
Abstract
The protein-coding regions of mRNAs have the information to make proteins and hence have been at the center of attention for understanding altered protein functions in disease states, including cancer. Indeed, the discovery of genomic alterations and driver mutations that change protein levels and/or activity has been pivotal in our understanding of cancer biology. However, to better understand complex molecular mechanisms that are deregulated in cancers, we also need to look at non-coding parts of mRNAs, including 3′UTRs (untranslated regions), which control mRNA stability, localization, and translation efficiency. Recently, these rather overlooked regions of mRNAs are gaining attention as mounting evidence provides functional links between 3′UTRs, protein functions, and cancer-related molecular mechanisms. Here, roles of 3′UTRs in cancer biology and mechanisms that result in cancer-specific 3′-end isoform variants will be reviewed. An increased appreciation of 3′UTRs may help the discovery of new ways to explain as of yet unknown oncogene activation and tumor suppressor inactivation cases in cancers, and provide new avenues for diagnostic and therapeutic applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Somatic and/or germ line mutations are universal characteristics of cancer. Discovery of mutations that activate oncogenes (e.g., RAS) or inactivate tumor suppressors (e.g., TP53, BRCA1) has been pivotal in our understanding of cancer biology, with great relevance to current clinical practices. Large-scale initiatives such as the International Cancer Genome Consortium (ICGC), The Cancer Genome Atlas (TCGA), and Genomics Evidence Neoplasia Exchange (GENIE) have revealed common genomic alterations (e.g., mutation hotspots, DNA copy variations etc.) in more than 30 different cancer types, generating novel insights into cancer biology. Currently, tumor-specific genetic defects are identified and can be used for treatment decisions (reviewed in [1]). Despite all these exciting developments, protein-coding sequences are only the 1.5–2% of mammalian genomes, whereas 80–90% of mammalian genomes are transcribed into non-coding (nc) RNAs, including microRNAs, long ncRNAs, siRNAs, piRNAs, tRNAs, rRNAs, snRNAs, and snoRNAs [2]. ncRNA genes are distributed throughout the genome, in between genes or within genes, with their own regulatory elements or they piggy-back on their host genes for expression. Many of ncRNAs are further processed by splicing or enzymatic cleavage into smaller and functional products that regulate other RNAs and/or proteins. Hence, ncRNAs provide an unprecedented functional richness and complexity to the concept of the central dogma. Consequently, with advances in RNA-sequencing methods, the discovery rate of ncRNAs and our understanding of the non-coding part of our genomes are increasing, providing novel insights into normal physiology and disease mechanisms including cancer (reviewed in [3, 4]).
Overall, together with protein-coding regions and ncRNAs, we have a more comprehensive understanding of the cancer cell. However, there is yet another group of sequences overlooked in mammalian genomes that represent both worlds; 3′ untranslated regions (3′UTRs), which are the non-coding sequences of protein-coding mRNAs. For a long time, 3′UTRs were known for their obscure role in regulating mRNA transport, mRNA stability, however were generally dismissed as merely accessory sequences next to the open reading frames. Now, accumulating evidence show 3′UTRs to be much more. This review addresses diverse roles of 3′UTRs, how 3′UTR variants form and discusses how advances in RNA biology are likely to shape our understanding of cancers from a 3′UTR perspective.
2 Roles of 3′UTRs
The coding region of an mRNA is the recipe to make a peptide; however, supporting role of the 3′UTR in this process is no less significant. 3′UTRs regulate mRNA metabolism in multiple aspects, including mRNA stability, mRNA localization, and mRNA translation efficiency, all of which control how much and when the coding sequence will be translated into the protein. The following sections will describe different aspects of mRNA metabolism from a 3′UTR perspective.
2.1 mRNA half-life and stability
mRNA half-lives range from minutes to hours. Pharmacological inhibition of transcription and more recently, metabolic labeling/sequencing approaches showed grouping of short and long-lived mRNAs according to the function of corresponding proteins. mRNAs of transcription factors and cell cycle regulators have short half-lives (less than 2 h), whereas mRNAs for housekeeping and biosynthetic proteins have much longer half-lives [5, 6]. This correlation is also present in yeast [7, 8], suggesting evolutionarily conserved mechanisms to control mRNA stability, which is regulated at the 3′UTRs.
3′UTRs harbor cis-elements that are recognized by trans-factors including RNA-binding proteins and microRNAs. Approximately 7.5% of all protein-coding genes are generally ubiquitously expressed and directly involved in RNA metabolism by binding to and/or processing RNA [9]. RNA-binding proteins generally bind to 3′UTR AU-rich elements through their RRM (RNA recognition motif), KH (hnRNP K homology), DEAD box helicase, and zinc finger domains [10]. There are also non-canonical RNA-binding domains including intrinsically disordered protein regions (reviewed in [11]). On the other hand, microRNAs bind to 3′UTRs through imperfect base pairing and negatively regulate gene expression at the posttranscriptional level [12]. Overall, binding of proteins and/or microRNAs to specific recognition sequences or secondary structures within mRNAs modulates translation or mRNA decay rates by recruiting specific enzyme complexes that perform the destruction processes. Indeed, availability of structural (e.g., stem loops) regions and/or cis-elements (e.g., AU-rich elements) correlates negatively with mRNA half-lives [6]. However, the availability of such cis-elements on a 3′UTR does not always enforce a specific biological destiny as mRNA stability can be modulated during biological processes including inflammatory response [13], cellular stress [14], and differentiation [15]. Possible ways to regulate mRNA stability are through different modes of binding of trans-factors; however, these are not necessarily mutually exclusive interactions (Fig. 1).

Modes of binding to cis-elements on 3′UTRs. mRNA cis-elements are recognized by RNA-binding proteins and/or microRNAs. These trans-factors regulate the mRNA fate by binding to their target sequences such as the AU-rich elements. Binding of a specific trans-factor may prevent the binding of others through competitive binding (A) and/or in a collaborative manner (B). Binding of trans-factors can be activated or inhibited (e.g., through phosphorylation) by upstream proteins (i.e., regulated binding, C). Binding of trans-factors to a given mRNA may deplete the trans-factor availability and indirectly affect other mRNAs (sponge effect, D)
2.1.1 Regulated binding for stability
Posttranslational modifications (PTMs) regulate the activity and/or subcellular localization of RNA-binding proteins. Lysine acetylation and tyrosine phosphorylation are among known PTMs, suggesting context-dependent regulation of RNA-binding proteins through various signaling routes. One of these signaling routes is the p38/MK2 pathway implicated in a variety of pathological processes including metastasis, apoptosis, and inflammation. In addition to known downstream effects, genotoxic stress-activated p38/MK2 pathway globally regulates RNA-binding protein and 3′UTR interactions, detected by a combination of transcriptomic and proteomic methods. For example, activated MK2 increases interleukin (IL-6) and TNF-α production by stabilizing their mRNAs through directly phosphorylating TTP (tristetraprolin), which is a destabilizing RNA-binding protein that otherwise binds to these mRNA 3′UTRs [16]. Activated p38/MK2 pathway also leads to phosphorylation HuR and regulates its cytoplasmic availability and binding to target mRNAs [17], one of which is COX-2 [18]. COX-2 is a prostaglandin synthase enzyme implicated in inflammation, cell growth, and tumorigenesis. Anti-inflammatory butyric acid produced in the gut, indirectly decreases the stability of inflammatory COX-2 mRNA. Butyrate decreases activity of p38/MK2, leading to less HuR binding to COX-2 3′UTR, and reduced mRNA and protein levels of this inflammatory gene [19].
Another target of the p38/MK2 is hnRNPA0, as part of an axis that is linked to TP53 and chemotherapy response. TP53 has key decisive roles in maintaining genomic integrity of cells by controlling cell-cycle arrest, apoptosis, and is one of the most frequently mutated tumor suppressor genes in cancers [20,21,22]. Upon DNA damage, p53 transcriptionally upregulates p21 and GADD45α transcription to regulate cell cycle arrest. However, in case of TP53 mutations, tumor cells become dependent on the p38/MK2 pathway to survive from topoisomerase inhibitors or platinum-based compounds [23, 24]. In response to DNA damage, MK2-phosphorylated hnRNPA0 binds to p21 and GADD45α 3′UTRs, leading to the stabilization of their mRNAs and inducing cell cycle arrest. This arrest provides time for DNA repair and thus allows development of resistance to cisplatin therapy in lung cancer cells [14].
2.1.2 Competitive binding for stability
More than one type of RNA-binding protein and/or microRNA can recognize the same or overlapping cis-elements on 3′UTRs. In certain instances, recognition of these cis-elements by a specific trans-factor instead of others determines the faith of the mRNA. For example, RNA binding-proteins including hnRNPD, hnRNPA/B, ZMAT3 (a.k.a. wild-type p53-induced gene 1; WIG1), HuR as well as miR-125b interact with the 3′UTR of TP53 mRNA and regulate TP53 protein levels [25, 26]. Binding of WIG1 and HuR to AU-rich elements on the TP53 3′UTR stabilizes the mRNA, leading to higher TP53 protein levels [26, 27]. On the other hand, binding of miR-125a and miR-125b to 3′UTR negatively regulates TP53 translation. HuR and miR-125b competitively and antagonistically control TP53 mRNA translation in response to genotoxic stress to fine-tune TP53 activity and hence modulate cell survival [28].
Another competition among trans-factors is for the 3′UTR of MYC. Destabilizer CELF1 (ELAV-like family member 1, also referred to as CUG-binding protein 1 (CUGBP1)) competes with stabilizer HuR to modulate MYC mRNA stability and translation during intestinal epithelial homeostasis [29]. Similarly, HuR and TTP bind to overlapping sites, with TTP acting as a destabilizing factor for various other transcripts encoding proteins related to immune function and cancer [30].
Accordingly, cell behavior and phenotypes are altered as in the case of apoptosis. B cell CLL/lymphoma 2 (BCL2), an anti-apoptotic protein has several mRNA isoforms. BCL2α mRNA contains the full-length 3′UTR. TRA2β, an SR-like protein, regulates apoptosis by binding to BCL2α 3′ UTR at the consensus binding motif (GAA) which is also the binding site for miR-204. TRA2β binding antagonizes the negative effects of miR-204 and causes stabilization of the BCL2α transcript and upregulation of BCL2 protein. Interestingly, BCL2 mRNA has another isoform, BCL2β that contains an alternatively spliced 3′UTR, whose sequence differs from that of the full-length 3′UTR and lack these cis-elements. TRA2β binding to BCL2 mRNA isoforms is functionally important as knockdown of TRA2β increases sensitivity to anticancer drugs [31]. As seen in case of TP53, MYC, and BCL2, transcripts can be recognized antagonistically by RNA-binding proteins and/or microRNAs. Hence, any shift in this balance of positive and negative regulators is likely to affect the fate of the mRNAs.
2.1.3 Collaborative binding for stability
mRNA cis-elements are often short, and cooperative binding of multiple RNA-binding proteins is required for specificity [32]. For example, RNPC1, an RNA-binding protein and a transcriptional target of TP53, regulates p21 mRNA stability together with HuR. RNPC1 directly interacts with HuR and enhances its RNA-binding activity. Then, both RNPC1 and HuR bind to upstream and downstream AU-rich elements in the p21 3′UTR and stabilize p21 mRNA [33].
On the other hand, mRNA repressor protein TRIM71 recognizes a structural RNA stem-loop motif on the 3′UTR of p21. Next, NMD (non-sense mediated decay) factors such as UPF1 assist TRIM71-mediated degradation of p21 mRNA [34], suggesting a new interplay between RNA-binding proteins and non-canonical NMD. Canonical NMD is generally activated when there is a premature stop codon (PTC) upstream of 3′UTR exon junction complexes (EJC). EJC complexes are deposited by the spliceosome on the mRNA approximately 20–24 nucleotides upstream of the splice junctions in the nucleus, thereby retaining the memory of the former location of the excised introns. In case of an upstream PTC, the ribosome cannot strip off EJCs during the first round of translation. These remaining EJCs initiate NMD, which is regulated by three main and highly conserved factors: UPF1, UPF2, and UPF3. NMD was initially recognized as a quality control mechanism to degrade mRNAs that have PTCs, which may otherwise be translated into truncated proteins. However, it is now clear that NMD is also active in embryogenesis and in normal cells as part of gene expression regulation independent of any non-sense mutations. Within this context, NMD degrades wild-type mRNAs to facilitate cellular responses [35]. Moreover, UPF1 is involved in additional mRNA decay pathways mediated by other RNA-binding proteins (e.g., staufen, stem-loop-binding protein, glucocorticoid receptor) and by microRNAs [36, 37]. Therefore, recruitment of UPF1 and/or other decay stimulating proteins to numerous target mRNAs will be of interest within the context of transformed cells as deregulated expression or activity of these trans-factors are likely to globally alter stabilities of cancer related mRNAs.
2.1.4 Sponge effect of 3′UTRs
Since their initial discovery, mounting evidence has illustrated the key regulatory roles for microRNAs in various developmental, differentiation, cell proliferation, and apoptosis pathways [38,39,40,41]. (Deregulated expression of microRNAs and their roles in cancer have been investigated extensively and reviewed elsewhere [42, 43]). In addition to direct binding of microRNAs to their target mRNAs, there are intricate and indirect relationships between microRNAs and how they recognize their targets on 3′UTRs.
As a well-examined example, miR-34 family was discovered as a direct transcriptional target of TP53 (reviewed in [44]), mediating induction of apoptosis, cell cycle arrest, EMT (epithelial–mesenchymal transition), and senescence [45]. This family is frequently down-regulated in various cancers and has tumor-suppressor roles through their mRNA targets, including MYC, CD44, MET, CDK4/6, NOTCH1, BCL2, and SNAIL (reviewed in [46]). Among numerous key targets of miR-34, CD44 and MYC represent a special case. Overexpression of MYC indirectly causes upregulation of CD44 expression because over-abundance of MYC 3′UTR acts as a miRNA sponge, decreasing the availability of miR-34a pool to bind to the 3′UTR of CD44 mRNA [47]. Since microRNAs recognize multiple mRNAs and a single mRNA can be targeted by more than one microRNA, similar sponge effects are likely to exist in a cancer type specific manner, further fine-tuning gene expression regulation.
Overall, role of 3′UTRs in regulating mRNA stability is probably the most investigated and understood aspect of RNA metabolism in cancer cells. It is clear that interaction of trans-factors with mRNA 3′UTRs are complex and cancer specific changes in the expression or regulation of trans-factors are likely to alter mRNA stabilities and later, protein levels of cancer related genes that are not altered at the DNA level.
2.2 Translation efficiency
Translation initiation is the rate-limiting step of protein synthesis. During initiation, 5′capped (m7GpppG) mRNAs associate with the small ribosomal subunit (40S) together with initiation factors (eIF1, eIF2, eIF3, and eIF4 complexes) to recruit the larger ribosomal subunit. During this assembly process, the eIF4 complex (consisting of eIF4A, eIF4B, eIF4E, and eIF4G) binds to the 5′cap of the mRNA. Next, the 5′cap and the 3′UTR poly(A) tail are circularized and brought together by eIF4G that interacts with the PABP (poly(A) binding protein). This circularization helps translation activation and possibly promotes ribosome recycling [48]. Given the significance of this mRNA conformation during translation initiation, most control elements that regulate the efficiency of translation are located within both 5′ and 3′UTRs. An interesting example is how translation of specific mRNAs is regulated under hypoxic conditions. Oxygen-regulated hypoxia-inducible factor 2α (HIF-2α) forms a complex with the 5′cap-binding protein eIF4E2 (an eIF4E homolog), and RBM4 (RNA binding motif protein 4) which binds to the 3′UTR of the EGFR mRNA. This complex along with other initiation factors direct EGFR mRNA to polysomes for translation under hypoxia when general protein synthesis is repressed [49]. Hence, binding of RBM4 to the 3’UTR of EGFR mRNA is pivotal in HIF-2α-mediated selective translation of EGFR mRNA under hypoxia.
eIF3, another factor for the 43S pre-initiation complex, associates with the eIF4F complex at the 5′cap of the mRNA and helps the recognition of translation initiator AUG codon [50]. Surprisingly, 3′UTR length can modulate eIF3 function, as demonstrated for the PTBP1 (polypyrimidine tract-binding protein) isoforms. eIF3 binds to both 5′ and 3′UTRs of PTBP1 mRNA. However, eIF3 binds differently to 3′UTR isoforms of PTBP1 transcripts, leading to translational regulation of PTBP1 in a cell cycle specific manner, which affects downstream regulation of alternative splicing events mediated by PTBP1 [51]. Another example is a multiprotein complex called GAIT (gamma interferon inhibitor of translation element) that recognizes a bipartite stem-loop structure in 3’UTRs. In myeloid cells, IFN-γ induces binding of GAIT proteins to 3′UTRs of multiple inflammation-related mRNAs, including CP (ceruloplasmin) and VEGFA, and represses their translation [52, 53]. A member of this complex, ribosomal protein L13a, binds to eIF4G to block ribosome recruitment, possibly through mRNA circularization that brings the 3′UTR-bound GAIT complex close to the 5′-end where it can effectively block 43S joining and translation-initiation. RNA secondary structure prediction algorithms predict presence of GAIT elements in other mRNA 3′UTRs in addition to CP and VEGFA mRNAs [54]. Other RBPs also interfere with ribosome recruitment and regulate translation efficiency of mRNAs. HNRNPK and HNRNPE1 bind to the 3′UTR of LOX mRNA and inhibit ribosome assembly and translation during erythroid cell differentiation [55].
While these examples depict the role of 3’UTRs in ribosome assembly and translation initiation, 3′UTRs can modulate the elongation step of translation as well. For example, PUF (PUMILIO and FBF)/AGO complexes bound to cis-elements on 3′UTRs inhibit GTPase activity of the elongation factor, eEF1A, and consequently inhibit peptide synthesis [56].
Another example is highly relevant to cancer related cellular processes. EMT related ILEI (interleukin-like EMT inducer) and DAB2 (Disabled-2) mRNAs are transcribed but are not translated unless the cells are activated by TGF-β [57, 58]. The underlying mechanism depends on an mRNA-protein complex that binds to the 3′UTRs of these transcripts. The so-called BAT (TGF-β activated translational) complex, containing hnRNPE1 inhibits the translation of these EMT related mRNAs by blocking eEF1A1 release during translation elongation. TGF-β signaling leads to phosphorylation of hnRNPE1, which disrupts the hnRNPE1-eEF1A1 interaction, allowing translation and triggering EMT in breast cells [59]. Based on these observations, mRNA circularization is an important step of ribosome function and 3’UTR cis-elements recognized by RNA-binding proteins have the potential to alter translation initiation and elongation dynamics.
2.3 mRNA localization and local protein synthesis
Subcellular enrichment of mRNAs enables accumulation of proteins at specific regions within cells. Polarized cells, such as neurons, are best-known examples for this type of local protein synthesis. mRNA 3′UTRs can facilitate local enrichment of mRNAs and consequently proteins. For example, BDNF (brain-derived neurotrophic factor) mRNA isoforms with different 3′UTR lengths have different localizations. The short 3′UTR isoform of BDNF localizes in soma of neurons, whereas the longer isoform is localized to dendrites. These isoforms have different translational rates and have different roles in the morphogenesis of dendritic spines [60]. Another case is Cdc42 (cell division cycle 42) which is a Rho-GTPase that regulates actin cytoskeleton and cellular morphology [61]. Two mRNA and protein isoforms of Cdc42 with distinct functions (i.e., axonogenesis and formation of dendritic spines) in neuronal polarity are differentially localized at neurites and soma of neurons [62]. These two isoforms differ in their C-terminal ten amino acids and in 3′UTR lengths. Interestingly, it is not the different amino acids but the alternative 3′UTR bound proteins that determine the differential protein localization between soma and neurites [62].
Similar cases are described in less polarized cells where localized mRNAs regulate cell behavior, including cell-cell adhesion and directed cell migration. For example, β-actin mRNA localization and local protein synthesis are enriched at focal adhesions, dynamically regulating the adhesive properties of migrating cells [63]. Deleting the 3′UTR from the β-actin mRNA results with delocalization of β-actin monomer synthesis and perturbs adherens junctions [64, 65]. Zip code binding protein 1 (ZBP1, also called IMP1 (IGF2 mRNA-binding protein 1)) binds to the 3′UTR of β-actin to regulate mRNA localization [66]. IMP1 first associates with β-actin mRNA in the nucleus and transports it to the cell edge in a translationally suppressed form [67]. Later, when an extracellular signal causes IMP1 phosphorylation by Src, β-actin mRNA is released from IMP1 and is translated at the cell edge [67]. IMP1 further facilitates the localization of E-cadherin, α-actinin, and actin-related protein 2/3 (ARP2/3) complex mRNAs, which are involved in cell-cell connections and focal adhesions. Interestingly, non-metastatic breast tumors express IMP1, but it is downregulated in metastatic cells [68]. Decreased expression of IMP1 causes delocalization of cell-motility-related mRNAs, increasing the growth and cell motility of metastatic breast cancer cells [69, 70].
Given these potential implications for protein function, localization, and cellular phenotypes, the question is, how are mRNAs localized or enriched at specific sites? Current evidence suggests two general mechanisms: guided mRNA transport and spatial mRNA degradation. These processes are both dependent on mRNA 3′UTRs.
For mRNA transport, motor proteins and/or RBPs bound to 3′UTRs can facilitate transportation through the cytoskeletal system. In addition to examples in yeast and Drosophila [71, 72], only a few cases have been reported in mammalian cells. For example, a group of mRNAs are anchored to the granule regions at the plus ends of microtubules in mammalian fibroblasts [73, 74]. Of further interest, these mRNAs involved in migration and metastatic progression, require APC (adenomatous polyposis coli) for their localization to cell protrusions in the NIH/3T3 cells spread on a fibronectin-coated surface [73]. Later, APC interacting other RNAs were identified in the brain. Interestingly over 200 mRNA targets were highly enriched for APC-related functions, including microtubule organization, cell motility, and cancer [75]. This unanticipated function for APC may have implications in motility and migration of cancer cells, where APC function is deregulated.
Alternative to active transport, spatial degradation is another way to enrich mRNAs at unique locations in cells. For this, 3′UTR cis-elements have a role in differentially regulating the stability of the mRNA. In Drosophila, Nanos mRNA is localized specifically at the posterior pole of the cytoplasm as it is targeted by 3′UTR-bound Smaug for deadenylation and degradation at other sites [76, 77]. Despite these interesting examples in different cell types, the extent of deregulated mRNA localization and consequences in cancers are not fully known. One reason could be that locally enriched mRNA and protein concentrations are technically difficult to quantify with conventional methods, especially in less polarized cells. Therefore, it is likely that local mRNA enrichment cases are under-estimated and currently, we have an insufficient view of spatial gene expression regulation in cancer cells. Hence, a better understanding of local protein synthesis guided by mRNA subcellular localization may be helpful to understand complex behaviors of cancer cells including focal adhesion dynamics, cell motility, EMT, and metastasis.
2.4 3′UTRs as hubs for protein-protein interactions
3′UTR bound RBPs can facilitate protein-protein interactions such as the CD47 example where protein localization of CD47 is regulated independent of its mRNA localization. CD47 has two mRNA isoforms that differ at their 3′UTR lengths. Only the long 3′UTR harboring mRNA can interact with HuR to recruit SET to its 3′UTR [78]. Next, SET interacts with the CD47 protein synthesized from the longer 3′UTR isoform and enhances plasma membrane localization of CD47, which is a “Don’t eat me” signal [78, 79]. SET transfers from the CD47 mRNA to the CD47 protein during synthesis. This transfer occurs within so-called TIS granules, which are reticular structures intertwined with the endoplasmic reticulum (ER). TIS granules are enriched for the RNA binding-protein TIS11B, and membrane protein-encoding mRNAs. Here, 3′UTR-mediated interaction of SET with membrane proteins occurs, allowing increased surface expression [80].
3 Ending the 3′-end: means to diversity
3′UTRs have pivotal roles in regulating protein abundance and function. With the advancements in RNA-sequencing methods tailored to detect 3′-ends of isoforms, we are just beginning to get a perspective of a 3′-end isoform diversity in cancers and how it is generated by different mechanisms.
3.1 Splicing and polyadenylation
Majority of eukaryotic genes contain long intervening sequences (introns) that disrupt the coding sequences (exons). The spliceosome regulates the removal of introns during pre-mRNA maturation [81]. Since the initial discovery of splicing more than 40 years ago [82, 83], mechanistic understanding of splicing has greatly improved through genetic and biochemical studies including cryo-EM of the spliceosome structure at atomic and near-atomic resolution (for a review, please see [84]). In addition to the spliceosome, numerous splicing regulators including splice enhancer and repressor proteins (e.g., SR proteins) contribute to the splicing decisions in a tissue and developmental stage-specific manner. With the advancements in RNA sequencing methods, it is becoming clear that alternative processing of mRNAs through splicing is the norm and not the exception for gene expression. As for cancer, given that genomic instability is a hallmark, it is not surprising to find a comparable deregulation at the transcriptome level. One aspect of this complexity is due to aberrant splicing in many cancers due to mutations and/or deregulated expression of spliceosome components or mutations in splicing regulating cis-elements such as splice acceptor and donor sites (Reviewed in [85, 86]). For example, RNA splicing machinery is frequently mutated in myeloid malignancies, whereas DNA copy number changes and deregulated expression of splicing factors are more widespread in solid tumors [87].
In fact, defects in splicing machinery are just the tip of the iceberg, with substantial downstream effects on many target transcripts, as was shown for a splicing factor, SRSF1 (serine and arginine-rich splicing factor 1). SRSF1 is amplified and upregulated in breast, lung, colon, and bladder cancers and generates potentially hundreds of pro-proliferative splice variants either through exon inclusion or skipping, enhancing tumorigenesis [88, 89]. For example, SRSF1 acts as an oncogene through regulating alternative splicing of the RON proto-oncogene (MSTR1), a tyrosine kinase receptor for the macrophage-stimulating protein [90]. SRSF1 regulated skipping of exon 11 generates the RONΔ11 isoform, which promotes cell motility and invasion [90]. SRSF1 overexpression also promotes the inclusion of exon 12a of BIN1 (bridging integrator 1) to generate the BIN1+12a isoform, which can no longer bind and inhibit MYC [91]. In addition, inclusion of an exon 13b of the kinase MKNK2 (MAP kinase-interacting serine/threonine-protein kinase 2) generates an isoform that enhances eIF4E phosphorylation [92]. Inclusion of exon 6 of DBF4B (regulatory subunit for CDC7, has a central role in DNA replication and cell proliferation) by SRSF1 promotes tumorigenesis of colon cancer cells [93]. As the SRSF1 case exemplifies the diverse downstream effects, mounting evidence points out to similar aberrant splicing cases and multilayered consequences in cancer cells.
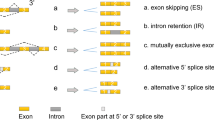
While it has become clear that aberrant splicing is widespread in cancers (aberrant splicing in cancers are discussed in detail in recent and excellent review articles [94,95,96]), general focus is on cassette exons and not so much on 3′-terminal exons. Nevertheless, alternative splicing generates new terminal exons that alter the 3′-end of the coding sequence and/or 3’UTR (Fig. 2). Alternative 3′-terminal exons are generally less conserved and expressed at lower levels than isoforms with the canonical last exons [97]; however, functionally they may be highly relevant to cancer biology.

Splicing leading to 3′UTR isoforms in a hypothetical model. a Removal of the last intronic sequence and use of a polyadenylation signal at the terminal exon marks the polyadenylation site and determines the 3′UTR length. b In case of alternative splicing and retention of a distal intron may change the 3′-end of the coding sequence and include the exonized intronic sequence as part of the 3′UTR. c Splicing causes inclusion of an alternative terminal exon (red), leading to changes in 3′-coding sequence and the 3′UTR sequence
A new terminal exon can be generated by intron retention, which is coupled to activation of an intronic polyadenylation site (Fig. 2b). Alternatively, an exon that is usually spliced out can be included. This new terminal exon may harbor a new stop codon and an exonic polyadenylation signal to be used to add the poly(A) tail (Fig. 2c). In both cases, use of a new terminal exon can potentially lead to mRNA variants that encode protein products with truncated C-terminus amino acids, along with alternate 3′UTR sequences. In case of C-terminus truncation, proteins may be nonfunctional, gain a new function and/or even modulate the function of the wild type protein in a dominant-negative manner. Therefore, the consequences would be unique to each transcript and its corresponding protein function. For example, the observation that dominant-negative and secreted variant isoforms of receptor tyrosine kinases (RTKs) exist as a result of intronic polyadenylation site usage [98], has potential implications and has to be explored in more depth in patient samples. In leukemia, intronic polyadenylation site usage causes inactivation of tumor suppressors including FOXN3, which is a transcriptional repressor involved in cell cycle regulation and tumorigenesis [99]. Alternatively, intronic polyadenylation cases in other genes including CARD11, MGA, and CHST11, promote oncogenic activity [99].
Other intriguing cases have been linked to DNA damage. Initial evidence came from budding yeast where DNA damage induces targeted, genome-wide variation of polyadenylation site usage [100]. Later in breast cancer cells, doxorubicin (DOXO), a TOP-II inhibitor, was shown to modulate use of alternative 3′-terminal exons, which use intronic polyadenylation sites, and globally mediate the genotoxic stress responses [97]. More evidence point out the significance of terminal exon shift in DNA repair pathways. For example, CDK12 phosphorylates C-terminal domain of RNA polymerase II to regulate transcription elongation, splicing, and polyadenylation [101]. CDK12 suppresses intronic polyadenylation and favors distal polyadenylation in mouse embryonic stem cells, enabling the production of full-length mRNAs, including the homologous recombination repair genes. Consequently, depletion of CDK12 causes attenuated DNA damage repair [102]. The functional importance of this finding was further extended with the use of a selective inhibitor (SR-4835) of CDK12 (and CDK13). SR-4835 triggered use of intronic polyadenylation sites (i.e., a total of 1824 sites) some of which impaired the expression/activity of DNA damage response proteins (BAP1, ATR, FANCL, WRN, BRCA1, BRCA2, FANCM, BRIP1, FANCD2, FANCI, BLM, FANCA, ATM), provoking a “BRCAness” phenotype that synergize with DNA-damaging chemotherapy and PARP inhibitors in triple-negative breast cancer cells [103]. Hence, these isoforms individually or collectively can affect treatment responses.
Current understanding is that at least ∼ 20% of human genes have one or more intronic polyadenylation events. Hence, it would be important to understand whether these isoforms are functional in cancer related phenotypes [104]. Of note, many cryptic poly(A) sites within proximal introns are suppressed by a mechanism called telescripting, based on U1 snRNP’s splicing independent role in full-length transcription [105]. Any role of deregulated telescripting in cancers is of yet unknown.
In addition to splicing induced changes to 3’UTRs, alternative polyadenylation also generates 3′-isoform diversity using polyadenylation sites within terminal exons, which are generally located within 3′UTRs. A multi-protein machinery that recognizes polyadenylation signals and neighboring conserved elements, carries out endonucleolytic cleavage of the pre-mRNA [106]. Alternative polyadenylation is the selection of different polyadenylation signals in a tissue-specific way and is globally regulated in response to cell proliferation and differentiation. Splicing independent alternative polyadenylation results with isoforms that have short or long 3′UTRs with identical coding sequences, as opposed to splicing coupled intronic polyadenylation (Fig. 3).

Polyadenylation sites. mRNA 3′UTRs may harbor more than one polyadenylation site (I, II, and III). Activation of a proximal polyadenylation site (II) results in shortening of the 3′UTR, resulting in loss of cis-elements. The resulting protein products from the short versus the long 3′UTR isoforms would be identical. However, in case of splicing coupled alternative polyadenylation (III), loss of amino acids and addition of few others coded by the exonized intron can change the protein C-terminus. Alternative terminal exon usage would also produce similar changes at the C-terminus of the protein
Global changes in 3′UTR lengths due to alternative polyadenylation were first demonstrated in activated T cells and cancer cells [107, 108]. In fact, 3′UTR shortening of growth-promoting mRNA transcripts was observed much earlier to correlate with cancer-related phenotypes. For example, CCND1 (cyclin D1) mRNA 3′UTR shortening prevents microRNA-mediated repression and leads to an additional increase in cyclin D1 protein levels in lymphoma cells and decreased overall survival of patients [109].
Recent 3′-end tailored transcriptomic methods and analysis tools reveal a global picture of alternative polyadenylation patterns that leads to 3′UTR shortening or lengthening cases in cancers [110, 111]. Moreover, individual investigation of 3′UTR isoforms provide functional relevance to increased proliferation, migration and metastasis of cancer cells. For example, 3′UTR shortening of insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) mRNA contributes more to the oncogenic transformation compared with the longer mRNA isoform [108]. Estradiol induces CDC6, a DNA replication regulator, mRNA upregulation. The shorter 3′UTR isoform of CDC6 is increased more than the longer 3′UTR isoform, enhancing S phase entry [112]. EGF (epidermal growth factor) induces similar 3′UTR shortening events in triple negative breast cancers with significant impact on relapse-free survival [113]. Alternative polyadenylation also allows NRAS and c-JUN to avoid posttranscriptional repression by PUMILIO in triple-negative breast tumors [114].
On the other hand, lengthening of 3′UTRs and reduced gene expression is seen in senescent cells. For example, RRAS2, a member of the Ras superfamily, is expressed as a longer 3′UTR isoform, leading to decreased expression in senescent cells [115]. Similarly, 3′UTR of HN1 (hematological and neurological expressed 1) is lengthened in senescent cells as opposed to cancer cells where expression of a shorter 3′UTR isoform is favored [116].
Overall, dynamic relationship between splicing and polyadenylation leads to an additional layer of transcriptome complexity at the 3′-ends of transcripts. While recent studies are accumulating to show functional relevance, it is obvious that the diversity of transcripts at the 3′-end is not fully known and appreciated. Unfortunately, we are not fully aware of these variants of even well-known cancer genes. For example, BRAF codes for a protein kinase functioning in the highly oncogenic RAS/RAF/MEK/ERK signaling pathway [117] and is mutated in about 50% of melanomas [118] as well as in other cancers, including colon cancers [119]. Despite being an attractive therapy target, information on BRAF isoforms are just beginning to surface. RNA-seq data of 4800 patients of different cancers including melanomas revealed the existence of BRAF mRNA isoforms, which differ in the last part of their coding sequences, as well as in the length and sequence of their 3′UTRs (BRAF: 76 nt; BRAF-X1 and BRAF-X2: up to 7 kb). Expression of these isoforms varies among cancers, contributing differently into BRAF protein levels [120]. Because 3′UTR sequences are different in these isoforms, regulation by microRNAs and/or RNA binding-proteins is very likely to be different in cancers [121]. Hence, differential regulation by trans-factors that bind to these BRAF isoforms may contribute to over activation of the BRAF kinase in a subset of patients who do not have DNA level alterations of BRAF.
3.2 Structural variations and 3′UTR diversity
In addition to splicing and polyadenylation, structural variations identified by long-read technologies can explain certain cases of altered 3′UTRs. Structural variations generally arise from chromosomal translocations that may partially or completely alter 3′UTR sequences. A breakpoint at 3′UTR of PDL1 is an intriguing example. PDL1 functions as a co-inhibitory molecule expressed on the surface of tumor cells to evade the immune system [122]. Increased expression of PDL1 aids immune evasion and is associated with worse prognosis in many tumors. Hence, immunotherapy targeting the PDL1/PD1 pathway is a promising strategy. Overexpression of PDL1 is a critical factor for blockade treatments; however, the current understanding of PDL1 deregulation in tumors is as of yet incomplete. Overexpression of PDL1 occurs through gene amplification and utilization of an ectopic promoter by translocation, as reported in a subset of Hodgkin, other B cell lymphomas and stomach adenocarcinomas [123]. Now, recent evidence is drawing attention to the 3′UTR of PDL1. The first evidence came from structural variation associated breakpoints that disrupt the 3′UTR of PDL1. These breakpoints included deletions, duplications, inversions and translocations, detected from whole-genome sequences of 49 adult T cell leukemia patients. Interestingly, all of the aberrant PDL1 transcripts were elevated; they retained the extracellular receptor-binding and transmembrane domains of PDL1 but not the 3′UTRs [124]. A group of 32 more cases (out of 10,210 samples) across 33 tumor panels from “The Cancer Genome Atlas” had aberrant 3′UTR of PDL1. This aberrant 3′UTR structure was also associated with elevated transcript expression levels in patients [124]. More recently, two groups simultaneously reported a secreted splice variant of PDL1 (secPDL1) which can negatively regulate T lymphocyte function [125, 126]. The secPDL1 has a different 3′UTR that harbors less AU-rich motifs than the full-length PDL1 mRNA. It remains to be seen whether these PDL1 mRNA 3′UTR isoforms have altered mRNA stabilities, and/or targeted differently by microRNAs, such as miR-17-5p [127], and miR-140 [128], and RNA-binding proteins. Based on these reports, 3’UTR cis-elements lost or gained through splicing or genomic alterations of PDL1 locus may have significant implications in immune checkpoint inhibitor treatments and possibly contribute to patient stratification efforts.
3.3 Mutations
A protein-focused view to identify mutations sometimes hinders discovery of less-obvious but potentially important mutations within non-coding parts of genes. For example, a frequent recurrent non-coding mutation in the 3′UTR of NOTCH1 (chr9: 139390152T > C) leads to a novel splicing event within the last exon of NOTCH1 in CLL patients. This isoform lacks the negative regulatory domain of NOTCH1, resulting in increased protein stability. Moreover, prognosis of patients with 3′UTR NOTCH1 mutations are similar to patients with coding mutations in NOTCH1, suggesting functionality [129]. A similar and frequent somatic mutation was reported in gastric cancer. A guanine-to-cytosine mutation in PDL1 3’ UTR leads to protein over-expression by disrupting binding of miR-570 [130]. Synonymous mutations can also alter 3′UTRs. For a long time, synonymous mutations within coding sequences were considered as silent and hence insignificant changes due to the degeneracy of codons. However, given that both DNA and RNA sequences are changed upon synonymous mutations, altered splicing patterns or misfolded RNA secondary structures are potentially significant outcomes of less obvious mutations.
3.4 RNA modifications
N6-methyladenosine (m6A) is a posttranscriptional RNA modification [131], most often found in proximity to stop codons, or within the 3′UTRs [132]. While the functional significance of these modifications is not completely understood, new evidence is beginning to suggest causality. In yeast, Rme1 (regulator of meiosis 1) is a DNA-binding protein that prevents meiosis in haploids. A functional m6A modification within the 3′UTR of Rme1 downregulates Rme1 mRNA levels to enable meiotic progression [133]. Considering the high homology in writer (METTL3/14, WTAP, RBM15/15B, and KIAA1429), reader (YTHDF1/2/3, IGF2BP1, and HNRNPA2B1), and eraser (FTO and ALKBH5) proteins, we are likely to discover functional consequences (e.g., altered RNA–protein interactions or RNA functions) due to m6A modifications within 3′UTRs. In cancer, functional evidence for RNA modifications within 3′UTRs is also starting to surface. For example, ALKBH5 demethylates an m6A residue on the 3′UTR of the NANOG mRNA, which encodes a pluripotency factor. Consequently, increased NANOG mRNA and protein expression is linked to breast cancer stem cell phenotype [134].
4 Conclusion
Recent omics approaches are paving the way to a more comprehensive understanding of cancer cells at genomic, transcriptomic and proteomic level. The tactic of searching for mutations in protein-coding regions through high throughput sequencing is the general basis of today’s personalized medicine efforts. However, whole exome sequencing and targeted gene sequencing are limited to the coding sequences and/or known clinically relevant genes. Therefore, discovery of other “not so obvious” mutations altering 3’UTRs is generally not the primary concern. In addition, conventional gene expression approaches are not tailored to reach 3’UTR ends and detect 3′-isoforms unless 3′-specific sequencing and analysis tools are used (reviewed in [135, 136]). This also hinders discovery of potentially important 3′-end isoforms.
Fortunately, some novel single-cell RNA sequencing (scRNA-seq) techniques (e.g., Drop-seq, Seq-Well, DroNC-seq, and SPLiT-seq) specifically sequence the 3′ end of transcripts, allowing exceptional depth to transcriptomic studies at a single cancer cell level. Accumulating evidence from these studies, strengthen the earlier findings, showing widespread expression of 3′-isoforms including the intronic polyadenylation events [137].
Recently, worm 3′ UTRome v2 resource became available in C. elegans, curated from high-quality 3′UTR data at ultradeep coverage from 1088 transcriptome datasets. This resource contains data for 23,084 3′UTR isoform variants corresponding to 14,788 protein-coding genes and is a novel resource to investigate alternative splicing, and polyadenylation patterns as well as trans-factors regulating these isoforms [138]. For humans, tissue and cancer-specific 3′UTRome is very much needed to identify “not so obvious” changes related to 3′UTRs, with substantial impact on protein function and cell phenotypes. Overall, discovery and characterization of cancer specific 3′UTR isoforms creates new possibilities for diagnosis and pharmacological intervention. Consequently, in the near future, findings in the RNA biology field are likely to provide truly novel insights into normal physiological events and disease mechanisms including cancer.
References
Nangalia, J., & Campbell, P. J. (2019). Genome sequencing during a patient’s journey through cancer. The New England Journal of Medicine, 381(22), 2145–2156. https://doi.org/10.1056/NEJMra1910138.
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., et al. (2005). The transcriptional landscape of the mammalian genome. Science, 309(5740), 1559–1563. https://doi.org/10.1126/science.1112014.
Huang, Q., Yan, J., & Agami, R. (2018). Long non-coding RNAs in metastasis. Cancer Metastasis Reviews, 37(1), 75–81. https://doi.org/10.1007/s10555-017-9713-x.
Anastasiadou, E., Jacob, L. S., & Slack, F. J. (2018). Non-coding RNA networks in cancer. Nature Reviews. Cancer, 18(1), 5–18. https://doi.org/10.1038/nrc.2017.99.
Yang, E., van Nimwegen, E., Zavolan, M., Rajewsky, N., Schroeder, M., Magnasco, M., & Darnell JE Jr. (2003). Decay rates of human mRNAs: Correlation with functional characteristics and sequence attributes. Genome Research, 13(8), 1863–1872. https://doi.org/10.1101/gr.1272403.
Sharova, L. V., Sharov, A. A., Nedorezov, T., Piao, Y., Shaik, N., & Ko, M. S. (2009). Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Research, 16(1), 45–58. https://doi.org/10.1093/dnares/dsn030.
Friedel, C. C., & Dölken, L. (2009). Metabolic tagging and purification of nascent RNA: Implications for transcriptomics. Molecular BioSystems, 5(11), 1271–1278. https://doi.org/10.1039/b911233b.
Eser, P., Demel, C., Maier, K. C., Schwalb, B., Pirkl, N., Martin, D. E., Cramer, P., & Tresch, A. (2014). Periodic mRNA synthesis and degradation co-operate during cell cycle gene expression. Molecular Systems Biology, 10, 717. https://doi.org/10.1002/msb.134886.
Gerstberger, S., Hafner, M., & Tuschl, T. (2014). A census of human RNA-binding proteins. Nature Reviews. Genetics, 15(12), 829–845. https://doi.org/10.1038/nrg3813.
Lunde, B. M., Moore, C., & Varani, G. (2007). RNA-binding proteins: Modular design for efficient function. Nature Reviews. Molecular Cell Biology, 8(6), 479–490. https://doi.org/10.1038/nrm2178.
Hentze, M. W., Castello, A., Schwarzl, T., & Preiss, T. (2018). A brave new world of RNA-binding proteins. Nature Reviews. Molecular Cell Biology, 19(5), 327–341. https://doi.org/10.1038/nrm.2017.130.
Lewis, B. P., Burge, C. B., & Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120(1), 15–20. https://doi.org/10.1016/j.cell.2004.12.035.
Paulsen, M. T., Veloso, A., Prasad, J., Bedi, K., Ljungman, E. A., Tsan, Y. C., Chang, C. W., Tarrier, B., Washburn, J. G., Lyons, R., Robinson, D. R., Kumar-Sinha, C., Wilson, T. E., & Ljungman, M. (2013). Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proceedings of the National Academy of Sciences of the United States of America, 110(6), 2240–2245. https://doi.org/10.1073/pnas.1219192110.
Cannell, I. G., Merrick, K. A., Morandell, S., Zhu, C. Q., Braun, C. J., Grant, R. A., Cameron, E. R., Tsao, M. S., Hemann, M. T., & Yaffe, M. B. (2015). A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell, 28, 623–637. https://doi.org/10.1016/j.ccell.2015.09.009.
Medford, R. M., Nguyen, H. T., & Nadal-Ginard, B. (1983). Transcriptional and cell cycle-mediated regulation of myosin heavy chain gene expression during muscle cell differentiation. The Journal of Biological Chemistry, 258(18), 11063–11073.
Hitti, E., Iakovleva, T., Brook, M., Deppenmeier, S., Gruber, A. D., Radzioch, D., Clark, A. R., Blackshear, P. J., Kotlyarov, A., & Gaestel, M. (2006). Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Molecular and Cellular Biology, 26(6), 2399–2407. https://doi.org/10.1128/MCB.26.6.2399-2407.2006.
Abdelmohsen, K., & Gorospe, M. (2010). Posttranscriptional regulation of cancer traits by HuR. Wiley Interdisciplinary Reviews RNA, 1(2), 214–229. https://doi.org/10.1002/wrna.4.
Sengupta, S., Jang, B. C., Wu, M. T., Paik, J. H., Furneaux, H., & Hla, T. (2003). The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. The Journal of Biological Chemistry, 278(27), 25227–25233. https://doi.org/10.1074/jbc.M301813200.
Torun, A., Enayat, S., Sheraj, I., Tunçer, S., Ülgen, D. H., & Banerjee, S. (2019). Butyrate mediated regulation of RNA binding proteins in the post-transcriptional regulation of inflammatory gene expression. Cellular Signalling, 64, 109410. https://doi.org/10.1016/j.cellsig.2019.109410.
Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell, 88(3), 323–331. https://doi.org/10.1016/s0092-8674(00)81871-1.
Hollstein, M., Sidransky, D., Vogelstein, B., & Harris, C. C. (1991). p53 mutations in human cancers. Science, 253(5015), 49–53. https://doi.org/10.1126/science.1905840.
Soussi, T., & Wiman, K. G. (2007). Shaping genetic alterations in human cancer: The p53 mutation paradigm. Cancer Cell, 12(4), 303–312. https://doi.org/10.1016/j.ccr.2007.10.001.
Morandell, S., Reinhardt, H. C., Cannell, I. G., Kim, J. S., Ruf, D. M., Mitra, T., Couvillon, A. D., Jacks, T., & Yaffe, M. B. (2013). A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Reports, 5(4), 868–877. https://doi.org/10.1016/j.celrep.2013.10.025.
Reinhardt, H. C., Hasskamp, P., Schmedding, I., Morandell, S., van Vugt, M. A., Wang, X., et al. (2010). DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Molecular Cell, 40(1), 34–49. https://doi.org/10.1016/j.molcel.2010.09.018.
Rosenstierne, M. W., Vinther, J., Mittler, G., Larsen, L., Mann, M., & Norrild, B. (2008). Conserved CPEs in the p53 3′ untranslated region influence mRNA stability and protein synthesis. Anticancer Research, 28(5A), 2553–2559.
Vilborg, A., Glahder, J. A., Wilhelm, M. T., Bersani, C., Corcoran, M., Mahmoudi, S., Rosenstierne, M., Grandér, D., Farnebo, M., Norrild, B., & Wiman, K. G. (2009). The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proceedings of the National Academy of Sciences of the United States of America, 106(37), 15756–15761. https://doi.org/10.1073/pnas.0900862106.
Mazan-Mamczarz, K., Galbán, S., López de Silanes, I., Martindale, J. L., Atasoy, U., Keene, J. D., et al. (2003). RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proceedings of the National Academy of Sciences of the United States of America, 100(14), 8354–8359. https://doi.org/10.1073/pnas.1432104100.
Ahuja, D., Goyal, A., & Ray, P. S. (2016). Interplay between RNA-binding protein HuR and microRNA-125b regulates p53 mRNA translation in response to genotoxic stress. RNA Biology, 13(11), 1152–1165. https://doi.org/10.1080/15476286.2016.1229734.
Liu, L., Ouyang, M., Rao, J. N., Zou, T., Xiao, L., Chung, H. K., Wu, J., Donahue, J. M., Gorospe, M., & Wang, J. Y. (2015). Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Molecular Biology of the Cell, 26(10), 1797–1810. https://doi.org/10.1091/mbc.E14-11-1500.
Mukherjee, N., Jacobs, N. C., Hafner, M., Kennington, E. A., Nusbaum, J. D., Tuschl, T., Blackshear, P. J., & Ohler, U. (2014). Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biology, 15(1), R12. https://doi.org/10.1186/gb-2014-15-1-r12.
Kuwano, Y., Nishida, K., Kajita, K., Satake, Y., Akaike, Y., Fujita, K., Kano, S., Masuda, K., & Rokutan, K. (2015). Transformer 2β and miR-204 regulate apoptosis through competitive binding to 3′ UTR of BCL2 mRNA. Cell Death and Differentiation, 22(5), 815–825. https://doi.org/10.1038/cdd.2014.176.
Hennig, J., & Sattler, M. (2015). Deciphering the protein-RNA recognition code: Combining large-scale quantitative methods with structural biology. Bioessays, 37(8), 899–908. https://doi.org/10.1002/bies.201500033.
Cho, S. J., Zhang, J., & Chen, X. (2010). RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Research, 38(7), 2256–2267. https://doi.org/10.1093/nar/gkp1229.
Torres-Fernández, L. A., Jux, B., Bille, M., Port, Y., Schneider, K., Geyer, M., Mayer, G., & Kolanus, W. (2019). The mRNA repressor TRIM71 cooperates with nonsense-mediated decay factors to destabilize the mRNA of CDKN1A/p21. Nucleic Acids Research, 47(22), 11861–11879. https://doi.org/10.1093/nar/gkz1057.
Kurosaki, T., Popp, M. W., & Maquat, L. E. (2019). Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nature Reviews. Molecular Cell Biology, 20(7), 406–420. https://doi.org/10.1038/s41580-019-0126-2.
Park, J., Seo, J. W., Ahn, N., Park, S., Hwang, J., & Nam, J. W. (2019). UPF1/SMG7-dependent microRNA-mediated gene regulation. Nature Communications, 10(1), 4181. https://doi.org/10.1038/s41467-019-12123-7.
Kim, Y. K., & Maquat, L. E. (2019). UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA, 25(4), 407–422. https://doi.org/10.1261/rna.070136.118.
Bartel, D. P. (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell, 116(2), 281–297.
Erson, A. E., & Petty, E. M. (2008). MicroRNAs in development and disease. Clinical Genetics, 74(4), 296–306. https://doi.org/10.1111/j.1399-0004.2008.01076.x.
Tuna, M., Machado, A. S., & Calin, G. A. (2016). Genetic and epigenetic alterations of microRNAs and implications for human cancers and other diseases. Genes, Chromosomes & Cancer, 55(3), 193–214. https://doi.org/10.1002/gcc.22332.
Gebert, L. F. R., & MacRae, I. J. (2019). Regulation of microRNA function in animals. Nature Reviews. Molecular Cell Biology, 20(1), 21–37. https://doi.org/10.1038/s41580-018-0045-7.
Peng, Y., & Croce, C. M. (2016). The role of MicroRNAs in human cancer. Signal Transduction and Targeted Therapy, 1, 15004. https://doi.org/10.1038/sigtrans.2015.4.
Teige, I., Mårtensson, L., & Frendéus, B. L. (2019). Targeting the antibody checkpoints to enhance cancer immunotherapy-focus on FcγRIIB. Frontiers in Immunology, 10, 481. https://doi.org/10.3389/fimmu.2019.00481.
Hermeking, H. (2007). p53 enters the microRNA world. Cancer Cell, 12(5), 414–418.
Zhang, L., Liao, Y., & Tang, L. (2019). MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. Journal of Experimental & Clinical Cancer Research, 38(1), 53. https://doi.org/10.1186/s13046-019-1059-5.
Slabáková, E., Culig, Z., Remšík, J., & Souček, K. (2017). Alternative mechanisms of miR-34a regulation in cancer. Cell Death & Disease, 8(10), e3100. https://doi.org/10.1038/cddis.2017.495.
Chen, P. C., Yu, C. C., Huang, W. Y., Huang, W. H., Chuang, Y. M., Lin, R. I., et al. (2019). c-MYC acts as a competing endogenous RNA to sponge. Cancers (Basel), 11(10). https://doi.org/10.3390/cancers11101457.
Szostak, E., & Gebauer, F. (2013). Translational control by 3'-UTR-binding proteins. Briefings in Functional Genomics, 12(1), 58–65. https://doi.org/10.1093/bfgp/els056.
Uniacke, J., Holterman, C. E., Lachance, G., Franovic, A., Jacob, M. D., Fabian, M. R., Payette, J., Holcik, M., Pause, A., & Lee, S. (2012). An oxygen-regulated switch in the protein synthesis machinery. Nature, 486(7401), 126–129. https://doi.org/10.1038/nature11055.
Szostak, E., García-Beyaert, M., Guitart, T., Graindorge, A., Coll, O., & Gebauer, F. (2018). Hrp48 and eIF3d contribute to msl-2 mRNA translational repression. Nucleic Acids Research, 46(8), 4099–4113. https://doi.org/10.1093/nar/gky246.
Arake de Tacca, L. M., Pulos-Holmes, M. C., Floor, S. N., & Cate, J. H. D. (2019). PTBP1 mRNA isoforms and regulation of their translation. RNA, 25(10), 1324–1336. https://doi.org/10.1261/rna.070193.118.
Mazumder, B., Sampath, P., & Fox, P. L. (2005). Regulation of macrophage ceruloplasmin gene expression: One paradigm of 3'-UTR-mediated translational control. Molecules and Cells, 20(2), 167–172.
Ray, P. S., & Fox, P. L. (2007). A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. The EMBO Journal, 26(14), 3360–3372. https://doi.org/10.1038/sj.emboj.7601774.
Arif, A., Yao, P., Terenzi, F., Jia, J., Ray, P. S., & Fox, P. L. (2018). The GAIT translational control system. Wiley Interdisciplinary Reviews RNA, 9(2), e1441. https://doi.org/10.1002/wrna.1441.
Ostareck, D. H., Ostareck-Lederer, A., Wilm, M., Thiele, B. J., Mann, M., & Hentze, M. W. (1997). mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell, 89(4), 597–606. https://doi.org/10.1016/s0092-8674(00)80241-x.
Friend, K., Campbell, Z. T., Cooke, A., Kroll-Conner, P., Wickens, M. P., & Kimble, J. (2012). A conserved PUF-Ago-eEF1A complex attenuates translation elongation. Nature Structural & Molecular Biology, 19(2), 176–183. https://doi.org/10.1038/nsmb.2214.
Waerner, T., Alacakaptan, M., Tamir, I., Oberauer, R., Gal, A., Brabletz, T., Schreiber, M., Jechlinger, M., & Beug, H. (2006). ILEI: A cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell, 10(3), 227–239. https://doi.org/10.1016/j.ccr.2006.07.020.
Chaudhury, A., Hussey, G. S., Ray, P. S., Jin, G., Fox, P. L., & Howe, P. H. (2010). TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nature Cell Biology, 12(3), 286–293. https://doi.org/10.1038/ncb2029.
Hussey, G. S., Chaudhury, A., Dawson, A. E., Lindner, D. J., Knudsen, C. R., Wilce, M. C., et al. (2011). Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Molecular Cell, 41(4), 419–431. https://doi.org/10.1016/j.molcel.2011.02.003.
Vicario, A., Colliva, A., Ratti, A., Davidovic, L., Baj, G., Gricman, Ł., Colombrita, C., Pallavicini, A., Jones, K. R., Bardoni, B., & Tongiorgi, E. (2015). Dendritic targeting of short and long 3' UTR BDNF mRNA is regulated by BDNF or NT-3 and distinct sets of RNA-binding proteins. Frontiers in Molecular Neuroscience, 8, 62. https://doi.org/10.3389/fnmol.2015.00062.
Melendez, J., Grogg, M., & Zheng, Y. (2011). Signaling role of Cdc42 in regulating mammalian physiology. The Journal of Biological Chemistry, 286(4), 2375–2381. https://doi.org/10.1074/jbc.R110.200329.
Ciolli Mattioli, C., Rom, A., Franke, V., Imami, K., Arrey, G., Terne, M., Woehler, A., Akalin, A., Ulitsky, I., & Chekulaeva, M. (2019). Alternative 3' UTRs direct localization of functionally diverse protein isoforms in neuronal compartments. Nucleic Acids Research, 47(5), 2560–2573. https://doi.org/10.1093/nar/gky1270.
Katz, Z. B., Wells, A. L., Park, H. Y., Wu, B., Shenoy, S. M., & Singer, R. H. (2012). β-Actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration. Genes & Development, 26(17), 1885–1890. https://doi.org/10.1101/gad.190413.112.
Rodriguez, A. J., Shenoy, S. M., Singer, R. H., & Condeelis, J. (2006). Visualization of mRNA translation in living cells. The Journal of Cell Biology, 175(1), 67–76. https://doi.org/10.1083/jcb.200512137.
Gutierrez, N., Eromobor, I., Petrie, R. J., Vedula, P., Cruz, L., & Rodriguez, A. J. (2014). The β-actin mRNA zipcode regulates epithelial adherens junction assembly but not maintenance. RNA, 20(5), 689–701. https://doi.org/10.1261/rna.043208.113.
Kislauskis, E. H., Zhu, X., & Singer, R. H. (1994). Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. The Journal of Cell Biology, 127(2), 441–451. https://doi.org/10.1083/jcb.127.2.441.
Hüttelmaier, S., Zenklusen, D., Lederer, M., Dictenberg, J., Lorenz, M., Meng, X., Bassell, G. J., Condeelis, J., & Singer, R. H. (2005). Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature, 438(7067), 512–515. https://doi.org/10.1038/nature04115.
Gu, W., Pan, F., & Singer, R. H. (2009). Blocking beta-catenin binding to the ZBP1 promoter represses ZBP1 expression, leading to increased proliferation and migration of metastatic breast-cancer cells. Journal of Cell Science, 122(Pt 11), 1895–1905. https://doi.org/10.1242/jcs.045278.
Gu, W., Katz, Z., Wu, B., Park, H. Y., Li, D., Lin, S., Wells, A. L., & Singer, R. H. (2012). Regulation of local expression of cell adhesion and motility-related mRNAs in breast cancer cells by IMP1/ZBP1. Journal of Cell Science, 125(Pt 1), 81–91. https://doi.org/10.1242/jcs.086132.
Nwokafor, C. U., Sellers, R. S., & Singer, R. H. (2016). IMP1, an mRNA binding protein that reduces the metastatic potential of breast cancer in a mouse model. Oncotarget, 7(45), 72662–72671. https://doi.org/10.18632/oncotarget.12083.
Niedner, A., Edelmann, F. T., & Niessing, D. (2014). Of social molecules: The interactive assembly of ASH1 mRNA-transport complexes in yeast. RNA Biology, 11(8), 998–1009. https://doi.org/10.4161/rna.29946.
Martin, K. C., & Ephrussi, A. (2009). mRNA localization: Gene expression in the spatial dimension. Cell, 136(4), 719–730. https://doi.org/10.1016/j.cell.2009.01.044.
Mili, S., Moissoglu, K., & Macara, I. G. (2008). Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature, 453(7191), 115–119. https://doi.org/10.1038/nature06888.
Wong, H. H., Lin, J. Q., Ströhl, F., Roque, C. G., Cioni, J. M., Cagnetta, R., et al. (2017). RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron, 95(4), 852–868.e8. https://doi.org/10.1016/j.neuron.2017.07.016.
Preitner, N., Quan, J., Nowakowski, D. W., Hancock, M. L., Shi, J., Tcherkezian, J., Young-Pearse, T. L., & Flanagan, J. G. (2014). APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell, 158(2), 368–382. https://doi.org/10.1016/j.cell.2014.05.042.
Bergsten, S. E., & Gavis, E. R. (1999). Role for mRNA localization in translational activation but not spatial restriction of nanos RNA. Development, 126(4), 659–669.
Zaessinger, S., Busseau, I., & Simonelig, M. (2006). Oskar allows nanos mRNA translation in Drosophila embryos by preventing its deadenylation by Smaug/CCR4. Development, 133(22), 4573–4583. https://doi.org/10.1242/dev.02649.
Berkovits, B. D., & Mayr, C. (2015). Alternative 3' UTRs act as scaffolds to regulate membrane protein localization. Nature, 522(7556), 363–367. https://doi.org/10.1038/nature14321.
Jaiswal, S., Jamieson, C. H., Pang, W. W., Park, C. Y., Chao, M. P., Majeti, R., et al. (2009). CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell, 138(2), 271–285. https://doi.org/10.1016/j.cell.2009.05.046.
Ma, W., & Mayr, C. (2018). A membraneless organelle associated with the endoplasmic reticulum enables 3'UTR-mediated protein-protein interactions. Cell, 175(6), 1492–1506.e19. https://doi.org/10.1016/j.cell.2018.10.007.
Grabowski, P. J., Seiler, S. R., & Sharp, P. A. (1985). A multicomponent complex is involved in the splicing of messenger RNA precursors. Cell, 42(1), 345–353. https://doi.org/10.1016/s0092-8674(85)80130-6.
Berget, S. M., Moore, C., & Sharp, P. A. (1977). Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proceedings of the National Academy of Sciences of the United States of America, 74(8), 3171–3175. https://doi.org/10.1073/pnas.74.8.3171.
Chow, L. T., Gelinas, R. E., Broker, T. R., & Roberts, R. J. (1977). An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell, 12(1), 1–8. https://doi.org/10.1016/0092-8674(77)90180-5.
Fica, S. M., & Nagai, K. (2017). Cryo-electron microscopy snapshots of the spliceosome: Structural insights into a dynamic ribonucleoprotein machine. Nature Structural & Molecular Biology, 24(10), 791–799. https://doi.org/10.1038/nsmb.3463.
Climente-González, H., Porta-Pardo, E., Godzik, A., & Eyras, E. (2017). The functional impact of alternative splicing in Cancer. Cell Reports, 20(9), 2215–2226. https://doi.org/10.1016/j.celrep.2017.08.012.
Hoyos, L. E., & Abdel-Wahab, O. (2018). Cancer-specific splicing changes and the potential for splicing-derived neoantigens. Cancer Cell, 34(2), 181–183. https://doi.org/10.1016/j.ccell.2018.07.008.
Anczuków, O., & Krainer, A. R. (2016). Splicing-factor alterations in cancers. RNA, 22(9), 1285–1301. https://doi.org/10.1261/rna.057919.116.
Anczuków, O., Rosenberg, A. Z., Akerman, M., Das, S., Zhan, L., Karni, R., Muthuswamy, S. K., & Krainer, A. R. (2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nature Structural & Molecular Biology, 19(2), 220–228. https://doi.org/10.1038/nsmb.2207.
Karni, R., de Stanchina, E., Lowe, S. W., Sinha, R., Mu, D., & Krainer, A. R. (2007). The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nature Structural & Molecular Biology, 14(3), 185–193. https://doi.org/10.1038/nsmb1209.
Ghigna, C., Giordano, S., Shen, H., Benvenuto, F., Castiglioni, F., Comoglio, P. M., Green, M. R., Riva, S., & Biamonti, G. (2005). Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Molecular Cell, 20(6), 881–890. https://doi.org/10.1016/j.molcel.2005.10.026.
Ge, K., DuHadaway, J., Du, W., Herlyn, M., Rodeck, U., & Prendergast, G. C. (1999). Mechanism for elimination of a tumor suppressor: Aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proceedings of the National Academy of Sciences of the United States of America, 96(17), 9689–9694. https://doi.org/10.1073/pnas.96.17.9689.
Scheper, G. C., Parra, J. L., Wilson, M., Van Kollenburg, B., Vertegaal, A. C., Han, Z. G., et al. (2003). The N and C termini of the splice variants of the human mitogen-activated protein kinase-interacting kinase Mnk2 determine activity and localization. Molecular and Cellular Biology, 23(16), 5692–5705. https://doi.org/10.1128/mcb.23.16.5692-5705.2003.
Chen, L., Luo, C., Shen, L., Liu, Y., Wang, Q., Zhang, C., Guo, R., Zhang, Y., Xie, Z., Wei, N., Wu, W., Han, J., & Feng, Y. (2017). SRSF1 prevents DNA damage and promotes tumorigenesis through regulation of DBF4B pre-mRNA splicing. Cell Reports, 21(12), 3406–3413. https://doi.org/10.1016/j.celrep.2017.11.091.
Dvinge, H., Kim, E., Abdel-Wahab, O., & Bradley, R. K. (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nature Reviews. Cancer, 16(7), 413–430. https://doi.org/10.1038/nrc.2016.51.
Urbanski, L. M., Leclair, N., & Anczuków, O. (2018). Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdisciplinary Reviews RNA, 9(4), e1476. https://doi.org/10.1002/wrna.1476.
Obeng, E. A., Stewart, C., & Abdel-Wahab, O. (2019). Altered RNA processing in cancer pathogenesis and therapy. Cancer Discovery, 9(11), 1493–1510. https://doi.org/10.1158/2159-8290.CD-19-0399.
Dutertre, M., Chakrama, F. Z., Combe, E., Desmet, F. O., Mortada, H., Polay Espinoza, M., Gratadou, L., & Auboeuf, D. (2014). A recently evolved class of alternative 3′-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nature Communications, 5, 3395. https://doi.org/10.1038/ncomms4395.
Vorlová, S., Rocco, G., Lefave, C. V., Jodelka, F. M., Hess, K., Hastings, M. L., et al. (2011). Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Molecular Cell, 43(6), 927–939. https://doi.org/10.1016/j.molcel.2011.08.009.
Lee, S. H., Singh, I., Tisdale, S., Abdel-Wahab, O., Leslie, C. S., & Mayr, C. (2018). Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature, 561(7721), 127–131. https://doi.org/10.1038/s41586-018-0465-8.
Graber, J. H., Nazeer, F. I., Yeh, P. C., Kuehner, J. N., Borikar, S., Hoskinson, D., & Moore, C. L. (2013). DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Research, 23(10), 1690–1703. https://doi.org/10.1101/gr.144964.112.
Davidson, L., Muniz, L., & West, S. (2014). 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes & Development, 28(4), 342–356. https://doi.org/10.1101/gad.231274.113.
Dubbury, S. J., Boutz, P. L., & Sharp, P. A. (2018). CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature, 564(7734), 141–145. https://doi.org/10.1038/s41586-018-0758-y.
Quereda, V., Bayle, S., Vena, F., Frydman, S. M., Monastyrskyi, A., Roush, W. R., et al. (2019). Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell, 36(5), 545–558.e7. https://doi.org/10.1016/j.ccell.2019.09.004.
Tian, B., Hu, J., Zhang, H., & Lutz, C. S. (2005). A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Research, 33(1), 201–212. https://doi.org/10.1093/nar/gki158.
Venters, C. C., Oh, J. M., Di, C., So, B. R., & Dreyfuss, G. (2019). U1 snRNP telescripting: Suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harbor Perspectives in Biology, 11(2). https://doi.org/10.1101/cshperspect.a032235.
Tian, B., & Manley, J. L. (2017). Alternative polyadenylation of mRNA precursors. Nature Reviews. Molecular Cell Biology, 18(1), 18–30. https://doi.org/10.1038/nrm.2016.116.
Sandberg, R., Neilson, J. R., Sarma, A., Sharp, P. A., & Burge, C. B. (2008). Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science, 320(5883), 1643–1647. https://doi.org/10.1126/science.1155390.
Mayr, C., & Bartel, D. P. (2009). Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell, 138(4), 673–684. https://doi.org/10.1016/j.cell.2009.06.016.
Rosenwald, A., Wright, G., Wiestner, A., Chan, W. C., Connors, J. M., Campo, E., Gascoyne, R. D., Grogan, T. M., Muller-Hermelink, H. K., Smeland, E. B., Chiorazzi, M., Giltnane, J. M., Hurt, E. M., Zhao, H., Averett, L., Henrickson, S., Yang, L., Powell, J., Wilson, W. H., Jaffe, E. S., Simon, R., Klausner, R. D., Montserrat, E., Bosch, F., Greiner, T. C., Weisenburger, D. D., Sanger, W. G., Dave, B. J., Lynch, J. C., Vose, J., Armitage, J. O., Fisher, R. I., Miller, T. P., LeBlanc, M., Ott, G., Kvaloy, S., Holte, H., Delabie, J., & Staudt, L. M. (2003). The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell, 3(2), 185–197.
Xia, Z., Donehower, L. A., Cooper, T. A., Neilson, J. R., Wheeler, D. A., Wagner, E. J., & Li, W. (2014). Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types. Nature Communications, 5, 5274. https://doi.org/10.1038/ncomms6274.
Begik, O., Oyken, M., Cinkilli Alican, T., Can, T., & Erson-Bensan, A. E. (2017). Alternative polyadenylation patterns for novel gene discovery and classification in Cancer. Neoplasia, 19(7), 574–582. https://doi.org/10.1016/j.neo.2017.04.008.
Akman, B. H., Can, T., & Erson-Bensan, A. E. (2012). Estrogen-induced upregulation and 3'-UTR shortening of CDC6. Nucleic Acids Research, 40(21), 10679–10688. https://doi.org/10.1093/nar/gks855.
Akman, H. B., Oyken, M., Tuncer, T., Can, T., & Erson-Bensan, A. E. (2015). 3 ' UTR shortening and EGF signaling: Implications for breast cancer. Human Molecular Genetics, 24(24), 6910–6920. https://doi.org/10.1093/hmg/ddv391.
Miles, W. O., Lembo, A., Volorio, A., Brachtel, E., Tian, B., Sgroi, D., Provero, P., & Dyson, N. (2016). Alternative polyadenylation in triple-negative breast tumors allows NRAS and c-JUN to bypass PUMILIO posttranscriptional regulation. Cancer Research, 76(24), 7231–7241. https://doi.org/10.1158/0008-5472.CAN-16-0844.
Chen, M., Lyu, G., Han, M., Nie, H., Shen, T., Chen, W., Niu, Y., Song, Y., Li, X., Li, H., Chen, X., Wang, Z., Xia, Z., Li, W., Tian, X. L., Ding, C., Gu, J., Zheng, Y., Liu, X., Hu, J., Wei, G., Tao, W., & Ni, T. (2018). 3' UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Research, 28, 285–294. https://doi.org/10.1101/gr.224451.117.
Jia, Q., Nie, H., Yu, P., Xie, B., Wang, C., Yang, F., et al. (2019). HNRNPA1-mediated 3' UTR length changes of HN1 contributes to cancer- and senescence-associated phenotypes. Aging (Albany NY), 11(13), 4407–4437. https://doi.org/10.18632/aging.102060.
Matallanas, D., Birtwistle, M., Romano, D., Zebisch, A., Rauch, J., von Kriegsheim, A., & Kolch, W. (2011). Raf family kinases: Old dogs have learned new tricks. Genes & Cancer, 2(3), 232–260. https://doi.org/10.1177/1947601911407323.
Cantwell-Dorris, E. R., O'Leary, J. J., & Sheils, O. M. (2011). BRAFV600E: Implications for carcinogenesis and molecular therapy. Molecular Cancer Therapeutics, 10(3), 385–394. https://doi.org/10.1158/1535-7163.MCT-10-0799.
Kim, A., & Cohen, M. S. (2016). The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opinion on Drug Discovery, 11(9), 907–916. https://doi.org/10.1080/17460441.2016.1201057.
Marranci, A., Jiang, Z., Vitiello, M., Guzzolino, E., Comelli, L., Sarti, S., Lubrano, S., Franchin, C., Echevarría-Vargas, I., Tuccoli, A., Mercatanti, A., Evangelista, M., Sportoletti, P., Cozza, G., Luzi, E., Capobianco, E., Villanueva, J., Arrigoni, G., Signore, G., Rocchiccioli, S., Pitto, L., Tsinoremas, N., & Poliseno, L. (2017). The landscape of BRAF transcript and protein variants in human cancer. Molecular Cancer, 16(1), 85. https://doi.org/10.1186/s12943-017-0645-4.
Marranci, A., Tuccoli, A., Vitiello, M., Mercoledi, E., Sarti, S., Lubrano, S., Evangelista, M., Fogli, A., Valdes, C., Russo, F., Monte, M. D., Caligo, M. A., Pellegrini, M., Capobianco, E., Tsinoremas, N., & Poliseno, L. (2015). Identification of BRAF 3'UTR isoforms in melanoma. The Journal of Investigative Dermatology, 135(6), 1694–1697. https://doi.org/10.1038/jid.2015.47.
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews. Cancer, 12(4), 252–264. https://doi.org/10.1038/nrc3239.
Shi, Y. (2018). Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunology, Immunotherapy, 67(10), 1481–1489. https://doi.org/10.1007/s00262-018-2226-9.
Kataoka, K., Shiraishi, Y., Takeda, Y., Sakata, S., Matsumoto, M., Nagano, S., Maeda, T., Nagata, Y., Kitanaka, A., Mizuno, S., Tanaka, H., Chiba, K., Ito, S., Watatani, Y., Kakiuchi, N., Suzuki, H., Yoshizato, T., Yoshida, K., Sanada, M., Itonaga, H., Imaizumi, Y., Totoki, Y., Munakata, W., Nakamura, H., Hama, N., Shide, K., Kubuki, Y., Hidaka, T., Kameda, T., Masuda, K., Minato, N., Kashiwase, K., Izutsu, K., Takaori-Kondo, A., Miyazaki, Y., Takahashi, S., Shibata, T., Kawamoto, H., Akatsuka, Y., Shimoda, K., Takeuchi, K., Seya, T., Miyano, S., & Ogawa, S. (2016). Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature, 534(7607), 402–406. https://doi.org/10.1038/nature18294.
Hassounah, N. B., Malladi, V. S., Huang, Y., Freeman, S. S., Beauchamp, E. M., Koyama, S., Souders, N., Martin, S., Dranoff, G., Wong, K. K., Pedamallu, C. S., Hammerman, P. S., & Akbay, E. A. (2019). Identification and characterization of an alternative cancer-derived PD-L1 splice variant. Cancer Immunology, Immunotherapy, 68(3), 407–420. https://doi.org/10.1007/s00262-018-2284-z.
Mahoney, K. M., Shukla, S. A., Patsoukis, N., Chaudhri, A., Browne, E. P., Arazi, A., Eisenhaure, T. M., Pendergraft III, W. F., Hua, P., Pham, H. C., Bu, X., Zhu, B., Hacohen, N., Fritsch, E. F., Boussiotis, V. A., Wu, C. J., & Freeman, G. J. (2019). A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunology, Immunotherapy, 68(3), 421–432. https://doi.org/10.1007/s00262-018-2282-1.
Audrito, V., Serra, S., Stingi, A., Orso, F., Gaudino, F., Bologna, C., et al. (2017). PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget, 8(9), 15894–15911. https://doi.org/10.18632/oncotarget.15213.
Xie, W. B., Liang, L. H., Wu, K. G., Wang, L. X., He, X., Song, C., Wang, Y. Q., & Li, Y. H. (2018). MiR-140 expression regulates cell proliferation and targets PD-L1 in NSCLC. Cellular Physiology and Biochemistry, 46(2), 654–663. https://doi.org/10.1159/000488634.
Puente, X. S., Beà, S., Valdés-Mas, R., Villamor, N., Gutiérrez-Abril, J., Martín-Subero, J. I., Munar, M., Rubio-Pérez, C., Jares, P., Aymerich, M., Baumann, T., Beekman, R., Belver, L., Carrio, A., Castellano, G., Clot, G., Colado, E., Colomer, D., Costa, D., Delgado, J., Enjuanes, A., Estivill, X., Ferrando, A. A., Gelpí, J. L., González, B., González, S., González, M., Gut, M., Hernández-Rivas, J. M., López-Guerra, M., Martín-García, D., Navarro, A., Nicolás, P., Orozco, M., Payer, Á. R., Pinyol, M., Pisano, D. G., Puente, D. A., Queirós, A. C., Quesada, V., Romeo-Casabona, C. M., Royo, C., Royo, R., Rozman, M., Russiñol, N., Salaverría, I., Stamatopoulos, K., Stunnenberg, H. G., Tamborero, D., Terol, M. J., Valencia, A., López-Bigas, N., Torrents, D., Gut, I., López-Guillermo, A., López-Otín, C., & Campo, E. (2015). Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature, 526(7574), 519–524. https://doi.org/10.1038/nature14666.
Wang, W., Sun, J., Li, F., Li, R., Gu, Y., Liu, C., Yang, P., Zhu, M., Chen, L., Tian, W., Zhou, H., Mao, Y., Zhang, L., Jiang, J., Wu, C., Hua, D., Chen, W., Lu, B., Ju, J., & Zhang, X. (2012). A frequent somatic mutation in CD274 3'-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Human Mutation, 33(3), 480–484. https://doi.org/10.1002/humu.22014.
Desrosiers, R., Friderici, K., & Rottman, F. (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proceedings of the National Academy of Sciences of the United States of America, 71(10), 3971–3975. https://doi.org/10.1073/pnas.71.10.3971.
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., Cesarkas, K., Jacob-Hirsch, J., Amariglio, N., Kupiec, M., Sorek, R., & Rechavi, G. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485(7397), 201–206. https://doi.org/10.1038/nature11112.
Bushkin, G. G., Pincus, D., Morgan, J. T., Richardson, K., Lewis, C., Chan, S. H., Bartel, D. P., & Fink, G. R. (2019). m6A modification of a 3' UTR site reduces RME1 mRNA levels to promote meiosis. Nature Communications, 10(1), 3414. https://doi.org/10.1038/s41467-019-11232-7.
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., He, X., & Semenza, G. L. (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proceedings of the National Academy of Sciences of the United States of America, 113(14), E2047–E2056. https://doi.org/10.1073/pnas.1602883113.
Erson-Bensan, A. E., & Can, T. (2016). Alternative Polyadenylation: Another foe in cancer. Molecular Cancer Research. https://doi.org/10.1158/1541-7786.MCR-15-0489.
Gruber, A. J., & Zavolan, M. (2019). Alternative cleavage and polyadenylation in health and disease. Nature Reviews. Genetics, 20(10), 599–614. https://doi.org/10.1038/s41576-019-0145-z.
Shulman, E. D., & Elkon, R. (2019). Cell-type-specific analysis of alternative polyadenylation using single-cell transcriptomics data. Nucleic Acids Research, 47(19), 10027–10039. https://doi.org/10.1093/nar/gkz781.
Steber, H. S., Gallante, C., O'Brien, S., Chiu, P. L., & Mangone, M. (2019). The C. elegans 3' UTRome v2 resource for studying mRNA cleavage and polyadenylation, 3'-UTR biology, and miRNA targeting. Genome Research, 29(12), 2104–2116. https://doi.org/10.1101/gr.254839.119.
Acknowledgments
The author sincerely apologizes to colleagues whose important work could not be cited in this review article because of space limitations, and thanks the laboratory members for critical reading of the manuscript. Funding for our laboratory’s work on 3′UTRs was from TUBITAK and METU.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Erson-Bensan, A.E. RNA-biology ruling cancer progression? Focus on 3′UTRs and splicing. Cancer Metastasis Rev 39, 887–901 (2020). https://doi.org/10.1007/s10555-020-09884-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09884-9