
Abstract
Purpose
Oxidative stress causes mitochondrial dysfunction in myocardial ischaemia/reperfusion (I/R) as well as in obesity. Mitochondrial depolarization triggers mitophagy to degrade damaged mitochondria, a process important for quality control. The aims of this study were to evaluate (i) the effect of I/R on mitochondrial oxidative phosphorylation and its temporal relationship with mitophagy in hearts from obese rats and their age-matched controls, and (ii) the role of oxidative stress in these processes using melatonin, a free radical scavenger.
Methods
Male Wistar rats were divided into 4 groups: control (normal diet ± melatonin) and high-fat sucrose diet (HFSD ± melatonin). Rats received melatonin orally (10 mg/kg/day). After 16 weeks, hearts were removed and subjected to 40-min stabilization, and 25-min global ischaemia/10-min reperfusion for preparation of mitochondria. Mitochondrial oxidative phosphorylation was measured polarographically. Western blotting was used for evaluation of PINK1, Parkin, p62/SQSTM1 (p62) and TOM 70. Infarct size was measured using tetrazolium staining.
Results
Ischaemia and reperfusion respectively reduced and increased mitochondrial QO2 (state 3) and the ox-phos rate in both control and HFSD mitochondria, showing no major changes between the groups, while melatonin pretreatment had little effect. p62 as indicator of mitophagic flux showed up- and downregulation of mitophagy by ischaemia and reperfusion respectively, with melatonin having no significant effect. Melatonin treatment caused a significant reduction in infarct size in hearts from both control and diet groups.
Conclusions
The results suggest that I/R (i) affects mitochondria from control and HFSD hearts similarly and (ii) melatonin-induced cardioprotection is not associated with reversal of mitochondrial dysfunction or changes in the PINK1/Parkin pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The significance of the mitochondrion in the cardiomyocyte cannot be overestimated: not only is this organelle responsible for generating ~ 90% of the ATP required for contraction, but it is also the major source of reactive oxygen species (ROS) in the heart. The deleterious effects of myocardial ischaemia/reperfusion (I/R) on mitochondrial oxidative phosphorylation function are well-established and attributed to oxidative stress [1, 2]. Ischaemia-induced mitochondrial depolarization triggers mitophagy, a process of autophagic degradation of dysfunctional or damaged mitochondria, which is important for mitochondrial quality control. The roles of proteins such as PTEN-induced kinase (PINK1), Parkin, the Tom complex and p62/SQSTM1 (p62) in mitophagy have recently been reviewed [3, 4] and it was suggested that upregulation of mitophagy during myocardial I/R injury is protective [5, 6].
In vitro studies confirmed the above proposal: overexpression of Parkin in isolated cardiomyocytes subjected to hypoxia-mediated cell death was associated with its increased translocation to the mitochondria and increased cell viability, while cardiomyocytes expressing Parkinson’s disease–associated mutants of Parkin failed to reduce hypoxia-mediated cell death [7]. It was shown that autophagy upregulation protected against simulated ischaemia-reperfusion in HL-1cells [8]. Upregulation of mitophagy also plays a role in ischaemic preconditioning (IPC) [9]: in both Langendorff perfused rat hearts and in vivo in mice subjected to regional IPC, Parkin and P62 translocate to the mitochondria and mediate mitophagy. However, in contrast to the above, there is evidence to suggest that the suppression of mitophagy may protect the heart from I/R [10].
Apart from the role of oxidative stress in mitochondrial dysfunction in I/R, an oversupply of reducing equivalents (i.e. electrons) to metabolic pathways, as occurs in obesity, may cause mitochondrial dysfunction [11,12,13]. Although detailed knowledge of underlying changes in mitochondrial dynamics in obesity is still unclear, it appears that alterations in mitochondrial fusion/fission and excess ROS are involved [14,15,16].
In view of the putative harmful effects of increased ROS production on mitochondrial dynamics, it is expected that removal of free radicals may affect mitochondrial behaviour. Thus melatonin, a highly effective free radical scavenger synthesized mainly by the pineal gland, may have beneficial effects on the above-mentioned processes. Indeed, the cardioprotective effects of melatonin treatment both in vivo and in vitro are well-established [17, 18]. Long- as well as short-term melatonin administration to control and hyperphagia-induced obese rats in vivo protected the hearts against I/R damage, as well as reduced body weight gain and visceral adiposity [19, 20]. Recent studies focused on the effects of melatonin on mitochondria [21,22,23]. Petrosillo and coworkers [23] showed that melatonin protects against I/R damage by inhibition of mitochondrial cardiolipin peroxidation, cytochrome c release as well as opening of the mitochondrial permeability transition pore (mPTP). Interestingly, the melatonin concentration in mitochondria greatly exceeds that in the blood [21]. However, the effects of long-term melatonin treatment on myocardial mitochondrial oxidative phosphorylation function as well as mitophagy in obesity have not been investigated.
In view of the above, the aims of the present study were to evaluate the following: (i) the effect of I/R on the oxidative phosphorylation function and its temporal relationship with mitophagy in mitochondria isolated from hearts of obese rats and their age-matched controls; (ii) the role of ROS in these processes by employing long-term in vivo melatonin administration to control and obese rats. Since our previous studies on the effects of melatonin were done on obese rats receiving a high-calorie sucrose diet [19, 20], we expanded our study to further evaluate the cardioprotective actions of melatonin by switching to a high-fat sucrose diet (HFSD). To allow comparison with our previous studies, the isolated perfused heart subjected to ischaemia/reperfusion was used throughout as experimental model.
Methods
Animals and Diet
Male Wistar rats were used as experimental animals. Their breeding, maintenance and feeding as well as food and water consumption and weight monitoring were all carried out in the Central Research Facility, Faculty of Health Sciences, Stellenbosch University. Rats (180–200 g; age, 1 month) were assigned to the following four groups: control rats (CON) ± melatonin (10 mg/kg/day in drinking water) and high-fat/high-sucrose diet (HFSD) rats ± melatonin (10 mg/kg/day in drinking water). These rats were fed ad libitum for a period of 16 weeks. Melatonin was given at onset of darkness. The HFSD consisted of 8.3% protein, 42% carbohydrate, 20.4% sucrose, 11.5% total fat, 7.6% saturated fat and 13% cholesterol. The normal chow diet contained 17.1% protein, 34.6% carbohydrate, 5.3% sucrose, 4.8% total fat, 0.9% saturated fat and 2.2% cholesterol. The study conformed to the revised South African National Standard for the Care and Use of Animals for Scientific Purposes (South African Bureau of Standards, SANS 10386, 2008) and was approved by the Committee for the ethical use of animals in research of the University of Stellenbosch (project number SU-ACUM14-00039).
Heart Perfusion Technique
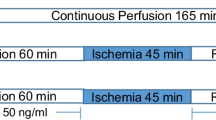
Sixteen weeks after initiation of the feeding programme, rats were anaesthetized by intraperitoneal injection of sodium pentobarbitone (160 mg/kg), and body weights as well as visceral fat determined. Hearts were perfused with modified Krebs-Henseleit bicarbonate buffer (KHB) as described before [24]. After mounting onto the aortic cannula, hearts were subjected to 40-min stabilization (STB) (10-min retrograde, 20-min working mode (preload 15 cm H2O, afterload 100 cm H2O) and 10-min retrograde perfusion). The following measurements were made during perfusion in the working mode: coronary flow, aortic flow, heart rate and peak systolic pressure (using a Statham pressure transducer (Transpac IV, Abbotts, Sligo, Ireland). Pressure signals were analysed using software developed by the Stellenbosch University Electronic Department (SED). For induction of ischaemia/reperfusion, two experimental models were used: (i) For mitochondrial studies, after stabilization, hearts were subjected to 25-min global ischaemia (temperature 36.5 °C), followed by 10-min retrograde reperfusion. Preference was given to global (rather than regional) ischaemia to ensure a high yield of mitochondria. Preliminary studies, performed on hearts subjected to 15–25-min global ischaemia and varying periods of reperfusion, indicated that 25-min ischaemia reliably induced significant mitochondrial dysfunction. Reperfusion for 10 min was chosen since this coincided with maximal activation of the RISK pathway in cardioprotection (data not shown). This model, however, did not allow measurement of functional recovery during reperfusion, since hearts were reperfused in the retrograde mode during this period. (ii) For evaluation of the effect of long-term melatonin treatment on myocardial susceptibility to I/R damage, four additional series of animals were studied where isolated hearts were stabilized for 40 min as described above, subjected to 35-min coronary artery ligation followed by 60-min reperfusion (10-min retrograde, 20-min working heart, 30-min retrograde). Infarct size was evaluated using the tetrazolium chloride staining method as described before [24]. The choice of 35-min regional ischaemia was also based on previous studies showing that this period of ischaemia elicited an infarct which could be measured accurately [24]. Myocardial function during perfusion in the working mode, was measured before (at 20-min perfusion during stabilization) and after coronary artery ligation (at 20- and 30-min reperfusion time).
Isolation of Mitochondria
At the end of either stabilization or ischaemia or reperfusion as described above, hearts were plunged in ice-cold mitochondrial isolation medium (KE, 0.18 M KCl/0.01 M EDTA; pH adjusted to 7.4 with 2 M Tris), the tissue cut finely with scissors and homogenized with a Polytron PT 10 homogenizer (2 × 4 s, 4 °C, setting 4). The mitochondria were isolated by differential centrifugation as described by Sordahl et al. [25]. The mitochondrial pellet was divided in two: one-half was suspended in KE medium for immediate measurement of mitochondrial function and the other half dissolved in lysis buffer (see below) and stored at − 80 °C for subsequent western blot analysis.
Mitochondrial Function
Mitochondrial oxidative phosphorylation was measured polarographically at 26 °C using an oxygraph (Hansatech Instruments, Bannan UK) containing a Clark electrode as described previously [26]. The protein content of mitochondrial samples was determined by the method of Lowry et al. [27].
The ability of mitochondria to recover their oxidative phosphorylation capacity after exposure to anoxia in vitro was assessed as described by Essop et al. [28]. An excess amount of ADP (10× the amount added previously to induce state 3 respiration) was added to the incubation chamber for a period of 20 min to deplete the medium of all oxygen. Thereafter, the mitochondrial suspension was reoxygenated for a period of 6 min and the mitochondrial respiration (state 3) expressed as a percentage of state 3 recorded before exposure to anoxia.
Western Blot Technique
Half of the mitochondrial pellet was suspended in 600–900 μl of lysis buffer containing (in mM) the following: Tris-HCl 20, p-nitrophenyl phosphate 20, EGTA 1.0, EDTA 1.0, NaCl 150, tetra-sodium-pyrophosphate 2.5, ß-glycerophosphate 1.0, sodium orthovanadate 1.0, phenylmethyl sulphonyl fluoride (PMSF) 1.0, aprotinin 10 μg/ml and leupeptin 10 μg/ml, Triton-X100 1% and pH 7.4. Zirconium oxide beads (0.5 mm, Next Advance, Biocom Biotech) were added to the lysates; whereafter, the solution was further lysed in a Bullet blender (Next Advance laboratory equipment) at 3 × 4 000 rpm (4 °C, for 1 min each with a 5-min resting period in between). Samples were then centrifuged at 1000×g for 10 min to obtain the supernatant; the protein content of which was determined using the Bradford technique [29]. Twenty-six well 5–20% pre-cast gradient gels and stain-free technology were used throughout this study. Depending on the protein of interest, 30 to 60 μg was loaded per lane. After separation, the image of the separated proteins on the gel was stored by the ChemiDoc software. The proteins were transferred to a PVDF membrane (Immobilon™ P, Millipore) and the image of the transferred proteins visualized and stored by the ChemiDoc. This image was used to calculate total transferred proteins per lane. Non-specific binding sites on the membranes were blocked with 5% fat-free milk in TBST (Tris-buffered saline + 0.1% Tween 20) for 1 to 2 h at room temperature. The membranes were incubated overnight at 4 °C with the primary antibodies and then incubated for 1 h at room temperature with a diluted horseradish peroxidase–labelled secondary antibody (Cell Signalling Technology®). The following primary antibodies were used: TOM-70 (Santa Cruz), PINK1 (Cell Signalling), Parkin (Abcam) and p62/SQSTM1 (p62) (Cell Signalling). After thorough washing with TBST, membranes were covered with ECL (enhanced chemiluminescence) detection reagents (Amersham, LIFE SCIENCE) and quantified using a Chemidoc-XRS imager (Bio-Rad). The intensity of bands detected by the ECL reaction was normalized to the total proteins that were transferred in each lane, negating the use of a loading control. Four samples/group were included on the same gel plus a sample prepared from a heart of an unperfused age-matched control animal to act as standard for normalization of all data.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, Inc.). Comparisons between the groups were performed using one- or two-way ANOVA (between all groups) or unpaired Student’s t test (between two groups) where appropriate. A p value of ≤ 0.05 was deemed statistically significant.
Results
Food and water consumption, weight gain and infarct size
Food consumption was higher and water intake lower in HFSD compared with control rats throughout the feeding period (Fig. 1). Melatonin-treated HFSD rats consumed more food and water than untreated HFSD rats. Interestingly, HFSD administration for 16 weeks (with and without melatonin) did not increase body weight, compared with their control counterparts (Fig. 2a). However, the diet caused a significant increase in intraperitoneal fat mass which was further increased by melatonin treatment (Fig. 2b).

Food (a) and water (b) consumption of control and HFSD rats (n = 15 rats/group): effect of melatonin (10 mg/kg/day). CON, control; HFSD, high-fat sucrose diet; MEL, melatonin. * p ≤ 0.05 vs control rats; **p ≤ 0.05 vs HFSD rats

Body weight (a) and intraperitoneal fat mass (% of body weight) (b) of control and HFSD rats. (n = 15/group): effect of melatonin (10 mg/kg/day). *p < 0.05 vs control rats; **p < 0.05 vs HFSD rats. CON, control; HFSD, high-fat sucrose diet; MEL, melatonin.
For both mitochondrial and infarct size studies, baseline myocardial function was measured before hearts were subjected to ischaemia. No significant changes in heart function (heart rate, peak systolic pressure, aortic flow, total work performance) between control and HFSD rats were observed (Fig. 3a–d). Treatment with melatonin significantly reduced peak systolic pressure, aortic flow and total work in HFSD rat hearts while in control rat hearts melatonin significantly reduced aortic flow compared with untreated controls (Fig. 3c). For the mitochondrial studies, the protocol did not allow evaluation of function during reperfusion since hearts were harvested for mitochondrial isolation after 10-min reperfusion only, when perfusion was still conducted in the retrograde mode.

Baseline function of working rat hearts (before exposure to ischaemia) (n = 6/group): effects of the HFSD and melatonin. *p < 0.05 vs control rats; **p < 0.05 vs HFSD rats. CON, control; HFSD, high-fat sucrose diet; MEL, melatonin.
Melatonin supplementation for a period of 16 weeks resulted in a significant reduction in infarct size after 35-min regional ischaemia in both control and HFSD groups (Fig. 4a). The percentage area at risk did not differ between the groups (n = 8 hearts/group) (Fig. 4b). Interestingly, melatonin administration to the HFSD group resulted in a more significant lowering in infarct size than in hearts from control rats. Despite the melatonin-induced reduction in infarct size, functional recovery during reperfusion did not differ between the groups: the untreated as well as treated control and HFSD groups recovered to a similar extent (data not shown).

Effect of HFSD diet and melatonin treatment on infarct size (a) and area at risk (b) of hearts after 35-min coronary artery ligation and 60-min reperfusion, n = 8/group. *p < 0.05 CON + MEL vs HFSD + MEL; **p < 0.01 vs CON; ***p < 0.001 vs HFSD
Blood Analyses
Blood analyses were not done in the present study. In a previous study, fasting blood sugar levels, serum insulin and the HOMA index were significantly elevated in the HFSD rats after 16-week feeding. Insulin resistance was further indicated by the observation that increasing insulin concentrations elicited a weak response in cardiomyocytes isolated from HFSD rats compared with those of control rats [30].
Mitochondrial Oxidative Phosphorylation Function
Substrate Glutamate/Malate (Fig. 5a–f)
In view of the many variables, the results obtained after stabilization, ischaemia and reperfusion are described separately.

Oxidative phosphorylation function of mitochondria isolated from hearts after 40-min stabilization, 25-min global ischaemia, 10-min reperfusion: effect of HFSD and melatonin treatment for 16 weeks. Substrates: glutamate/malate (n = 5/group). ADP/O ratio: nmoles ATP produced/nAtom oxygen consumed; QO2 S-3: nAtoms oxygen taken up in presence of ADP/mg mitochondrial protein/min; QO2 S-4: nAtoms oxygen taken up in absence of ADP/mg mitochondrial protein/min; ox-phos rate: ADP/O ratio X QO2 S-3 (nmoles ATP produced /mg mitochondrial protein/min). *p < 0.05 vs corresponding control rats. #p < 0.05 vs corresponding HFSD rats (ANOVA and Student’s t test). AMC, age-matched control; CON, control; HFSD, high-fat sucrose diet; MEL, melatonin; STB, stabilization; ISC, ischaemia; RP, reperfusion; S, state; OX-PHOS, oxidative phosphorylation rate; RCI, respiratory control index
Stabilization
After stabilization for 40 min, no differences in mitochondrial function were observed between control and HFSD rats except for an increase in ADP/O ratio (Fig. 5a) in the HFSD group. Melatonin treatment was without effect during stabilization in control mitochondria, while it significantly increased the ADP/O ratio and reduced the RCI in mitochondria from HFSD hearts.
Ischaemia
Exposure to ischaemia resulted in very similar responses in mitochondrial function, namely a significant reduction in QO2 S-3, RCI and ox-phos rate, and an increase in QO2 S-4 compared with their corresponding stabilization groups, with no significant differences between control and HFSD groups. Melatonin treatment for 16 weeks had no effect on mitochondrial function and similar responses were observed in both control and HFSD groups.
Reperfusion
Reperfusion after 25-min global ischaemia had very similar effects on mitochondria from control and HFSD groups, namely an increase in RCI, a reduction in QO2 S-4, without significant changes in ADP/O ratio, QO2 S-3 and ox-phos rate. At this stage, melatonin treatment did improve the ADP/O ratio in the control and HFSD groups, without significant differences between the control and HFSD groups.
No significant changes were observed in the percentage recovery in QO2 (state 3) after reoxygenation between mitochondria isolated from control and HFSD hearts during the perfusion protocol, with melatonin having no effect.
Substrate Palmitoyl-l-Carnitine/Malate (Fig. 6a–f)
Stabilization
After stabilization, the ADP/O ratio was significantly reduced, while the QO2 S-3, QO2 S-4 and ox-phos rate were significantly higher in HFSD rats compared with their controls. Melatonin treatment had different effects on mitochondrial behaviour during stabilization: in the control mitochondria, it reduced the ADP/O ratio and increased both QO2 S-3 and QO2 S-4, while in HFSD mitochondria, it reduced the QO2 S-3, the ox-phos rate and percentage recovery after reoxygenation.

Oxidative phosphorylation function of mitochondria isolated from hearts after 40-min stabilization, 25-min global ischaemia and 10-min reperfusion: effect of HFSD and melatonin treatment for 16 weeks. Substrates: palmitoyl-l-carnitine/malate (n = 5/group). *p < 0.05 vs corresponding control rats. #p < 0.05 vs corresponding HFSD rats (ANOVA and Student’s t test). AMC, age-matched control; CON, control; HFSD, high-fat sucrose diet; MEL, melatonin; STB, stabilization; ISC, ischaemia; RP, reperfusion; S, state; OX-PHOS, oxidative phosphorylation rate; RCI, respiratory control index
Ischaemia
The pattern of changes observed after exposure of the hearts to ischaemia was, with the exception of QO2 S-3, very similar in mitochondria from control and HFSD hearts. After exposure to ischaemia, the ADP/O ratio, RCI, and S-3 ox-phos rate were significantly reduced, compared with stabilization in both groups, while QO2 S-4 was increased. The only exception was that control mitochondria did not show the characteristic reduction in QO2 S-3 as exhibited by HFSD mitochondria. Treatment with melatonin had no effect on the ischaemia-induced reduction in ADP/O ratio, RCI and ox-phos rate in both groups. However, in the case of control mitochondria, melatonin reduced both QO2 S-3 and QO2 S-4, compared with its untreated counterparts.
Reperfusion
As with glutamate/malate, reperfusion also had marked effects with palmitoyl-l-carnitine/malate as substrates. HFSD mitochondria showed marked increases in ADP/O ratio while in the case of the control mitochondria, the changes were not significant (when compared with their ischaemic counterparts). In both groups reperfusion did not affect QO2 S-3 values, while reducing QO2 S-4. Upon reperfusion, melatonin increased the ADP/O ratio in both groups, having no effect on the other parameters of mitochondrial function.
With palmitoyl-l-carnitine/malate, the pattern of recovery after reoxygenation differed from those obtained with glutamate/malate. Interestingly, the percentage recovery of mitochondria, isolated from both control and HFSD rat hearts after stabilization, averaged ~ 150%, while with melatonin treatment, it was significantly less (~ 100%). After exposure to ischaemia, the recovery potential of both groups of mitochondria was significantly lower, improving after reperfusion. Apart from lowering the percentage recovery after stabilization in HFSD mitochondria, the effect of melatonin was negligible (Fig. 6f).
Mitophagy (Fig. 7)
As in the study of mitochondrial function, mitochondria were isolated from hearts subjected to either 40-min stabilization or 25-min global ischaemia, or 25-min ischaemia/10 min reperfusion.


a Representative Western blot analysis of mitochondria after isolated from hearts from control and HFSD rats after 40-min stabilization, 25-min global ischaemia and 10-min reperfusion (n = 4/group). Abbreviations on blot: C, control; CM, control + melatonin: H, HFSD; HM, HFSD + melatonin. b Comparison between mitochondria from CON and HFSD rats. c Comparison between melatonin-treated and untreated HFSD rats. d Comparison between melatonin-treated and untreated control rats. *p ≤ 0.05 vs corresponding control rats. #p ≤ 0.05 vs corresponding HFSD rats (analysis of variance followed by Student’s t test). AMC, age-matched control; CON, control; HFSD, high-fat sucrose diet; MEL, melatonin; STB, stabilization; ISC, ischaemia; RP, reperfusion; S, state; OX-PHOS, oxidative phosphorylation rate; RCI, respiratory control index
Stabilization
The expression of TOM70, PINK1 and PARKIN was similar in mitochondria from control and HFSD hearts after stabilization, the only exception being reduced p62 levels in HFSD.
The perfusion protocol had variable effects on each marker. While expression of Parkin did not change upon exposure of the control hearts to ischaemia ± reperfusion, the other three markers showed significant changes: p62 and PINK1 levels were significantly reduced by both ischaemia and reperfusion, while TOM 70 was increased by ischaemia and further increased by reperfusion. Mitochondria isolated from HFSD hearts showed a similar pattern for p62 as the controls namely a reduction after exposure to ischaemia but a significant increase upon reperfusion. TOM 70 was increased significantly by exposure to ischaemia and remained high during reperfusion, while PINK1 and Parkin expression were unchanged.
Melatonin treatment of control rats for a period of 16 weeks had no significant effects on the expression of mitophagy marker proteins during the experimental protocol and patterns very similar to those of untreated controls were seen (see Figs. 7c, d). The only significant changes observed were increased levels of PINK1 during reperfusion, as well as a reduction in Parkin in the stabilization group of HFSD rats.
Discussion
Experimental Model
In the present follow-up study to delineate the effects of long-term melatonin treatment in obesity, we made two major changes in the protocol, namely employing a high-fat sucrose diet (instead of a high-sucrose diet), while increasing the dosage of melatonin from 4 to 10 mg melatonin/kg/day (see refs 19, 20). Surprisingly, changing the diet to a HFSD prevented the significant increase in body weight (Fig. 2), characteristic of the high sucrose diet [19, 20] and occurred despite an increase in food consumption (Fig. 1), suggesting a change in whole body metabolism. However, as with the high-sucrose diet, the HFSD diet did cause a significant increase in fat mass (expressed as a percentage of body weight), as was also observed by Everson and coworkers [31]. Other similarities between the two models were an elevation in fasting blood glucose (slight), serum insulin levels (very significant) and development of insulin resistance [19, 20, 30].
In order to determine whether increasing the melatonin dosage would enhance its effects, we increased the long-term melatonin treatment from 4 to 10 mg/kg/day [19,20]. Previous studies in this regard used melatonin ranging in concentrations from 1 mg/kg/min [32] to 30 mg/kg/day [33], while several workers used 10 mg/kg/day [34,35], administered for varying periods of time. Interestingly, treatment with melatonin (10 mg/kg/day) in the present study further increased the visceral fat mass of these rats, when compared with untreated HFSD rats (see Fig. 2), a finding in contrast with previous studies where melatonin reduced body weight and intra-abdominal adiposity [19, 20, 36,37,38]. The increased food intake following melatonin administration at dark onset (Fig. 1) may contribute to the increase in visceral fat. Angers et al. [39] reported that dark onset melatonin administration increased food intake in Wistar rats, probably due to a reinforcement of the eating behaviour which usually accompanies melatonin peak secretion. Although we do not have a ready explanation for the different outcomes in body weight obtained in the effects of melatonin on rats receiving a HFSD or a sucrose high-calorie diet, it could possibly be attributed to the metabolic effects of the HFSD. This possibility, however, as well as the metabolic changes induced by a HFSD, compared with a high sucrose diet, need to be further investigated.
As also observed in previous studies, basal myocardial function of HFSD rats was similar to those of rats receiving normal rat chow [40,41], suggesting that increased fat combined with high calorie intake did not induce cardiac dysfunction ex vivo. Little, however, is known about the long-term effects of melatonin administration on the myocardial function of control and obese rats. We previously showed that long-term melatonin administration of 4 mg/kg/day did not cause significant changes in aortic output of hearts from rats on a high-sucrose diet [19]. In contrast, in the present study, long-term administration of 10 mg/kg/day caused a small but significant reduction in the ex vivo aortic output of hearts from both control and HFSD rats (Fig. 3). Since these effects of melatonin were observed in both control and HFSD hearts, the divergent observations could be due to the increase in melatonin dosage. The exact mechanism whereby long-term melatonin affects cardiac output is not clear yet, but it may be due to the blood pressure–lowering effects of melatonin [42].
The significant protective effect of melatonin against I/R damage [17,18] was again confirmed in the present study: long-term melatonin administration (10 mg/kg/day) caused a significant reduction in infarct size after ischaemia/reperfusion, being more effective in the HFSD group (Fig. 4). This may be due to an additive effect of melatonin to the protection induced by a HFSD per se: a previous study from our laboratory showed that hearts from rats on a HFSD were more resistant than their corresponding controls to ischaemia/reperfusion damage [40]. The lack of improvement in functional recovery during reperfusion is probably due to stunning during early reperfusion (results not shown).
Mitochondrial Function: Effect of Diet, Ischaemia/Reperfusion and Melatonin
Interesting observations were made regarding the effects of ischaemia per se as well as reperfusion on mitochondrial function: (i) relatively few major changes were observed in the in vitro behaviour of mitochondria isolated from control and HFSD rat hearts. Apart from an increase in the baseline function of HFSD mitochondria, no differences were seen between control and HFSD groups, during I/R; thus, administration of a HFSD diet concomitant with development of insulin resistance did not exacerbate the deleterious effects of I/R on mitochondrial ox-phos function; (ii) ischaemia/reperfusion had the expected effects on the mitochondrial oxidative phosphorylation process, namely a reduction in ADP/O, QO2 S-3 and ox-phos rate, and an increase in QO2 S-4 after exposure to ischaemia in both groups, with an increase in RCI and reduction in QO2 S-4 during reperfusion, regardless of the substrate. This indicated that exposure to ischaemia had lasting effects on mitochondrial behaviour, persisting when incubated under ideal conditions; (iii) reperfusion reversed several of the harmful effects of ischaemia on mitochondrial function per se, although not to pre-ischaemic levels. This improvement in mitochondrial function upon reperfusion is in accordance with the improvement in ultrastructure observed under these conditions [43].
Contrary to these results using an insulin-resistant prediabetic rat model, convincing evidence exits of impaired baseline mitochondrial function in hearts from diabetic, insulin-resistant animals [44,45,46]. For example, in a mouse model of type 2 diabetes (db/db), a reduction in subsarcolemmal mitochondrial state 3 respiration, electron transport chain and ATP synthase activities were reported [47]. However, in accordance with our observations, Ferdinandy and coworkers [48] reported no major differences in mitochondrial oxygen uptake, Ca uptake or membrane potential in hearts from a prediabetic model. Similarly, oxygen uptake by heart mitochondria isolated from mice on a high-fat diet was not changed substantially [49]. However, these contradictory findings may be due to the differences in experimental models employed.
The deleterious effects of prior I/R on mitochondrial function are well-established [50] and it is generally accepted as being a critical factor in contributing to myocardial injury [51, 52]. It is also known that I/R leads to decreased activity of complexes I [53] and III [54] and that the latter is a dominant ROS generator [55], confirming that the ETC is a major source of mitochondrial injury during I/R.
Melatonin Effects
Long-term melatonin treatment had slight and variable effects on the oxidative phosphorylation function of mitochondria isolated from hearts of control and HFSD rat hearts, particularly with glutamate/malate as substrates: melatonin had no effect on any of the parameters of oxidative phosphorylation in controls during the perfusion protocol, while in mitochondria from HFSD hearts, it increased the ADP/O and reduced the RCI ratios during baseline conditions. With palmitoyl-l-carnitine as substrates, melatonin reduced baseline QO2 state 3 and the ox-phos rate, without having other major effects during I/R.
This lack of significant beneficial effects of prolonged melatonin treatment on mitochondrial function after exposure of the heart to ischaemia ± reperfusion was surprising in view of the marked cardioprotective effects of melatonin against I/R injury (Fig. 4). It is also surprising since melatonin can scavenge free radicals (for reviews see refs 21, 22) and can be taken up, synthesized and metabolized by these organelles. The conclusion that the reduction in infarct size by long-term melatonin treatment is not due to reversal of I/R mitochondrial damage was confirmed by the finding that addition of melatonin directly to the perfusate of ex vivo perfused I/R hearts also did not result in an improvement in mitochondrial oxidative phosphorylation function, despite a significant reduction in infarct size [26].
Failure to link the cardioprotective effects of long-term melatonin to an improvement in mitochondrial function is in contrast to the results obtained by Petrosillo et al. who reported that direct administration of melatonin to perfused hearts inhibited mitochondrial permeability transition and cytochrome c release during I/R [23, 56]. These events elicited by administration of melatonin prior to ischaemia have been linked to activation of the prosurvival SAFE pathway and import of STAT-3 into mitochondria [57, 58]. Whether these discrepancies can be attributed to the high melatonin dosage used by Petrosillo et al. (50 μM) (as opposed to long-term administration of 10 mg/kg/day) remains to be elucidated. The health status of the experimental animal may also be important: Yu et al. [59] showed that melatonin (10 mg/kg, administered 5 days before experimentation) to type 1 diabetic rats preserved mitochondrial function by reducing mitochondrial oxidative stress and enhancing its biogenesis. Clearly further carefully controlled studies are required to solve these problems.
Mitophagy: Effects of Diet, I/R and Melatonin
Although evaluation of mitophagy at different time intervals during a perfusion protocol (“snap-shot approach”) does not indicate flux, expression of p62 has been regarded as a reliable indicator of autophagic flux [60]. In the present study, the snap-shot approach was used to (i) evaluate of the effects of I/R on mitochondrial mitophagy; (ii) investigate the temporal relationship between I/R-induced changes in mitochondrial oxidative phosphorylation and mitophagy; and (iii) to compare the effects of a long-term high-fat diet and melatonin treatment on mitophagy. In addition to p62, we focused on the well-established PINK1/PARKIN pathway as indicators of mitophagy.
Effects of I/R
Exposure to ischaemia per se caused a significant reduction in p62 and PINK1 levels, an increase in TOM70 with no effect on Parkin levels. Apart from p62, reperfusion had surprisingly little effects on these parameters. These changes coincided with the reduction in mitochondrial QO2 state 3 and ox-phos rate and increased QO2 state 4, changes which were expected to be associated with increased mitophagy. Interestingly, p62 levels were lower in both control and HFSD mitochondria isolated after exposure to ischaemia, which may suggest upregulation of the mitophagy process during ischaemia, aimed at removal of dysfunctional mitochondria. However, the increase in p62 expression seen after reperfusion (to a larger extent in mitochondria isolated from HFSD hearts) was suggestive of downregulation of mitophagy, a surprising observation, particularly in view of the generation of ROS known to occur during reperfusion [3, 4]. This may be due to the recovery of reversibly damaged mitochondria during reoxygenation (as evidenced by the improvement in mitochondrial oxidative phosphorylation during the reperfusion phase (Figs. 5 and 6) as well as ultrastructure [43]). Increased expression of p62 after exposure of the heart ischaemia/reperfusion was also observed by others [61].
The reduction in PINK1 levels is difficult to explain, since mitochondrial depolarization during ischaemia interrupts the degradation process of PINK1, which could lead to its accumulation and phosphorylation of its substrate proteins, triggering mitophagy [3, 4]. However, the reduction in PINK1 after exposure to 25-min global ischaemia may indicate upregulation of mitophagy during this phase of the protocol. It has been shown that loss of this protein increases the vulnerability of the heart to I/R injury, worsening mitochondrial function [62]. The ischaemia-induced reduction in PINK1 levels observed in the present study is in contrast with the results obtained by Ji and coworkers [10] who demonstrated a significant increase in these proteins after in vivo exposure of the heart to 30-min ischaemia/120-min reperfusion, conditions however, which differed markedly from those in the present study.
Expression of Parkin was found to be unchanged by I/R in mitochondria isolated from both control and HFSD rats. Since Parkin ubiquinates p62 and promotes its binding to LC3-II, recruiting damaged mitochondria to the phagophore [3, 9], its unchanged expression observed throughout our study is difficult to explain, particularly in view of the concomitant increases in p62 levels at reperfusion and may be due to the fact that these measurements were done in the snap-shot mode and do not indicate flux.
In contrast to Parkin, both ischaemia and reperfusion affected the expression of TOM70 in the heart. In view of its role in the transport of PINK1 [63, 64] into mitochondria, it is expected to be affected by processes initiating mitophagy, for example depolarization of the mitochondrial membranes. Its upregulation by ischaemia per se as well as by I/R confirms its participation in subsequent events and suggests that these changes may precede the onset of mitophagy. These findings differ from those by Pei and workers [64] who reported a significant depression in TOM70 expression in response to MI. However, the latter study was performed in rats after 5-day permanent coronary artery ligation, a protocol which differed from the one used in the present study.
Apart from a significant increase in p62 expression during reperfusion in HFSD mitochondria, the response of these two groups to I/R was very similar. These observations are not unexpected since there were relatively few major changes in the oxidative phosphorylation process of mitochondria isolated from control and HFSD rat hearts, under baseline conditions as well as after I/R.
Ferdinandy and coworkers [48], using a mouse model of prediabetes, also showed that although early dysregulation of mitochondrial fusion and autophagy (shown by increased MFN2 and reduced BNIP3 expression) occurred, the other canonical markers of mitophagy and autophagy were unchanged at baseline. A number of proteomic studies were done in recent years on hearts of prediabetic db/db mice [47] and mice fed a high-fat diet [65] which showed changes in the levels of electron transport proteins and ATP synthase, but unfortunately the proteins involved in mitophagy were not mentioned. Clearly, this approach should also be applied in hearts subjected to ischaemia/reperfusion.
It became clear in evaluation of the data obtained that the effects of ischaemia/reperfusion on mitophagy need to be further investigated with a focus on flux rather than using a snap-shot approach.
Effects of Long-Term Melatonin Treatment on Mitophagy
Melatonin had minor effects on the parameters of mitophagy used and the patterns observed during the various stages of the perfusion protocol were very similar to those of the untreated groups, for example Parkin, p62 and TOM70 levels. However, a previous study from our laboratory suggested that melatonin has profound effects on an alternative non-conventional mitophagy pathway [26], suggesting to be the predominant form of mitophagy during stress [66] which may play an important role in its cardioprotective actions.
In conclusion, the results obtained in this study show that the characteristic changes observed in the electron transport and oxidative phosphorylation processes of mitochondria isolated from hearts subjected to global ischaemia followed by reperfusion coincide with changes in mitophagy, as indicated by the PINK1/Parkin pathway. Changes in the expression of p62 after exposure of hearts to I/R suggest increased mitophagic flux during ischaemia and downregulation during reperfusion. Interestingly, feeding of a HFSD diet for 16 weeks had no harmful effects on mitochondrial behaviour while the melatonin-induced cardioprotection was not associated with reversal of I/R-induced changes in mitochondrial oxidative function or the PINK/Parkin pathway.
Summary
Long-term melatonin administration maintains its cardioprotective effects in rats regardless of their diet. However, changing from a high-calorie sucrose diet to a HFSD affected the response to melatonin in a number of ways, namely (i) having no effect on body weight (as opposed to the reduction seen with a high-calorie sucrose diet), but increasing the fat mass (as a percentage of body weight) which was attributed to a change in whole body metabolism induced by the HFSD; (ii) increasing cardioprotection by further reducing infarct size, compared with melatonin-treated controls. Infarct size reduction by long-term melatonin was not associated with improvement in mitochondrial function after exposure of hearts from control and HFSD rats to either ischaemia or I/R while not having a major effect on mitophagy as indicated by Pink1, Parkin and p62 expression.
References
Ferrari R. Importance of oxygen free radicals during ischemia and reperfusion in the experimental and clinical setting. Oxygen free radicals and the heart. Am J Cardiovasc Pathol. 1992;4:103–14.
Watts JA, Kline JA. Bench to bedside: the role of mitochondrial medicine in the pathogenesis and treatment of cellular injury. Acad Emerg Med. 2003;10:985–97.
Anzell AR, Sanderson TH. Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol Neurobiol. 2018;55:2547–64.
Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477–90.
Hamacher-Brady A, Brady N, Loque SE, Saven MR, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57.
Lu W, Sun J, Yoon JS, Zhang Y, Zheng L, Murphy E, et al. Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis. PLoS One. 2016;11:e0147792.
Kubli DA, Zhang G, Lee Y, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–26.
Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87.
Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975.
Ji W, Wei S, Hao P, et al. Aldehyde dehydrogenase 2 has cardioprotective effects on myocardial ischaemia/reperfusion injury via suppressing mitophagy. Front Pharmacol. 2016;7:101.
Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071–7.
Putti R, Sica R, Migliaccio V, Lionetti L. Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. 2015;6:109.
Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes. 2015;64:3135–45.
Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41:1846–54.
Lahera V, de Las HN, López-Farré A, Manucha W, Ferder LL. Role of mitochondrial dysfunction in hypertension and obesity. Curr Hypertens Rep. 2017;19:11.
Che Y, Wang Z-P, Yuan Y, Zhang N, et al. Autophagy in a model of obesity: a ong-term high fat diet induces cardiac dysfunction. Mol Med Rep. 2015;18:3251–61.
Lochner A, Marais E, Huisamen B. Melatonin and cardioprotection against ischaemia/reperfusion injury: what’s new. A review. J Pineal Res. 2018;65:e12490.
Reiter RJ, Tan DX. Melatonin: a novel protective agent against oxidative injury of the ischemic/reperfused heart. Cardiovasc Res. 2003;58:10–9.
Nduhirabandi F, Du Toit EF, Blackhurst D, Marais D, Lochner A. Chronic melatonin consumption prevents obesity-related metabolic abnormalities and protects the heart against ischemic and reperfusion injury in a prediabetic model of diet-induced obesity. J Pineal Res. 2011;50:171–82.
Nduhirabandi F, Huisamen B, Strijdom H, Blackhurst D, Lochner A. Short-term melatonin consumption protects the hearts of obese rats independent of body weight and visceral adiposity. J Pineal Res. 2014;57:317–32.
Reiter RJ, Rosales-Corral S, Tan DX, et al. Melatonin as a mitochondria-targeted anti-oxidant: one of nature’s best ideas. Cell Mol Life Sci. 2017;74:3853–81.
Tan DX, Manchester LC, Qin L, Reiter RJ. Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci. 2016;17:e2124.
Petrosillo G, Colantuono G, Moro N, Ruggiero FM, Tiravanti E, di Venosa N, et al. Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am J Phys Heart Circ Physiol. 2009;297:H1487–93.
Lochner A, Genade S, Moolman JA. Ischemic preconditioning: infarct size is a more reliable endpoint than functional recovery. Basic Res Cardiol. 2003;98:337–46.
Sordahl LA, Besch HR Jr, Allen JC, Crow C, Lindenmayer GE, Schwartz A. Enzymatic aspects of the cardiac muscle cell: mitochondria, sarcoplasmic reticulum and noncovalent cation active transport system. Methods Achiev Exp Pathol. 1971;5:287–346.
Dube K, Dhanabalan K, Salie R, Blignaut M, Huisamen B, Lochner A. Melatonin has profound effects on mitochondrial dynamics in myocardial ischaemia/reperfusion. Heliyon. 2019;5:e02659.
Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–75.
Essop MF, Chan WY, Valle A, Garcia-Palmer FJ, du Toit EF. Impaired contractile function and mitochondrial respiratory capacity in response to oxygen deprivation in a rat model of prediabetes. Acta Physiol (Oxford). 2009;197:289–96.
Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Smit SE, Johnson R, Van Vuuren MA, Huisamen B. Myocardial clearance by aspalathin treatment in young, mature and obese insulin-resistant rats. Planta Med. 2018;84:75–82.
Everson F, Genis F, Ogundipe O, et al. Treatment with a fixed dose combination antiretroviral therapy drug containing tenofovir, emtricitabine and efavirenz is associated with cardioprotection in high calorie diet-induced obese rats. PLoS One. 2018;13:e0208537.
Hussein MR, Ahmed OG, Hassan AF, Ahmed MA. Intake of melatonin is associated with amelioration of physiological changes, both metabolic and morphological pathologies associated with obesity: an animal model. Int J Exp Pathol. 2007;88:19–29.
Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, et al. Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology. 2003;144:5347–2.
Aqil A, Rosado I, Ruiz, et al. Melatonin improves glucose homeostasis in young Zucker diabetic fatty rats. J Pineal Res. 2012;52:203–10.
Aqil A, Reiter RJ, Jimenez-Aranda A, et al. Melatonin ameliorates low-grade inflammation and oxidative stress in young Zucker diabetic fatty rats. J Pineal Res. 2013;54:381–8.
Nduhirabandi F, Du Toit EF, Lochner A. Melatonin and the metabolic syndrome: a tool for effective therapy in obesity-associated abnormalities? Acta Physiol Acta. 2012;205:209–23.
Bartness TJ, Wade GN. Body weight, food intake and energy regulation in exercising and melatonin-treated Siberian hamsters. Physiol Behav. 1985;35:805–8.
Wolden-Hanson T, Mitton DR, McCants RL, Yellon SM, Wilkinson CW, Matsumoto AM, et al. Daily melatonin administration to middle- aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology. 2000;141:487–97.
Angers K, Haddad N, Selmaoui B, Thibault L. Effect of melatonin on total food intake and macronutrient choice in rats. Physiol Behav. 2003;80(1):9–18.
Salie R, Huisamen B, Lochner A. High carbohydrate and high fat diets protect the heart against ischaemia/reoerfusion injury. Cardiovasc Diabetol. 2014;13:109.
Donner D, Headrick JP, Peart JN, du Toit EF. Obesity improves myocardial ischemic tolerance and RISK signalling in insulin-insensitive rats. Dis Mod Mech. 2013;6:457–66.
Pandi-Perumal SR, BaHammam AS, Ojike NI, Akinseye OA, Kendzerska T, Buttoo K, et al. Melatonin and human cardiovascular disease. J Cardiovasc Pharmacol Ther. 2017;22:122–32.
Edoute Y, van der Merwe E, Sanan D, Kotzé JC, Steinmann C, Lochner A. Normothermic ischemic arrest of the isolated working rat heart. Effects of time and reperfusion on myocardial ultrastructural, mitochondrial oxidative function and mechanical recovery. Circ Res. 1983;53:663–78.
Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta. 1813;2011:1351–9.
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME. Abel ED (2005) reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–95.
Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, et al. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol. 2011;106:447–57.
Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–40.
Koncsos G, Varga ZV, Baranyi T, et al. Diastolic dysfunction in prediabetic male rats: role of mitochondrial oxidative stress. Am J Physiol Heart Circ Physiol. 2016;311:H927–43.
Abdurrahchim D, Ciapaite J, Wessels B, et al. Cardiac diastolic dysfunction in high fat-fed mice is associated with lipotoxcicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta. 1842;2014:1525–37.
Lesnefsky EJ, Chen Q, Moqhaddas S. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–7.
Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–43.
Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia/reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–89.
Chen Q, Younus M, Thompson J, Hu Y, Hollander JM, Lesnefsky EJ. Intermediate metabolism and fatty acid oxidation: novel targets of electron transport chain-driven injury during ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2018;314:H787–95.
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–12.
Chen Q, Vasques EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–31.
Petrosillo G, Moro N, Ruggiero FM, Paradies G. Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome release. Free Rad Biol Med. 2009;47:969–74.
Lamont K, Nduhirabandi F, Adam T, Thomas DP, Opie LH, Lecour S. Role of melatonin, melatonin receptors, and STAT3 in the cardioprotective effect of chronic and moderate consumption of red wine. Biochem Biophys Res Comm. 2015;465:719–24.
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, et al. JAK2/STAT3 activation by melatonin attenuates mitochondrial oxidative damage induced by ischemia/reperfusion injury. J Pineal Res. 2013;55:275–86.
Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1a-SIRT3 signaling. Sci Rep. 2017;7:41337.
Gottlieb RA, Andres AM, Sin J, Taylor DPJ. Untangling autophagy measurements all fluxed up. Circ Res. 2015;116:504–14.
Xie H, Xu Q, Jia J, et al. Hydrogen sulphide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun. 2015;458:632–8.
Siddall HK, Yellon DM, Ong S-B, Mukherjee UA, Burke N, Hall AR, et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS One. 2013;8:e62400.
Kato H, Lu Q, Rapaport D, Kozjak-Pavlovic V. Tom 70 is essential for Pink 1 import into mitochondria. PLoS One. 2013;8:e58435.
Pei H-F, Hou J-N, Wei F-P, Xue Q, Zhang F, Peng CF, et al. Melatonin attenuates postmyocardial infarction injury via increasing Tom70 expression. J Pineal Res. 2017;62:e12371.
Cruz-Topete D, List EO, Okada S, Kelder B, Kopchick JJ. Proteomic changes in the heart from diet-induced pre-diabetic mice. J Proteome. 2011;74:716–27.
Saito T, Nah J, Oka S, et al. An alternative mitophagy mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129:801–19.
Funding
The project was funded by the South African National Research Foundation, Grant no 93579.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
Male Wistar rats were used as experimental animals. The breeding, maintenance and feeding of these rats as well as food and water consumption, and weight monitoring were all carried out in the Central Research Facility, Faculty of Health Sciences, Stellenbosch University The study conformed to the revised South African National Standard for the Care and Use of Animals for Scientific Purposes (South African Bureau of Standards, SANS 10386, 2008) and was approved by the Committee for the ethical use of animals in research of the University of Stellenbosch (project number SU-ACUM14-00039).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dhanabalan, K., Mzezewa, S., Huisamen, B. et al. Mitochondrial Oxidative Phosphorylation Function and Mitophagy in Ischaemic/Reperfused Hearts from Control and High-Fat Diet Rats: Effects of Long-Term Melatonin Treatment. Cardiovasc Drugs Ther 34, 799–811 (2020). https://doi.org/10.1007/s10557-020-06997-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-020-06997-9