
Experimental methods for determining the limiting activity coefficients that are relevant in the approbation of theoretical approaches to solve the problem of gas solubility in a liquid are described. Traditional methods realized on a single installation are preferred in order to obtain consistent information. The necessity of additional purification of problem samples of the studied substances is demonstrated. A developed and created low-temperature laboratory column is presented, for which the degree of separation in cases with a coil and packed rectification section, as well as the relative fugacity of the pentane — fluorodichloroethane test mixture are found. Based on the conducted experiments, it can be concluded that it is possible to efficiently use both a coil and a packed column for the purification of substances and enhancement of the measuring effect, and the possibility of obtaining reliable data on limiting solubility from the experimental installation. The gravimetric static method for measuring the concentration of the components in binary mixtures is promising for the study of little-researched and rare substances and is applicable in combination with a small volume cell.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In the simulation of various low-temperature technology and chemical production, when controlling harmful impurities in working fluids and coolants, it is often necessary to determine the solubility of a gas in a liquid. The laws of dilute solutions are usually applied to determine solubility.
The nature of real dilute solutions is more complicated than the nature of regular solutions, and lots of data for non-stratifying systems are satisfactorily described (accurately within 1%) separately by different concentration dependences. At limiting dilution (the concentration of components in the liquid phase is X < 0.002–0.005, the molecules of the solute are surrounded by the solvent), this is Raoult’s law for the solvent and Henry’s law for the dissolved component. In the case of highly dilute solutions (X < 0.03), a linear correction is added to the equations for calculating the parameters of the dissolved component. In dilute solutions (X < 0.2), the parameters of both components are described by equations containing quadratic corrections to the laws of infinitely dilute solutions [1].
In real solutions, the deviation from ideal behavior is accounted for by a correction function — the activity coefficient γ , i.e., everything that distinguishes a solution from an ideal one (interactions between solvent molecules, the effect of hydration) is formally considered involving various correlations of experimental data (Margules, Redlich–Kister, and others). These equations should be considered, first of all, as a means for processing the corresponding experimental data [2], their extrapolation properties have not been studied, there is no information on the use of these ratios to predict the solubility of a gas in a liquid.
More promising for solving the problem of calculating the solubility of a gas in a liquid seems to be an approach using a unified equation of state (UEOS) and general conditions of liquid–vapor phase equilibrium. This approach is strictly thermodynamically justified for describing any type of phase equilibrium of a mixture.
The use of the UEOS presents the ability to calculate the thermodynamic properties of substances and to model multiphase equilibria [3].
This approach is used mainly for calculating the properties of gas-condensate mixtures [3, 4], extends to systems of complex chemical nature, high molecular weight, and related substances.
The main difficulties in applying the approach are associated with the accuracy of determining the compositions of the coexisting phases, which is due to the problem of the adequacy of the equation of state used.
The data on the limiting values of the activity coefficients of the components are important for solving the problems of separation of substances and make it possible to estimate the degree of imperfection of the system, the range of fulfillment of Henry’s law. These data can be used to describe vapor-liquid equilibria in the entire concentration range.
Among the many methods for obtaining reliable experimental information [5,6,7], it is necessary to note the traditional methods based on the study of phase equilibria and volumetric relationships, which can be used to develop computational approaches and to study the thermodynamic properties of mixtures at high pressures in a wide range of compositions.
On the experimental setup created by the authors of this work [8], it was possible to combine several methods for determining the thermal properties of substances under conditions of heterogeneous and homogeneous equilibria, which make it possible to study the solubility of a gas in a liquid and determine the limiting activity coefficients. First of all, it is a static method for studying vapor-liquid equilibrium with measuring the total vapor pressure and determining the concentration of the vapor and liquid phases (the content of components in the selected samples is measured by the relative volumetric method, the gravimetric static method [9] and, if necessary, by gas chromatographic analysis). The following were also implemented: a static method for the study of solubility with synthetic specification of the total composition of the heterogeneous mixture and the composition of the liquid phase, determination of the thermal properties of substances at the border of the heterogeneous region; constant volume piezometer method for measurements in single-phase liquid and gas; flow method for measuring the vapor phase composition of highly dilute solutions.
In addition, to determine the activity coefficient at the installation, it is possible to implement differential methods, the cryoscopic method, continuous gas extraction method (Rayleigh distillation method).
With the use of gas chromatographic equipment, it is possible to apply the dynamic method of an inert gas jet and the solubility method. Measurements by different methods can provide an increase in the reliability of the study while achieving consistent results.
To ensure the reliability of measurements in a wide range of state parameters of naturally rare investigated pure substances and their mixtures, the following are provided:
-
small ballast volume of the measuring cell;
-
reliable temperature control of the cell in a wide range of temperatures T with good dynamic properties of the cryostat;
-
isothermal and isobaric conditions of measuring sensors and systems;
-
conditions for the minimum influence of distorting factors when determining the volume of the piezometer, the volumes of phases and filling lines, during sampling, mixing, filling;
-
the possibility of checking measuring instruments during the study.
Due to high purity of the substances used in the study, the requirements for the experimental technique, for differential measurements (limited capabilities of standard equipment) have increased.
To implement new possibilities, pre-instrumental amplifiers have been created based on AD620A microcircuits. A line for analyzing the composition of equilibrium vapor has been created. The suspension of the measuring volume has been improved.
In the case of investigating little-studied, rare, new or aggressive samples, it is preferable to determine the concentrations of impact on the sample, which do not require constant calibrations and a lot of calibration gas mixtures, or with a pre-set composition.
The developed method for the implementation of the volumetric method for measuring the concentration with the appropriate sensitivity of the measuring device meets these requirements and is suitable for prospecting work. The instrument, created using the method for measuring the concentration in a two-component gas mixture, includes an incompressible measured volume (150 mL) of a neutral resistant material (glass), heat stabilized together with strain gauges in a sealed casing in a thermostated environment. To ensure the reliability of a verified measuring system, only one calibration is required for pure components and, if necessary, for a mixture of equimolar composition. Measurements are available at pressures significantly below atmospheric (5–10 kPa). The concentration measurement accuracy increases in proportion to the mass (pressure) of the sample and the difference in molecular weights of the components. However, there are also significant disadvantages of the method. The resolution of the concentration determination decreases with a decrease in the difference in the molecular weights of the components (up to the loss of measurement capabilities in the case of isomers). The method is sensitive to impurities that differ significantly in molecular weight. The content of such volatile substances increases in the vapor phase, which significantly affects the results of measurements of the composition and total pressure, while the error in determining the limiting solubility significantly increases. The accompanying volatile impurities can be easily removed by flash evaporation at a temperature close to the triple point of the substance, but in the case of an impurity with a low separation coefficient with the main product, this is not feasible or leads to large losses of the substance.
This problem can be solved by creating and using a rectification column comparable in volume to the equilibrium chamber for the study of dilute solutions. When using such a not very efficient column (8–10 theoretical plates), it is possible to expand the concentration range of studies of the solubility of mixtures (with a low separation coefficient α ≅ Y/X ≈ 1, where Y is the composition of the vapor, X is the composition of the liquid) to the region of limiting dilutions and azeotropic composition [10].
The simplest option is to use a coil column: although the surface of contact between liquid and vapor is small in comparison with a packed column of the same volume, its advantages for multipurpose use over other types of columns are ease of manufacture, a minimum of stagnant zones, and stable operating modes at low thermal loads.
A diagram of such a measuring rectification column for a sufficiently effective concentration of volatile impurities from samples of a bottoms liquid at a temperature of 90–400 K is shown in Fig. 1.

Diagram of the measuring rectification column: 1 — electric heater; 2 — heating condenser coil; 3 — still; 4 — rectification part of the column; 5 — casing; 6 — freon evaporator; 7 — nitrogen evaporator; 8 — container with liquid nitrogen; 9 — adjustable thermal bridge; 10 — nitrogen evaporator valve; 11 — reflux condenser; 12 — heat exchanger; 13 — filter; 14 — bypass valve; 15 — capacitor; 16 — compressor; 17 — temperature sensor; 18 — multi-junction differential thermocouple.
The still 3 with a capacity of 0.7 L is detachably coupled with two copper coils (∅12 mm × 1 mm × 2500 mm, ∅10 mm × 1 mm × 2500 mm) or the nozzle section (∅28 mm × 1.5 mm × 650 mm, Raschig rings ∅3 mm × 3 mm with a baffle, made of corrosion-resistant mesh) with a reflux condenser 11 equipped with a container 8 for liquid nitrogen.
The assembled structure is hermetically insulated with two separated layers of basalt wool 5, pressurized with gasified nitrogen. Bottom liquid is heated by an electric heater or a flat spiral coil 2 — a refrigerant condenser installed in the lower part of the still volume.
The heat of condensation is removed from the reflux condenser by a cryoagent (at high temperatures it is water) through the thermal bridge 9, by gasification in the heat exchanger 7, and by a refrigerant in the evaporator 6.
In this case, compressed refrigerant vapors of the refrigeration unit with a recuperative heat exchanger 12 and condenser 15 are simultaneously passed through coils 2 and 6, which cools the reflux condenser 11 to 230 K.
In a wider temperature range, the reflux condenser is cooled by removing heat to boiling liquid nitrogen through a tubular thermal bridge controlled by the position of the boiling nitrogen partition wall.
High heat loads are provided by boiling liquid nitrogen in the coil 7 with the opening of the vapor relief valve 10.
The temperature regimes of the operating column are indicated by a platinum resistance thermometer 17 and a differential 18 copper-constantan thermocouple installed in the still, reflux condenser and on the turns of the upper coil. The pressure in the column is measured by a pressure gauge in the blow-off line, the pressure drop in the rectification section is estimated by measuring the vapor pressure in the still.
The efficiency of the column was determined by studying the solubility of the azeotropic [11] mixture R141b in pentane. The mixture is distinguished by a small relative volatility (α) of the components, a significant difference in molecular weights and is convenient for studying the characteristic operating modes of the column at temperatures T = 284 K, 315 K. Experiments with coil and packed columns were carried out at the minimum possible pressure of 0.04 MPa (stable modes were observed in a wide range of loads) and the minimum operating pressure of 0.1 MPa. Pentane with 2–3% R141b admixture was charged into the column still. At low temperatures, volatile impurities (air) were removed, then the concentrations of the components in vapor (Y) and liquid (X), in the reflux condenser (d) and the still (c) of the column were measured in nonsampling stationary automatic mode (see Table 1).
The relationship between the relative volatility α, the number of separation plates n, the stationary degree of separation K, the measured values of Y , X is determined by the Fenske–Underwood equation [12]
When fully diluted, K∞ = Xd/Xc or ln K∞ = n ln α .
The relative volatility is determined from the expression
The concentrations of R141b in the equilibrium vapor are measured according to the results of measurements in a thermostated still at temperatures T1 = 284, T2 = 315 K and the content of R141b in the liquid Xc1 = 0.029, Xc2 = 0.06 mol/mol.
The values of relative volatilities were determined:
Their close to linear isothermal concentration dependences were estimated.
The value of αav, characteristic for the concentration range of the operating column, was calculated at the corresponding temperature (T1 or T2) from the arithmetic mean of the liquid compositions in the still and in the reflux condenser.
The measurements were carried out sampling 50–100 mg of gas along thin (2 mm × 0.75 mm) analysis lines into an evacuated measuring volume. The mass of the measured volume with and without a gas sample was recorded by a strain gauge and a digital voltmeter with a resolution of 60 μV/mg, the pressure — with a resolution of 1 μV/Pa, which corresponds to a resolution of 200 ppm in the concentration of components in this mixture. An accuracy of more than 0.002 mol/mol is ensured by taking into account the small calibrated positive deviations (parabolic shape) of the actual values characteristic of the measuring device from the calculated ones, reaching the greatest discrepancy of 0.0025 mol/mol at an equimolar composition.
Air and other low-temperature impurities were monitored during the evacuation of samples with a lowvolume 0.15 L cryogenic pump and indication of the residual pressure by a strain gauge with a resolution of 14 μV/Pa. Even a small admixture of air can significantly affect the measurement process and distort the column mode data. The bottom row of Table 1 illustrates the mode with insufficient elimination of a volatile impurity with a relatively low molecular weight. The most balanced mode in the region of limiting dilutions is confirmation of the possibility of measuring the relative volatility in the column reflux condenser and refining the compositions in the still with a known number of separation plates (Yc = 0.0299; Xc = 0.0200; α = 1.49).
It should be noted that the indicators of the coil rectification segment of this form (n ≈ 7.5) are rather high in comparison with the packed one (n ≈ 9) when their volumes are the same.
Due to the small difference between the pressure in the still and the pressure in the reflux condenser of the operating column (ΔPp < ΔPc < 0.005MPa), it is possible to effectively extract volatile impurities in the initial samples by approaching the triple point of the substance. When the pressure in the reflux condenser is 0.02 MPa, the concentration of R141b (as an impurity) can be reduced to 100 ppm, which was confirmed by unchanged subsequent readings of the impurity content and their correspondence to the pure solvent.
At pressures below 0.03 MPa, the coil column is less sensitive to regime disruptions, which makes it possible to concentrate the impurity while maintaining a minimum amount of reflux in the working section and to protect the bottom liquid from contamination after stopping the process. Using the column, air impurities were removed from the butane sample and the total content of H2, N2, CH4 impurities in ethylene was reduced to 200 ppm.
The results of studies of the vapor-liquid equilibrium of the ethylene–butane system with a low solute content have been specified and supplemented. Activity coefficients in the ethylene–butane mixture on isotherms 273 K, 283 K were determined by different static methods:
1. From the solubility data P, T , X, obtained by the static method with a synthetic specification of the composition:
where index 2 corresponds to the dissolved component, index 1 — to the solvent, index 0 — to the properties of pure components, index ∞ is the limiting value at infinite dilution.
2. Static method from the P, T , X , Y data:
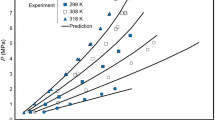
The limiting values of the activity coefficients were determined by method 1 (Fig. 2): for \( T=283\kern1em \mathrm{K}\kern1em {\upgamma}_2^{\infty }=0.82, \) for \( T=273\kern1em \mathrm{K}\kern1em {\upgamma}_2^{\infty }=0.79; \) method 2: for \( T=283\kern1em \mathrm{K}\kern1em {\upgamma}_2^{\infty }=0.84, \) for \( T=273\kern1em \mathrm{K}\kern1em {\upgamma}_2^{\infty }=0.83. \)

Extrapolation of pressures at a boiling point of 283 K to determine the activity coefficients at infinite dilution.
The proximity of the calculation results with an error of \( \partial {\upgamma}_2^{\infty }=4-7\% \) indicates the necessary consistency and the possibility of using methods 1 and 2 under these conditions.
Conclusions
The use of a column with a coil rectification segment (as well as with a packed rectification segment) allows to provide the necessary additional purification of starting materials from impurities and to carry out studies of extremely dilute solutions with a multiply and reproducibly enhanced measuring effect.
When preparing samples using the described unit, it is possible to obtain consistent solubility data by different methods, which increases the reliability of the results.
The gravimetric static method for measuring the concentration of the components of binary mixtures is promising for the study of little-studied and rare substances and is applicable in combination with a smallvolume measuring cell.
References
A. M. Rosen, “Regularities of dilute solutions of non-electrolytes” [in Russian], Zh. Fiz. Khim., No. 43, Issue I, 169–179 (1969).
A. G. Morachevsky, N. A. Smirnova, and E. M. Piotrovskaya, Thermodynamics of Liquid-Vapor Equilibrium [in Russian], Khimiya, Leningrad (1989).
A. I. Brusilovsky, Phase Transformations in the Development of Oil and Gas Fields [in Russian], Graal’, Moscow (2002).
R. Reid, J. Prausnitz, and T. L. Sherwood, The Properties of Gases and Liquids [in Russian], Khimiya, Leningrad (1982).
A. G. Morachevsky, N. A. Smirnova, I. M. Balashova, and I. B. Pukinsky, Thermodynamics of Dilute Solutions of Non-Electrolytes [in Russian], Khimiya, Leningrad (1982).
D. S. Tsiklis, Technique of Physicochemical Research at High and Ultrahigh Pressures [in Russian], Khimiya, Moscow (1976).
V. A. Durov and E. P. Ageev, Thermodynamic Theory of Solutions [in Russian], textbook, Librokom, Moscow (2010).
V. L. Bondarenko, A. V. Valyakina, A. V. Borisenko, A. V. Trotsenko, and V. N. Valyakin, “Vapor-Liquid Equilibrium of the Ethylene–Butane Mixture,” Chem. Petrol. Eng., 53, Nos. 11–12, 778–787 (2018).
Pat. No. 71993 Ukraine, IPC G01N 9/02, G01N 27/407. “Gravimetric static method for determining the concentration of twocomponent gas mixtures” [in Russian], V.N. Valyakin, A. V. Trotsenko, and A. V. Valyakin (2012).
I. N. Bushmakin and E. D. Voeikova, “Liquid–vapor equilibrium in the benzene–CCl4 system” [in Russian], Zh. Obshch. Khim., 19, Issue 9, 1615–1626 (1949).
E. W. Lemmon, M. L. Huber, and M. O. McLinden, “NIST Reference Fluid Thermodynamic and Transport Properties Database (REFPROP): Version 9.1. NIST Standard Reference Database 23, National Institute of Standards and Technology, AC&MD, Gaithersburg (2013).
Ya. D. Zelvensky, A. A. Titov, and V. A. Shalygin, Rectification of Dilute Solutions [in Russian], Khimiya, Leningrad (1974).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimicheskoe i Neftegazovoe Mashinostroenie, Vol. 56, No. 6, pp. 12–16, June, 2020.
Rights and permissions
About this article

Cite this article
Valyakina, A.V., Valyakin, V.N., Bondarenko, V.L. et al. Methods and Experimental Techniques to Research Solubility of Gases in Liquids. Chem Petrol Eng 56, 440–447 (2020). https://doi.org/10.1007/s10556-020-00792-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10556-020-00792-x