
Abstract
Background
Periprocedural myocardial infarction (MI) is a frequent complication of percutaneous coronary intervention (PCI). Statins might reduce its incidence. The aims of the present study are to assess whether such benefit is a class-effect or whether differences exist between various lipid-lowering strategies and whether cardioprotection is exerted by increasing circulating endothelial progenitor cells (EPCs).
Methods
The REMEDY study will enroll a total of 1080 patients submitted to elective PCI. Eligible patients will be randomized into 4 groups: 1) placebo; 2) atorvastatin (80 mg + 40 mg before PCI); 3) rosuvastatin (40 mg twice before PCI); and 4) rosuvastatin (5 mg) and ezetimibe (10 mg) twice before PCI. Peri-procedural MI is defined as an elevation of markers of cardiac injury (either CK-MB or troponin I or T) values >5x the upper reference limit estimated at the 99th percentile of the normal distribution, or a rise >20 % in case of baseline values already elevated. EPCs will be assessed before, at 24 h and - in a subset of diabetic patients - at 3 months after PCI (EPC-substudies). The primary endpoint of the main REMEDY study is the rate of peri-procedural MI in each of the 4 treatment arms. Secondary endpoints are the combined occurrence of 1-month major adverse events (MACE, including death, MI, or the need for unplanned revascularization); and any post-procedural increase in serum creatinine. Endpoints of the EPC-substudies are the impact of tested regimens on 1) early (24-h) and 3-month EPC levels and functional activity; 2) stent strut re-endothelialization and neointimal hyperplasia; 3) 1-year MACE. REMEDY will add important information on the cardioprotective effects of statins after PCI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Periprocedural myocardial infarction (MI) is a frequent complication of percutaneous coronary intervention (PCI), occurring in up to 40 % of cases [1, 2]. Although most patients remain asymptomatic and with no changes in cardiac function, even a mild release of creatine kinase-MB isoenzyme (CK-MB) is associated with higher mortality during the follow-up [2]. Randomized studies have demonstrated that HMG-CoA reductase inhibitors (statins) might reduce the incidence of periprocedural MI in both stable and acute settings [3–10]. At present, however, few data exist on whether this myocardial protection should be considered as a class-effect and whether one statin is superior to others [11–15]. In addition, mechanisms responsible of this beneficial effect have not been elucidated, although many so-called “pleiotropic” effects have been advocated. The anti-inflammatory properties of statins may play an important role, because of the demonstration that the benefit appears to be higher in patients with high C-reactive protein [16–24]. Statin administration also rapidly improves endothelial function [25]. Indeed, even short-term treatment with statins (unable to provide LDL reduction persistent enough to decrease the atherosclerotic burden) may have important effects on endothelial function [25–27]. In the present Rosuvastatin For RE duction Of M yocardial D amag E During Coronary Angioplast Y (REMEDY) trial we will assess whether the beneficial effect of statins in reducing periprocedural MI 1) is a class-effect or, on the contrary, differences exist between rosuvastatin, atorvastatin and the rosuvastatin/ezetimibe combination, and 2) is due to their effects on endothelial progenitor cells (REMEDY-EPC studies).
Methods
Patient Population
This is a multicenter, prospective, randomized, double-blind, double-dummy, placebo-controlled trial on all-comers with coronary artery disease (CAD) candidate to an elective PCI, both treated or untreated with statins. Inclusion and exclusion criteria are reported in Table 1. Written informed consent for participation to the study will be obtained from each enrolled subject before randomization. The current study protocol is an Investigator-Initiated study (ISS), proposed and endorsed by the Italian Federation of Cardiology Gruppo di Studio Aterosclerosi, Trombosi e Biologia Vascolare (ATBV Study Group), partially funded by AstraZeneca and by a Programma Operativo Nazionale (PON) Ricerca e Competitività 2007–2013 PON01_02342, and will be performed through a network of hospitals belonging to the two main Italian National Societies, Società Italiana di Cardiologia, Associazione Nazionale Medici Cardiologi Ospedalieri, as well as the Gruppo Italiano Studi Emodinamici, on a voluntary basis (Appendix 1). The trial will be conducted according to the principles of the Declaration of Helsinki and Good Clinical Practice, and has been approved by the Ethic Committees of each participating center. The trial has been registered with Europeac Clinical Trials Register (EudraCT Number: 2009–013,622-17).
Randomization
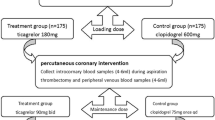
Randomization will be performed by a central randomization, concealed with the use of a web-based system. Randomization sequence will be computer-generated and blocked. Randomization will be 1:1:1:1. Outcome evaluation will be performed on patients randomized and receiving a PCI. Patients admitted to the study will be randomized into 4 parallel groups (Fig. 1): 1) standard background treatment (i.e., performing PCI on the background of standard treatment, without any change of the therapy received by the patient, according to local practice) + placebo twice immediately before the PCI; 2) standard background treatment + atorvastatin, (given 80 mg + 40 mg before PCI (7); 3) standard background treatment + rosuvastatin (40 mg twice before PCI); and 4) standard background treatment + rosuvastatin (5 mg) and ezetimibe (10 mg) twice before PCI (dosages expected to be equipotent, in terms of LDL cholesterol reduction, to the rosuvastatin regimen, but testing a largely HMG-CoA reductase inhibition-independent way of reducing LDL cholesterol). The first dosage of the tested drug will be given within a time interval of 24–12 h before the planned procedure. The second dosage will be given immediately (within a time interval of 1 h-10 min) before the start of the PCI. The protocol includes two substudies to be conducted at specific sites, the REMEDY-EPC early substudy (performed as a single-center substudy at the University Cardiology Division, SS. Annunziata Hospital, Chieti, Italy), enrolling consecutive patients with stable coronary artery disease (CAD) (Fig. 2); and the REMEDY-EPC late substudy (performed as a single-center substudy at the Clinica Mediterranea, Naples, Italy), enrolling consecutive patients with stable coronary artery disease (CAD) and diabetes mellitus (Fig. 3).

Study design of the REMEDY trial. 1. standard background treatment (i.e., performing PCI on the background of standard treatment, without any change of the therapy received by the patient, according to local practice) + placebo twice immediately before the PCI; 2. standard background treatment + atorvastatin, given 80 mg + 40 mg before PCI; 3. standard background treatment + rosuvastatin 40 mg twice before PCI; 4. standard background treatment + rosuvastatin 5 mg + 10 mg ezetimibe twice before PCI. Additional determinations performed in the REMEDY-EPC late sub-study, are detailed in Fig. 3

Study design of the REMEDY-EPC early substudy. CAD patients enrolled will be randomized in 4 groups according to the treatment strategy reported in Fig. 1. Sampling for EPC levels will be performed at the time of randomization to placebo or lipid-lowering treatments (sample R), and 1 h before the diagnostic angiography and PCI at time of treatment reload (placebo or lipid-lowering treatments, sample T0).

Study design of the REMEDY-EPC late sub-study. Diabetic patients enrolled will be randomized in 4 groups according to the treatment strategy reported in Fig. 1. Sampling for EPC levels will be performed before the administration of the tested treatments, at 24 h and 3-month after PCI. At discharge, diabetic patients will be further randomized in 2 groups: 1) receiving standard background treatment and high (80 mg) atorvastatin dose for 3 months; and 2) receiving standard background treatment and low (20 mg) atorvastatin dose for 3 months. Patients will be scheduled for re-hospitalization at 3-month in order to assess 1) EPC levels, and b) degree of DES re-endothelialization and neointimal hyperplasia by OCT evaluation
Percutaneous Coronary Intervention
Stents will be implanted according to current clinical practice. In order to limit additional confounders, all diabetic patients enrolled in the REMEDY-EPC late substudy will be treated by the same drug-eluting stent (DES) type, specifically the Cre8™ Amphilimus sirolimus eluting stent (Cre8 AES; Alvimedica, Istanbul, Turkey) 1) to prevent confounding effects due to differences between various DES, and 2) to follow the promising preliminary results of this DES in diabetic patients [28]. Cre8™ is a polymer-free sirolimus-eluting stent with a thin (80 μm) L605 cobalt-chromium alloy, integrally coated by an ultra-thin (0.3 μm) passive carbon coating (i-Carbofilm™). Angiographic success is defined as a final angiographic residual stenosis of <20 % by visual estimation. Procedural success is considered in cases of angiographic success and the absence of any in-hospital major complication (acute MI, need for bypass surgery or repeat PCI, or death). Angiographic complications include: minor/major side branch compromise or occlusions; abrupt intra-procedural vessel closure; major arterial dissection; thrombus formation; transient and/or prolonged slow-no reflow; distal embolization; coronary perforation. All patients will receive daily aspirin (100 mg/day) and a thienopyridine [usually clopidogrel 75 mg/day if on chronic (>3 day) treatment, or 600 mg loading at least 6 h before the procedure]. All patients will receive an intravenous bolus of unfractionated heparin (70 IU/kg) or bivalirudin (bolus of 0.75 mg/kg prior to the start of the intervention, followed by infusion of 1.75 mg/kg/h) for the duration of the procedure, according to the operator’s preference. Additional heparin or bivalirudin boluses will be given to maintain an activated clotting time > 250 s. For the REMEDY-EPC late substudy, the activated clotting time will be measured on the ACT PLUS® Automated Coagulation Timer System (Medtronic Vascular Inc., Santa Rosa, CA) 5 min after heparin administration. Glycoprotein IIb/IIIa inhibitors will be administered according to operators’ preference.
Postprocedure Management and Follow-up
Aspirin (100 mg/day), and clopidogrel (75 mg daily for 30 days in case of bare metal stents or 6–12 months in case of DES) will be prescribed after PCI. All patients will be discharged on statins according to the local practice and preference. Diabetic patients enrolled into the REMEDY-EPC late substudy will be further randomized into 2 groups: 1) receiving standard background treatment and high (80 mg/day) atorvastatin dose for 3 months (Atorvastatin High-Dose Group); and 2) receiving standard background treatment and low (20 mg/day) atorvastatin dose for 3 months (Atorvastatin Low-Dose Group). These subgroups of patients will be scheduled for re-hospitalization at 3 months in order to assess 1) EPC levels, 2) glyco-metabolic control, and 3) the degree of stent strut re-endothelialization and intrastent neointima hyperplasia evaluated by optical coherence tomography (OCT) in the 2 treatment groups (high- versus low-dose atorvastatin) (Fig. 3).
Study Endpoints
The primary endpoint of the REMEDY study is the rate of periprocedural MI in each of the 4 treatment arms. Periprocedural MI is defined as an elevation of troponin (either I or T) values >5 x 99th percentile upper reference limit (URL) in patients with normal baseline values (≤99th percentile URL) or a rise of troponin values >20 % if the baseline values were elevated and are stable or falling [29]. Secondary endpoints are: 1) the combined occurrence of 1-month major adverse cardiovascular events (MACE, including death, MI, stroke or the need for unplanned revascularization) in the 4 treatment arms. 2) any post-procedural increase in serum creatinine or decrease in creatinine clearance (calculated by the Cockcroft-Gault formula) in the 4 treatment arms, in order also to assess possible beneficial effects of treatments on contrast-induced acute kidney injury (CI-AKI).
Endpoints of the REMEDY-EPC substudies are 1) to examine whether preprocedural exposure to different statins can acutely (within 24 h) increase EPC levels and functional activity (with potential differences between treatments) compared with a standard treatment in patients with stable CAD undergoing successful PCI; 2) to examine whether statin-induced changes in EPC levels and function are quantitatively related, and therefore potentially causally linked, with the acute effects on postprocedural MI; 3) for the REMEDY-EPC late substudy: to assess whether statin dose (atorvastatin 80 mg/day versus 20 mg/day) induces different 3-month changes in EPC levels in diabetic patients treated by DES implantation; 4) to assess whether EPC levels may influence stent struts re-endothelialization and neointimal hyperplasia, as assessed by OCT at the 3-month follow-up; and 5) to assess the impact of EPC levels at different times (pre-PCI, 24 h and 3-month after PCI) on the 12-month occurrence of MACE.
Biochemical Parameters
Blood samples will be taken twice before (possibly at admission and immediately before the PCI, in any case at least at 6 h distance) and then at 6, and 24 h after the procedure, to assay CK-MB (measured by mass assay) and/or cardiac troponin (TnI or TnT), and serum creatinine. Total cholesterol, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides, high-sensitivity C-reactive protein (CRP), aspartate and alanine transaminases (safety evaluation) will be assessed at admission. Additional determinations, such as those in the proposed REMEDY-EPC substudies, will be performed at the discretion of participating investigators and are detailed separately (Appendix 2 ). Sampling at 48 h will be optional, in case of abnormal markers in previous samples. Measurements of CK-MB mass and/or TnI or TnT will be performed locally [30]. Upper normal limits are defined as the 99th percentiles of the normal population, with a total imprecision of <10 %, according to the Joint European Society of Cardiology/American College of Cardiology guidelines [31]. Serum creatinine will be measured by standard clinical chemistry techniques.
Clinical Follow-up
Clinical follow-up at one, 3, 6 and 12 months will be obtained by office visit or telephone call in all patients to assess the occurrence of MACE. An electrocardiogram and a detailed medical history of any event occurred during the follow-up will be obtained. Additional testing will be left to the discretion of the investigators. Diabetic patients enrolled into the REMEDY-EPC late substudy will be scheduled for a 3-month re-hospitalization in order to assess 1) EPC levels and 2) the degree of stent struts endothelization and neointimal hyperplasia at 3 months by OCT evaluation (Fig. 3). Details on the OCT analysis are reported in Appendix 2. Serious events and any other safety issues will be reviewed by an independent Data Monitoring and Safety Committee. All events will be adjudicated by a Clinical Events Committee (CEC), blinded to treatment assignment. At least 2 members of the CEC review clinical data and relevant documentation and determine whether endpoints have occurred according to the study definitions. In case of disagreement between reviewers, a third member of the CEC will adjudicate the event, and data will be considered by the entire committee if 2 of the 3 reviewers do not agree.
Power Calculations and Statistical Analysis
A total of 1080 patients will be enrolled (270 per arm). Treatment with atorvastatin in both stable and unstable coronary syndromes has been associated with a 50–90 % reduction of periprocedural MI [6, 8]. Sample size calculation has been performed to test the hypothesis of a therapeutic relative gain over placebo =50 % for at least one out of the 3 tested active treatments. Additional details on statistical analysis for the REMEDY-EPC late substudy are reported in Appendix 2. Descriptive statistic of the baseline characteristics will be presented separately for each treatment arm. Continuous variables will be reported as mean (± standard deviation), median (25th–75th percentiles), minimum and maximum. Counts and percentage will be computed for categorical variables. All analyses will be intention-to-treat. The population analysis will include all randomized patients. An on-treatment sensitivity analysis of the primary outcome will be performed, including patients with no major deviations from the protocol. The percent of patients reaching the primary outcome in each active treatment arm will be compared with the placebo arm, and/or other treatment arms, with the Fisher exact test. Difference in proportions and 95 % confidence intervals will be computed. The hierarchical process for testing hypotheses reported above will be applied to control for the overall type I error. A secondary multivariable analysis of the primary endpoint will be performed to adjust treatment effect for potential confounders. Adjusted odds ratios and 95 % confidence intervals will be computed through a logistic model, and adjusted differences in proportions and 95 % confidence intervals will be computed through a loglinear model with identity link. Secondary outcomes on a categorical scale will be analyzed as the primary outcome (Fisher exact test; difference in proportions and 95 % confidence interval); secondary outcomes on a continuous scale will be compared with the Student t test or the Mann Whitney U test. Mean differences and 95 % confidence intervals will be computed. All statistical analyses will be performed using the SPSS 20.0 (SPSS Inc., Chicago, Illinois) and P values <0.05 will be considered significant.
Discussion
The REMEDY study aims at answering the following unresolved clinical questions about the periprocedural use of statins in patients undergoing PCI:
-
1)
Can the results previously shown with atorvastatin (a lipophilic statin) be reproduced with rosuvastatin (a hydrophilic statin)? This question will be answered by the comparison of the rosuvastatin with the placebo arm. In the Protective Effect of Rosuvastatin and Antiplatelet Therapy On contrast-induced acute kidney injury and myocardial damage in patients with Acute Coronary Syndrome (PRATO-ACS) study [10] early high-dose rosuvastatin (40 mg on admission followed by 20 mg/day) did not show cardioprotective effects when administered in addition to high-dose clopidogrel in statin-naïve non-ST-elevation acute coronary syndrome patients scheduled for early invasive strategy. Accordingly, the ROSEMARY study [28] suggests that early high-dose rosuvastatin therapy (40 mg before treatment plus maintenance for 7 days) in patients with ST elevation MI undergoing primary PCI did not improve periprocedural myocardial perfusion or reduce infarct volume measured by magnetic resonance imaging compared with the conventional low-dose rosuvastatin regimen. On the contrary, in the ROMA trial, high loading dose of rosuvastatin (40 mg) within 24 h before elective PCI decreased the incidence of periprocedural MI compared to the standard treatment [29]. Furthermore, Takano et al. [30] reported that the incidence of periprocedural MI was reduced more effectively by high-dose than by low-dose rosuvastatin in statin-näive patients.
-
2)
Can a dose of rosuvastatin, more potent in terms of LDL cholesterol reduction and HMG-CoA reductase inhibition, further improve on the results of the ARMYDA-ACS study [8]? This question will be answered by the comparison of the atorvastatin with the rosuvastatin arm. This issue has been previously addressed in the ROMA II trial [12] comparing a reloading dose of rosuvastatin (40 mg) and atorvastatin (80 mg) administered within 24 h before PCI in reducing the rate of periprocedural MI and major cardiac and cerebrovascular events in stable patients on chronic statin treatment undergoing elective PCI. Periprocedural MI occurred more frequently in the control group than in the rosuvastatin group and in the atorvastatin group. However, there was no difference between the rosuvastatin group and atorvastatin group in terms of peri-procedural MI and major adverse cardiac and cerebrovascular events at follow-up.
-
3)
Are the results obtained with statins related to their ability to impact on LDL-C or to some of the putative “pleiotropic” effects (independent of LDL-C reduction and related to the many pathways affected by HMG-CoA reductase inhibition)? Such a question will be tentatively answered by the comparison of the rosuvastatin arm with the rosuvastatin low dose + ezetimibe arm. If the rosuvastatin/ezetimibe combination will produce a similar reduction of necrosis marker release versus the rosuvastatin high-dose arm, it is likely that the effect is not due specifically to HMG-CoA reductase inhibition and possibly attributable to LDL-C reduction. We do not expect a large reduction in LDL-C for the short treatments tested here, and it is also possible that there will be no significant differences among treatment tested in terms of LDL-cholesterol reduction. We do not expect a significant impact on the primary study outcome of the low-dose rosuvastatin/ezetimibe arm, but the confirmation of such expectation will be anyhow worthwhile.
-
4)
In addition to these clinical questions, blood samples taken during the study will allow to investigate the effects of the study treatments on CI-AKI and on and EPC levels (REMEDY-EPC substudies) in the context of periprocedural myocardial damage. Randomized clinical trials evaluating high-dose versus usual-dose statin therapy in patients with stable CAD have shown that intensive lipid lowering results in additional reduction of vascular events [31–33]. Despite that, reluctance still exists on high-dose statin therapy, due to several reasons, including 1) concern about higher rates of side-effects; 2) need for closer clinical follow-up; and 3) cost issues [28]. However, the absolute cardiovascular risk reduction achieved by high-dose statin therapy is proportional to the absolute baseline risk [34]. Due to this uncertainty, different strategies in the usage of high-dose statins in patients with stable CAD have been adopted [35]. The expected benefit afforded by such an intensive statin regimen is a 5 % proportional reduction in the incidence of major cardiovascular events per 10 mg/dl of LDL-C reduction [35, 36]. The present study will clarify whether a higher-than-expected clinical benefit can be explained by the non-lipid-related pleiotropic effects, such as the increase in circulating EPC levels. Indeed, a low number of circulating EPCs has been associated with an increased risk for cardiac death and all other coronary events [32, 37] reviewed in [38]. Enhancement of EPCs has been considered one of the most promising therapeutic alternatives for cardiovascular disease [39]. As an example, it has been reported that for every 10 CFU increase in EPCs, a patient’s likelihood for multivessel coronary disease declined by 20 % [40]. The process of EPC mobilization leads to accelerated re-endothelization. Several pharmacological pathways may mobilize and increase EPCs [41, 42], and statin therapy is the most studied so far [43, 44]. The REMEDY-EPC substudies are the first randomized studies assessing whether a dose–response relationship exists between statin therapy and EPC levels. A result in this direction will support an important additional mechanism by which intensive statin dose may improve outcome after PCI. Furthermore, it will offer an additional marker (beside LCL-C levels) to assess the benefit of statin therapy after PCI.
References
Califf RM, Abdelmeguid AE, Kuntz RE, et al. Myonecrosis after revascularization procedures. J Am Coll Cardiol. 1998;31:241–51.
Lindsey JB, Marso SP, Pencina M, et al. Prognostic impact of periprocedural bleeding and myocardial infarction after percutaneous coronary intervention in unselected patients: results from the EVENT (evaluation of drug-eluting stents and ischemic events) registry. JACC Cardiovasc Interv. 2009;2:1074–82.
Chan AW, Bhatt DL, Chew DP, et al. Early and sustained survival benefit associated with statin therapy at the time of percutaneous coronary intervention. Circulation. 2002;105:691–6.
Ellis SG, Chew D, Chan A, et al. Death following creatine kinase-MB elevation after coronary intervention: identification of an early risk period: importance of creatine kinase-MB level, completeness of revascularization, ventricular function, and probable benefit of statin therapy. Circulation. 2002;106:1205–10.
Herrmann J, Lerman A, Baumgart D, et al. Preprocedural statin medication reduces the extent of periprocedural non-Q-wave myocardial infarction. Circulation. 2002;106:2180–3.
Briguori C, Colombo A, Airoldi F, et al. Statin administration before percutaneous coronary intervention: impact on periprocedural myocardial infarction. Eur Heart J. 2004;25:1822–8.
Pasceri V, Patti G, Nusca A, et al. Randomized trial of atorvastatin for reduction of myocardial damage during coronary intervention: results from the ARMYDA (atorvastatin for reduction of MYocardial damage during angioplasty) study. Circulation. 2004;110:674–8.
Patti G, Pasceri V, Colonna G, et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol. 2007;49:1272–8.
Briguori C, Visconti G, Focaccio A, et al. Novel approaches for preventing or limiting events (Naples) II trial: impact of a single high loading dose of atorvastatin on periprocedural myocardial infarction. J Am Coll Cardiol. 2009;54:2157–63.
Leoncini M, Toso A, Maioli M, et al. Early high-dose rosuvastatin and cardioprotection in the protective effect of rosuvastatin and antiplatelet therapy on contrast-induced acute kidney injury and myocardial damage in patients with acute coronary syndrome (PRATO-ACS) study. Am Heart J. 2014;168:792–7.
Patti G, Cannon CP, Murphy SA, et al. Clinical benefit of statin pretreatment in patients undergoing percutaneous coronary intervention: a collaborative patient-level meta-analysis of 13 randomized studies. Circulation. 2011;123:1622–32.
Sardella G, Lucisano L, Mancone M, et al. Comparison of high reloading ROsuvastatin and atorvastatin pretreatment in patients undergoing elective PCI to reduce the incidence of MyocArdial periprocedural necrosis. The ROMA II trial. Int J Cardiol. 2013;168:3715–20.
Arsenault BJ, Barter P, DeMicco DA, et al. Prediction of cardiovascular events in statin-treated stable coronary patients of the treating to new targets randomized controlled trial by lipid and non-lipid biomarkers. PLoS One. 2014;9:e114519.
Benjo AM, El-Hayek GE, Messerli F, et al. High dose statin loading prior to percutaneous coronary intervention decreases cardiovascular events: a meta-analysis of randomized controlled trials. Catheter Cardiovasc Interv. 2015;85:53–60.
Marenzi G, Cosentino N, Werba JP, et al. A meta-analysis of randomized controlled trials on statins for the prevention of contrast-induced acute kidney injury in patients with and without acute coronary syndromes. Int J Cardiol. 2015;183C:47–53.
Albert MA, Danielson E, Rifai N, et al. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70.
Chan AW, Bhatt DL, Chew DP, et al. Relation of inflammation and benefit of statins after percutaneous coronary interventions. Circulation. 2003;107:1750–6.
Di Napoli P, Antonio Taccardi A, Grilli A, et al. Simvastatin reduces reperfusion injury by modulating nitric oxide synthase expression: an ex vivo study in isolated working rat hearts. Cardiovasc Res. 2001;51:283–93.
Field KM. Effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors on high-sensitivity C-reactive protein levels. Pharmacother. 2005;25:1365–77.
Jones SP, Lefer DJ. Cardioprotective actions of acute HMG-CoA reductase inhibition in the setting of myocardial infarction. Acta Physiol Scand. 2001;173:139–43.
Notarbartolo A, Davi G, Averna M, et al. Inhibition of thromboxane biosynthesis and platelet function by simvastatin in type IIa hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:247–51.
Pasceri V, Cheng JS, Willerson JT, et al. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation. 2001;103:2531–4.
Patti G, Di Sciascio G, D'Ambrosio A, et al. Prognostic value of interleukin-1 receptor antagonist in patients undergoing percutaneous coronary intervention. Am J Cardiol. 2002;89:372–6.
Walter DH, Fichtlscherer S, Sellwig M, et al. Preprocedural C-reactive protein levels and cardiovascular events after coronary stent implantation. J Am Coll Cardiol. 2001;37:839–46.
Wassmann S, Faul A, Hennen B, et al. Rapid effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition on coronary endothelial function. Circ Res. 2003;93:e98–103.
Correia LC, Sposito AC, Lima JC, et al. Anti-inflammatory effect of atorvastatin (80 mg) in unstable angina pectoris and non-Q-wave acute myocardial infarction. Am J Cardiol. 2003;92:298–301.
Patti G, Chello M, Pasceri V, et al. Protection from procedural myocardial injury by atorvastatin is associated with lower levels of adhesion molecules after percutaneous coronary intervention: results from the ARMYDA-CAMs (atorvastatin for reduction of MYocardial damage during angioplasty-cell adhesion molecules) substudy. J Am Coll Cardiol. 2006;48:1560–6.
Fox CS, Golden SH, Anderson C, et al. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation. 2015;132:691–718.
Sardella G, Conti G, Donahue M, et al. Rosuvastatin pretreatment in patients undergoing elective PCI to reduce the incidence of myocardial periprocedural necrosis: the ROMA trial. Catheter Cardiovasc Interv. 2013;81:E36–43.
Takano H, Ohba T, Yamamoto E, et al. Usefulness of rosuvastatin to prevent periprocedural myocardial injury in patients undergoing elective coronary intervention. Am J Cardiol. 2013;111:1688–93.
Cholesterol Treatment Trialists C, Kearney PM, Blackwell L, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–25.
LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35.
Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294:2437–45.
Cholesterol Treatment Trialists C, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Dorresteijn JA, Boekholdt SM, van der Graaf Y, et al. High-dose statin therapy in patients with stable coronary artery disease: treating the right patients based on individualized prediction of treatment effect. Circulation. 2013;127:2485–93.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Briguori C, Testa U, Riccioni R, et al. Correlations between progression of coronary artery disease and circulating endothelial progenitor cells. FASEB J. 2010;24:1981–8.
Madonna R, De Caterina R. Circulating endothelial progenitor cells: do they live up to their name? Vasc Pharmacol. 2015;67-69:2–5.
Moreno PR, Sanz J, Fuster V. Promoting mechanisms of vascular health: circulating progenitor cells, angiogenesis, and reverse cholesterol transport. J Am Coll Cardiol. 2009;53:2315–23.
Kunz GA, Liang G, Cuculi F, et al. Circulating endothelial progenitor cells predict coronary artery disease severity. Am Heart J. 2006;152:190–5.
Fukuda D, Sata M. The renin-angiotensin system: a potential modulator of endothelial progenitor cells. Hypertens Res. 2007;30:1017–8.
Werner C, Kamani CH, Gensch C, et al. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes. 2007;56:2609–15.
Ricottini E, Madonna R, Grieco D, et al. Effect of high-dose atorvastatin reload on the release of endothelial progenitor cells in patients on long-term statin treatment who underwent percutaneous coronary intervention (from the ARMYDA-EPC study). Am J Cardiol. 2016;117:165–71.
Walter DH, Rittig K, Bahlmann FH, et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–24.
Acknowledgments
The REMEDY Study is partially funded by AstraZeneca Italy (to RDC) and a Programma Operativo Nazionale (PON) Ricerca e Competitività 2007-2013 PON01_02342 (to CB and GC).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(PDF 724 kb)
Rights and permissions
About this article

Cite this article
Briguori, C., Madonna, R., Zimarino, M. et al. Rosuvastatin for Reduction of Myocardial Damage during Coronary Angioplasty - the Remedy Trial. Cardiovasc Drugs Ther 30, 465–472 (2016). https://doi.org/10.1007/s10557-016-6672-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-016-6672-3