 30 m × 0.32 mm) and a FID.
The carbon balance and mass balance were 100 ± 5 %.
3 Results and Discussion
3.1 Textural Properties of Catalysts
The textual properties (BET surface area, pore volume and average pore diameter) and elemental compositions of the catalysts are shown in Table 1. BET surface area of all samples after hydrothermal treatment is lower than that untreated. Obviously, the catalyst surface area depends on the K content almost linearly in each series. Dry et al. [9] mentioned that the potassium could improve the agglomeration of the FeOOH precursor and further enlarge the crystallite size of α-Fe2O3 after being calcined, which would induce the decrease in surface area. In the preparation of the CP-xK catalysts, the FeOOH precursor is small and the dangling hydroxyl group is rich on the surface. In contrast, the FeOOH precursor in HY-xK catalysts is large and the dangling hydroxyl group is less. The term “dangling hydroxyl group” in this paper is defined as “hydroxyl groups located on the surface of FeOOH precursors that easily lead to agglomeration of FeOOH precursors via oxygen bridge mechanism”. The pore distribution of catalysts prepared by different methods is shown in Fig. 1. CP-xK catalysts exhibit a relatively narrow pore size distribution. HY-xK catalysts show a bimodal pore distribution with maxima at 3 and 40 nm, respectively. In addition, Table 1 shows that the elemental composition of CP-xK catalysts is similar to the corresponding HY-xK catalysts, thus providing a comparison benchmark.

The pore size distribution of catalysts as-prepared
3.2 Crystallite Structure of Catalysts
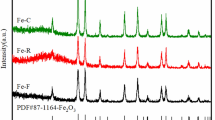
XRD patterns of catalysts as-prepared with different methods are presented in Fig. 2a. The only detectable phase in the diffraction patterns of the catalysts with different preparation methods is hematite. It is obvious that the CP-xK catalysts display broad diffraction peaks at 2θ of 35° and 64°, which is the characteristic of amorphous hematite with small crystallite diameters (about 4 nm). In contrast, iron phases in HY-xK catalysts are well-crystallized hematite with sharp diffraction peaks at 2θ angle of 24.2°, 33.1°, 35.6°, 40.8°, 49.52°, 54.0°, 57.6°, 62.5° and 64.0°. Interestingly, comparison with the standard stick pattern (JCPDS Card No. 033-0664) of pure hematite, the peak of the (110) plane is anomaly higher than that of the (104) plane. This result indicates that hydrothermal treatment remarkably influences the growth orientation of hematite nanocrystals, resulting in the preferential exposure of the (110) plane. Moreover, it is found that the intensity of α-Fe2O3 characteristic peak increases monotonously with the content of potassium for both series catalysts. In summary, the hydrothermal treatment and potassium addition promote the growth of α-Fe2O3 crystallite. This is in agreement with the decrease in the catalyst surface area.

XRD pattern of catalysts: (a) as-prepared and (b) after reaction: filled triangle quartz; filled circle magnetite; filled diamond Fe5C2
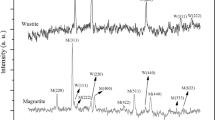
The XRD patterns of the used catalysts are shown in Fig. 2b. There are feature lines assigned to dilute quartz in all samples. Two series of catalysts, are almost dominated by diffractions at 2θ of 35.5°, 43.1°, 57.0° and 62.6°, suggesting that magnetite (Fe3O4) formed during FTS is highly crystallized, which is consistent with the observations by Bukur et al. [29]. It is well known that although magnetite is the most active phase for the water–gas-shift (WGS) reaction on iron-based catalysts [30], this phase is not active for FTS, as Huang et al. [31] reported that magnetite alone showed no initial activity when being exposed to syngas. The peaks at 39.3° and 43.5° are attributed to the characteristic diffraction of iron carbide (χ-Fe5C2) which is the active phase for FTS [32]. The relatively stronger diffraction peak of iron carbide for the catalysts after hydrothermal treatment, compared with that for co-precipitated catalysts, conforms with the higher FTS activity of the catalysts after hydrothermal treatment. Because K has been found to be the most efficient promoter for iron-based catalysts among all of the additives studied to date, their promoting effects on the FTS performance of the two series of catalysts are also investigated in the present work aiming to improve the FTS selectivity. If the K content is similar, the iron phase is comparable for two preparation methods. In addition, the XRD patterns indicate that the diffraction peak intensities of iron carbide are enlarged and the Fe3O4 peak decreases with the increase of K content in CP-xK catalysts. This meant that the K is helpful to stable the carbide by preventing from steam oxidation during FTS. Particularly, the peak intensity of iron carbide in HY-1.32K catalyst is higher than that of the CP-1.38K catalyst and magnetite of HY-1.32K catalyst is not detected in XRD patterns. In order to further determine iron phase compositions of catalysts, the used catalysts (CP-1.38K and HY-1.32K) were measured by MES. Figure 3 shows the Mössbauer spectra of catalysts. MES parameters of various phases and their relative contributions to spectral absorption areas are summarized in Table 2. The subspectra with Hhf in the range of 476–504 and 445–467 kOe are contributed to ferrimagnetic Fe3O4 phases. The subspectra with 0.20 mm/s <IS <0.40 mm/s and 102 kOe <Hhf <256 kOe correspond to the stoichiometric χ-Fe5C2 [33]. As can be seen, the CP-1.38K catalyst is composed of Fe3O4 and χ-Fe5C2, while the HY-1.32K catalyst is only composed of χ-Fe5C2. This is in agreement with XRD results. However, in HY-1.32K catalyst, content of iron carbide is higher than that in CP-1.38K catalyst. The high iron carbide content indicates that the catalyst after hydrothermal treatment can be easily reduced into active carbide phases in FTS reaction.

Mössbauer spectra of catalysts after reaction
XRD patterns of catalysts after different time on stream (TOS) are presented in Fig. 4. It is found that the diffraction peak intensities of iron carbide of CP-1.38K catalyst after running 72 h are higher than that after running 480 h. However, the diffraction peak intensities of iron carbide of HY-1.32K catalyst after running 72 h are lower than that after running 480 h. It indicates that the stability of iron carbide of HY-1.32K catalyst is improved in FTS reaction. This improved stability may be ascribed to the stabilization effect caused by catalyst structure after hydrothermal treatment. Under typical FTS conditions where H2O is present, the iron carbide can be oxidized to Fe3O4, leading to the deactivation of the catalysts [34]. The catalyst structure after hydrothermal treatment has broad pore size distribution and uniform particles which facilitate the diffusion of gas and mass transport. Notably, H2O carried with gas in the FTS will be removed quickly. For the catalyst after hydrothermal treatment, the iron carbide will be slower oxidized by H2O than that catalyst untreated. It indicates that hydrothermal treatment improves the stability of catalyst.

XRD patterns of catalysts after different time on stream: filled triangle quartz; filled circle magnetite; filled diamond Fe5C2
3.3 SEM and TEM Results
The morphology and microstructure of the as-prepared CP-1.38K and HY-1.32K catalysts were elucidated by means of SEM and TEM. The SEM images of CP-1.38K and HY-1.32K catalysts are shown in Fig. 5. CP-1.38K catalyst is plane and exposes irregular facet. The images reveal that the primary particle size of CP-1.38K catalyst is small (Fig. 5a, b). This suggests that the catalyst may be composed of numerous small iron oxide particles, bound together via oxygen bridge connection (Fig. 5b). The primary particles are so close to each other that accelerate the particle sintering rate, as observed by BET. The HY-1.32K catalyst shows shale-like shape and is regular, attributed to the particle reforming after hydrothermal treatment (Fig. 5c, d). In high magnification (Fig. 5d), the HY-1.32K bulk is packed with nanoparticles. By reference to XRD, the nanoparticle is crystallite.

SEM images of catalysts as-prepared: (a, b) CP-1.38K; (c, d) HY-1.32K
TEM and HRTEM images of CP-1.38K and HY-1.32K catalysts are shown in Fig. 6. It can be seen that the as-prepared CP-1.38K catalyst shows irregular agglomeration of 4 nm nanoparticles (Fig. 6a), while the HY-1.32K catalyst is composed of uniform particles, which are about 11 nm in diameter (Fig. 6c). The HRTEM images further illustrate that the exposed crystal planes of CP-1.38K catalyst are dominated by (104) plane of α-Fe2O3 (Fig. 6b), while HY-1.32K catalyst mainly exposes (110) plane (Fig. 6d). This is in agreement with the phenomenon in XRD. As suggested by Li et al. [35], precipitated Fe–Mo Fischer–Tropsch catalysts were dominated by (104) plane of α-Fe2O3. Cornell et al. [36] reported that the (110), (100), (012) and (113) planes were fringed with singly coordinated hydroxyls in pairs, whereas (104) and (001) planes were covered with bi-coordinated hydroxyl, which was a stronger ligand. In fact, this feature is utilized to preferably adsorb H2PO4 −, thus makes the (104) plane growth halt, and to control the morphology of Fe2O3 powder [37]. The CP-1.38K catalyst is rich in (104) plane and bi-coordinated hydroxyl group, so the particle tends to merge via hydroxyl bridge.

TEM and HRTEM images of catalysts as-prepared: (a, b) CP-1.38K; (c, d) HY-1.32K
3.4 Reduction Behaviors of Catalysts
The reduction behaviors of catalysts were studied by H2-TPR. The reduction profiles and H2 consumption for the catalysts are presented in Fig. 7 and Table 3, respectively. It is reasonable to assume that only the Fe2O3 phase was primarily present after calcination based on the similar TPR profiles for all the catalysts. Figure 7 reveals that all catalysts show multiple reduction peaks, which can be assigned to two different reduction stages of iron oxides. The first stage (lower-temperature reduction peak) is assigned to the transformations of CuO → Cu and Fe2O3 → Fe3O4, whereas the second stage (two higher-temperature reduction peaks) represents the transformation of Fe3O4 → Fe [38]. The sum of H2 consumption for first stage (0.15–0.19 mol H2/mol M), consistent with the theoretical value of CuO → Cu and Fe2O3 → Fe3O4 (0.208 mol H2/mol M), verifies the above statement.

H2-TPR profiles of catalysts
For the CP-xK catalysts, the reduction peaks for the first stage are sawtooth shaped and not smooth. However, for the HY-xK catalysts, the reduction peaks for the first stage are smooth, meaning that the iron is mixed well with copper in the latter and the promotion of Cu is more efficient. There appears to be better synergistic interaction between the copper promoter and the iron in HY-xK catalysts so that copper is more effective to the reduction of catalysts. It suggests that HY-xK catalysts form a uniform Fe–Cu matrix after hydrothermal treatment, while CP-xK catalysts form inhomogeneous Fe–Cu matrix. Comparison of the H2-TPR profiles of the CP-xK and HY-xK catalysts with similar K content clearly shows that the reduction of CP-xK catalysts starts at slightly lower temperature, but the reduction of HY-xK catalysts finishes at lower temperature. When K content is low, both CP-xK and HY-xK catalysts display comparable reduction temperature (Fig. 7). When K content is high, the peak of first stage is shifted to higher temperature [39]. Rankin and Bartholomew [40] have reported that the interaction of iron oxide and potassium oxide could cause a delay in the reduction of iron oxide due to the suppressed adsorption of H2.
3.5 TGA
TGA profile of catalysts after calcination is not shown. In the TGA curve of HY-1.32K, there is a small weight loss of about 4.3 % due to desorption of physisorbed water molecules from 100 to 300 °C. No further weight loss and no thermal effect are observed after 300 °C. But in the TGA curve of CP-1.38K, there is weight loss of about 8 % due to desorption of physisorbed water molecules from 100 to 300 °C. In addition, a significant weight loss of about 9.8 % is observed in the temperature range of 300–520 °C, due to the condensation dehydration of surface hydroxyl groups [41]. It indicates that HY-1.32K catalyst displays less hydroxyl amounts, in accordance with HRTEM characterization.
3.6 FTS Performance
The activities of the two series of catalysts in the temperature range of 220–290 °C are shown in Fig. 8. As expected, all catalysts show increased CO conversion with increasing temperatures from 220 to 290 °C. The activity of HY-xK catalysts is higher than that of CP-xK catalysts if the K content is comparable. Also, turnover frequencies (TOFs) of HY-xK catalysts are higher than that of CP-xK catalysts at the same temperature. Ignoring consideration of the BET area, the close activity of two series catalysts is astonishing. Possibly, the texture is transformed into a final state no matter what the initial structure is. Mahajan et al. [42] reported that the final state of nanometer iron was similar to that of micronmeter iron in FTS. The CO conversion increases significantly with increasing K content and passes through a maximum at 1.32 wt% for HY-xK catalysts and 1.38 wt% for CP-xK catalysts. Further increase in K content leads to a decrease in CO conversion for both catalysts [14]. The FTS activity depends on both the K content and the preparation method. The effect of K content on the FTS performance of the catalysts observed here is in agreement with the studies on other iron-based catalysts [14]. Miller and Moskovits [10] found that there was a competition between dissociative CO chemisorption and H2 adsorption on the active sites of catalysts, which leaded to a maximum in conversion as a function of alkali-metal content. When the K content is even higher, CO dissociation proceeded faster than carbon hydrogenation, leading to an excessive carbon deposition that eventually deactivates the catalyst [43], which explains the lower activity of the catalysts with higher K content. The catalysts after hydrothermal treatment are more sensitive to K content, probably due to the uniform dispersion of Cu or the regular crystallite.

Effects of potassium content and temperature on carbon monoxide conversion of catalysts
The methane and high-molecular-weight hydrocarbon (C5 +) selectivities of catalysts under different temperatures are shown in Fig. 9. Hydrocarbon product distributions of catalysts are summarized in Table 4. The methane selectivity of HY-xK catalysts is slightly lower than the corresponding CP-xK catalysts. The C5 + selectivity of HY-xK catalysts is slightly higher than the corresponding CP-xK catalysts. It indicates that hydrothermal treatment can decrease methane selectivity and increase C5 + selectivity of the iron-based catalyst in FTS reaction.

Effects of potassium content and temperature on CH4 and C5 + selectivity of catalysts
The influence of preparation method on catalyst stability is presented in Fig. 10. The CO conversion of CP-1.38K catalyst quickly decreases from 67 % at the beginning of the reaction to about 30 % at 480 h. However, the steady CO conversion of HY-1.32K catalyst is quickly stabilized at a relatively high level (58 %). It is known that the interconversion between iron oxides and iron carbide is dynamic and reversible depending upon the environment in FTS. When the H2O partial pressure is high, iron carbide will be oxidized to Fe3O4, which leads to the deactivation of iron-based FTS catalysts [34]. In FTS, a large amount of H2O will be produced, which in turn oxidizes the iron carbide. The structure of HY-1.32K catalyst can quickly remove H2O by the broad pore size distribution and uniform particles; thus, oxidization of iron carbide is suppressed, which stabilizes the iron carbide. This result discloses the unique merits of the hydrothermal treatment in promoting the iron-based FTS catalysts.

CO conversion as a function of time on stream of catalysts (CP-1.38K: reaction temperature 235 °C; HY-1.32K: reaction temperature 230 °C)
4 Conclusions
In this study, two series of Fe/Cu/K catalysts were prepared by co-precipitation and by co-precipitation accompanied with hydrothermal treatment, respectively. The catalysts after hydrothermal treatment, have relative low BET surface area, uniform morphology and Fe–Cu matrix. In addition, hydrothermal treatment remarkably influences the growth orientation of hematite nanocrystals, resulting in the preferential exposure of the (110) plane.
In FTS reaction, the catalysts after hydrothermal treatment exhibit higher activity than the catalysts prepared by co-precipitation, which is attributed to the more iron carbide during the FTS process. In addition, the stability of catalysts after hydrothermal treatment is very stable, while the stability of catalyst prepared by co-precipitation is very poor during a period of 480 h. It is probably due to the broad pore size distribution and uniform particles of the catalysts after hydrothermal treatment.
References
Bukur DB, Lang X (1999) Ind Eng Chem Res 38:3270
Dry ME (1990) Catal Lett 7:241
Dry ME (2002) Catal Today 71:227
Zhang CH, Yang Y, Teng BT, Li TZ, Zheng HY, Xiang HW, Li YW (2006) J Catal 237:405
O’Brien RJ, Davis BH (2004) Catal Lett 94:1
Dry ME, Shingles T, Boshoff LJ, Oosthuizen GJ (1969) J Catal 15:190
Arakawa H, Bell AT (1983) Ind Eng Chem Process Des Dev 22:97
Bukur DB, Mukesh D, Patel SA (1990) Ind Eng Chem Res 29:194
Dry ME, Oosthuizen G (1968) J Catal 11:18
Miller DG, Moskovits M (1988) J Phys Chem 92:6081
O’Brien RJ, Xu L, Spicer RL, Davis BH (1996) Energy Fuels 10:921
Benziger J, Madix R (1980) Surf Sci 94:119
Li S, Li A, Krishnamoorthy S, Iglesia E (2001) Catal Lett 77:197
Yang Y, Xiang HW, Xu YY, Bai L, Li YW (2004) Appl Catal A 266:181
Bukur DB, Lang X, Mukesh D, Zimmerman WH, Rosynek MP, Li C (1990) Ind Eng Chem Res 29:1588
Davis BH, Tungate FL (1991) DOE, Liquefaction contractors meeting, p 275
Shultz JF, Hofer LJE, Karn FS, Anderson RB (1962) J Phys Chem 66:501
Liu KK, Suo HY, Zhang C, Xu J, Yang Y, Xiang HW, Li YW (2010) Catal Commun 12:137
Schwuger M-J, Stickdorn K, Schomaecker R (1995) Chem Rev 95:849
Liu C, Zou B, Rondinone AJ, Zhang ZJ (2000) J Phys Chem B 104:1141
Calderone VR, Shiju NR, Curulla-Ferré D, Chambrey S, Khodakov A, Rose A, Thiessen J, Jess A, Rothenberg G (2013) Angew Chem Int Ed 52:4275
Dong HH, Xie MJ, Xu J, Li MF, Peng LM, Guo XF, Ding WP (2011) Chem Commun 47:4019
Kölbel H, Ralek M (1980) Catal Rev Sci Eng 21:225
Boudart M, McDonald MA (1984) J Phys Chem 88:2185
Feng S, Xu R (2000) Chem. Res 34:239
Mohapatra SK, John SE, Banerjee S, Misra M (2009) Chem Mater 21:3048
Qu J, Yin YX, Wang YQ, Yan Y, Guo YG, Song WG (2013) ACS Appl Mater Interfaces 5:3932
Sun JQ, Zheng SK, Zhang K, Song DC, Liu YT, Sun XD, Chen JG (2014) J Mater Chem 2:13116
Bukur DB, Koranne M, Lang X, Rao KRPM, Huffman GP (1995) Appl Catal A 126:85
Lox ES, Froment GF (1993) Ind Eng Chem Res 32:71
Huang CS, Xu L, Davis BH (1993) Fuel Sci Technol Int 11:639
Shroff MD, Kalakkad DS, Coulter KE, Kohler SD, Harrington MS, Jackson NB, Sault AG, Datye AK (1995) J Catal 156:185
Sirimanothan N, Hamdeh HH, Zhang Y, Davis BH (2002) Catal Lett 82:181
Ning WS, Koizumi N, Chang H, Mochizuki T, Itoh T, Yamada M (2006) Appl Catal A 312:35
Cui XJ, Xu J, Zhang CH, Yang Y, Gao P, Wu BS, Li YW (2011) J Catal 282:35
Cornell RM, Schwertmann U (1996) The iron oxides: structure, properties, reactions, occurences and uses. Second edition. VCH, Weinheim
Liu Z, Lv BL, Wu D, Sun YH, Xu Y (2012) Eur J Inorg Chem 2012:4076
Suo HY, Wang SG, Zhang CH, Xu J, Wu BS, Yang Y, Xiang HW, Li YW (2012) J Catal 286:111
Lohitharn N, Goodwin JG Jr (2008) J Catal 260:7
Rankin JL, Bartholomew CH (1986) J Catal 100:533
Kavitha MK, John H, Gopinath P (2014) Mater Res Bull 49:132
Mahajan D, Gütlich P, Stumm U (2003) Catal Commun 4:101
Herranz T, Rojas S, Perez-Alonso F, Ojeda M, Terreros P, Fierro J (2006) J Appl Catal A 311:66
Acknowledgments
The authors are indebted to the support from the National Natural Science Foundation of China (No. 21373254). This work is also supported by Wuhan Kaidi General Research Institute of Engineering & Technology Co., Ltd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, C., Chen, J. Effect of Hydrothermal Treatment on Precipitated Iron Catalyst for Fischer–Tropsch Synthesis. Catal Lett 145, 702–711 (2015). https://doi.org/10.1007/s10562-014-1457-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1457-4