
Abstract
Over the last two decades, a novel subgroup of serine proteases, the cell surface–anchored serine proteases, has emerged as an important component of the human degradome, and several members have garnered significant attention for their roles in cancer progression and metastasis. A large body of literature describes that cell surface–anchored serine proteases are deregulated in cancer and that they contribute to both tumor formation and metastasis through diverse molecular mechanisms. The loss of precise regulation of cell surface–anchored serine protease expression and/or catalytic activity may be contributing to the etiology of several cancer types. There is therefore a strong impetus to understand the events that lead to deregulation at the gene and protein levels, how these precipitate in various stages of tumorigenesis, and whether targeting of selected proteases can lead to novel cancer intervention strategies. This review summarizes current knowledge about cell surface–anchored serine proteases and their role in cancer based on biochemical characterization, cell culture–based studies, expression studies, and in vivo experiments. Efforts to develop inhibitors to target cell surface–anchored serine proteases in cancer therapy will also be summarized.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The class of serine proteases contains 175 predicted members in humans of which the vast majority are secreted proteases [1]. A subgroup of serine proteases is directly anchored to plasma membranes. These cell surface–anchored serine proteases are tethered to the plasma membrane either via a carboxy-terminal transmembrane domain (type I), an amino-terminal proximal signal anchor that functions as a transmembrane domain (type II), or a carboxy-terminal hydrophobic region that functions as a signal for membrane attachment via a glycosyl-phosphatidylinositol linkage (GPI-anchored). The type I transmembrane tryptase γ1 (also known as PRSS31, transmembrane tryptase, and transmembrane protease γ1) is expressed in cells of hematopoietic origin and has been studied most extensively in mast cells [2]. To our knowledge, no studies on this protease in cancer have been published, and it will therefore not be discussed further. The catalytic domain of type II transmembrane serine proteases belongs to the S1 family of serine proteases which includes the prototypic chymotrypsin and trypsin [3] . One exception is fibroblast activation protein (FAP or seprase) which is a type II transmembrane serine protease of the peptidase S9b family, a prolyl oligopeptidase subfamily, with post-proline dipeptidyl peptidase and endopeptidase enzymatic activity (for reviews, see [4, 5]). Though FAP has been implicated in cancer, this review will focus on the S1 membrane–anchored serine proteases, and from here on, type II transmembrane serine proteases (TTSPs) will refer to the S1 family members.
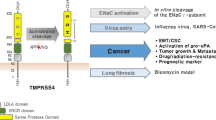
The TTSP family encompasses 17 members in humans. This review will focus on 10 TTSPs (indicated with asterisks in Fig. 1a) which have been implicated in cancer. TTSPs share a conserved N-terminal signal anchor that functions as the transmembrane domain, a “stem region” that is composed of a variable number of domains that belong to one of six conserved motifs, and a C-terminal serine protease domain (Fig. 1a). The serine protease domain has a histidine, aspartate, and serine triad of residues necessary for catalytic activity. TTSPs are divided into subfamilies based on the composition of the domains in the stem region, the phylogenetic relationship of the serine protease domain, and the chromosomal location of their genes [3, 6,7,8,9,10,11,12,13,14] (Fig. 1a). TTSPs are synthesized as zymogens and require specific cleavage in a conserved activation loop resulting in a structural rearrangement leading to the formation of a fully functional protease. Several TTSPs, including matriptase [15], matriptase-2 [16], hepsin [17], TMPRSS2 [18], TMPRSS3 [19], TMPRSS4 [20], and TMPRSS13 [21], are capable of auto-activation which is suggestive of a basal activity for their respective zymogens. Indeed, the rat and human matriptase zymogens have been shown to harbor activity in vitro [22,23,24,25,26]. Furthermore, knock-in mice expressing only a noncleavable form of matriptase (zymogen-locked) are viable, unlike matriptase-null mice, suggesting that matriptase zymogen is biologically active and capable of executing developmental and homeostatic functions of the protease [27]. Regulation of TTSP proteolytic activity is attributed to shedding of the protease from the cell surface upon complex formation with membrane-associated or membrane-secreted serine protease inhibitors or by internalization followed by lysosomal degradation [8].

Overview of human membrane–anchored serine proteases and cognate inhibitors. a The type II transmembrane serine protease (TTSP) family members are attached to the membrane via a signal anchor (SA) located close to the N terminus. TTSPs are phylogenetically divided into four subfamilies: (1) matriptase, (2) hepsin/transmembrane protease, serine (TMPRSS), (3) human airway trypsin-like (HAT)/differentially expressed in squamous cell carcinoma gene (DESC), and (4) corin. Asterisks indicate proteases included in this review. b Hepatocyte growth factor activator inhibitor type 1 (HAI-1) and HAI-2 are type I transmembrane serine protease inhibitors. They have two extracellular Kunitz-type serine proteinase inhibitor domains (KD1 and KD2): a single-pass transmembrane domain near the carboxyl terminus and a short intracytoplasmic domain. Two major splicing variants (isoforms a and b) of HAI-2 are known where the b isoform lacks KD1. HAI-2a is the predominant form in humans. c Prostasin and testisin are composed of a single protease domain linked to a glycosylphosphatidylinositol (GPI) anchor that is added posttranslationally to the C terminus and attaches the proteases to the outer leaflet of the plasma membrane. Domains: SA = signal anchor; LDLA = low-density lipoprotein receptor class A; SRCR = group A scavenger receptor cysteine-rich; SP = serine protease; SEA = sea urchin sperm protein, enteropeptidase, agrin; CUB = Cls/Clr, urchin embryonic growth factor, bone morphogenetic protein-1; MAM = meprin, A5 antigen, receptor protein phosphatase μ. TM = transmembrane; KD1 = Kunitz-type serine proteinase inhibitor domain 1; KD2 = Kunitz-type serine proteinase inhibitor domain 2; PKD = polycystic kidney disease (PKD)-like; MANEC = motif at N terminus with eight cysteines; GPI = glycosylphosphatidylinositol anchor
The two cell surface Kunitz-type serine protease inhibitors hepatocyte growth factor activator inhibitor-1 (HAI-1; SPINT1) and HAI-2 (SPINT2) were initially identified in a human gastric cancer cell line, and cDNA cloning revealed that they are both type I transmembrane proteins [28, 29]. They have two extracellular Kunitz-type serine proteinase inhibitor domains (KD1 and KD2): a single-pass transmembrane domain near the carboxyl terminus and a short intracytoplasmic domain. In addition, the amino terminus of HAI-1 has a Motif At N terminus with Eight Cysteines (MANEC) domain and a polycystic kidney disease (PKD)-like domain, as well as a low density lipoprotein (LDL)-receptor class A domain between KD1 and KD2 [30] (Fig. 1b). Two major splicing variants (isoforms a and b) are known for HAI-2 where the b isoform lacks KD1 [30].
Prostasin (PRSS8) is a serine protease with trypsin-like substrate specificity that was first isolated from seminal fluid [31]. Later, it was reported that prostasin is GPI anchored to the cell surface and is released from the cell upon GPI-anchor cleavage by phospholipase C (Fig. 1c) [32]. The Kunitz-type inhibitor HAI-1 was also found to form stable inhibitor complexes with prostasin [33,34,35]. Testisin (PRSS21) was first cloned and characterized in human eosinophils [36] and characterized as a new human serine proteinase in the testis [37]. It was later demonstrated that testisin is tethered to the cell surface via a GPI-anchor (Fig. 1c) [38]. Both testisin and prostasin expression are epigenetically regulated by gene methylation [39, 40].
2 Role of cell surface–anchored serine proteases in cancer
In this section, studies implicating cell surface–anchored serine proteases in cancer are summarized (see Table 1 and Fig. 2). The expression and function of cognate inhibitors will also be examined in various cancer types.

Roles of cell surface–anchored serine proteases in cancer development and progression. Cell surface–anchored serine proteases are involved in the development/initiation of primary tumors (determined through genetic mouse models), progression of primary tumors (determined through apoptosis/proliferation assays cell culture assays and/or xenograft models), cancer cell migration/invasion (determined through invasion assays in vitro), and formation of metastatic lesions (determined through metastasis development in genetic mouse models and/or xenograft models). Upward-facing arrows indicate proteases that are positively associated with tumor development/progression (pro-oncogenic), and downward-facing arrows indicate proteases negatively associated with tumor development/progression (tumor-suppressive)
2.1 Skin and head & neck squamous cell carcinomas
2.1.1 Matriptase
Matriptase is both an initiator and a strong tumor-promoter in squamous cell carcinoma (SCC). A transgenic mouse model was generated in which matriptase expression is under the control of the keratin-5 (K5) promoter and thus overexpressed in the epidermis [41]. In contrast to wild-type (WT) mice which did not develop tumors, 100% of transgenic mice developed spontaneous epidermal neoplasia [41]. Furthermore, after exposure to a single topical dose of the chemical carcinogen 7,12-dimethylbenzanthracene (DMBA), nearly all the matriptase transgenic mice developed SCC within 40 weeks. Importantly, when the cognate matriptase inhibitor HAI-1 was simultaneously overexpressed with matriptase in the epidermis, development of SCC following DMBA exposure was abrogated, indicating that HAI-1 expression is sufficient to protect mice from matriptase-induced SCC [41]. Moreover, while matriptase is detected in complex with HAI-1 in normal epidermis, it is mainly present in its noncomplexed form in human epidermal SCC samples, indicating that the balance between matriptase and its endogenous inhibitor is dysregulated upon SCC transformation [42]. Doxycycline-inducible expression of another cognate inhibitor, HAI-2, in the K5-matriptase transgenic mice caused significant regression in the size and number of established DMBA-induced epidermal tumors [43]. This indicates that continued dysregulation of matriptase activity is necessary for SCC progression and that inhibition of matriptase activity in established tumors may be an avenue for therapeutic intervention [43]. Induction of HAI-2 expression led to decreased intratumoral infiltration of inflammatory cells, suggesting that matriptase-mediated SCC progression is mediated in part by tumor-promoting inflammation [43]. It was suggested in a 2009 study that protease-activated receptor 2 (PAR-2) is a substrate of matriptase in the skin based on their co-localization by immunohistochemistry (IHC) in human epidermis and the activation of PAR-2 by matriptase in human keratinocyte cell culture models [42]. These findings are in alignment with a later study using genetic mouse models to investigate the role of PAR-2 in SCC development [44]. K5-matriptase transgenic mice crossed to PAR-2 null mice were protected against the development of epidermal hyperplasia or dysplasia that is normally associated with matriptase overexpression in the epidermis, indicating that pre-malignant transformation into SCC is dependent on PAR-2 expression [44]. This was further confirmed by the inability of transgenic matriptase to potentiate DMBA-induced tumors in the absence of PAR-2 [44]. Matriptase exerts its pro-oncogenic properties to induce epidermal SCC through the activation of PAR-2–NF-κB signaling as well as through the phosphatidylinositol-3-kinase (PI3K)–Akt–mTor pathway [41, 44].
It has also been demonstrated by IHC analysis that matriptase is highly expressed in head and neck SCC (HNSCC) including carcinomas of the tongue, lip, larynx, and gingiva, and that the protease is frequently co-expressed with the receptor tyrosine kinase c-Met [45]. This would allow for activation of c-Met and downstream proliferative signaling of epithelial cells upon matriptase-mediated activation of the c-Met ligand hepatocyte growth factor (HGF) [45]. To test the functional connection between matriptase and HGF/c-Met, the K5-matriptase transgenic mouse described above was crossed into a mouse model with c-Met ablated in the basal keratinocytes of the epidermis (K14-Cre+/0;Hgfrfl/−) [45]. Loss of c-Met impaired matriptase-induced SCC formation, demonstrating an essential role for c-Met signaling for the pro-oncogenic properties of matriptase [45]. Another study specifically investigating the role of HAI-1 in oral SCC (OSCC) cell lines, including those of the gingiva and tongue, found that silencing of HAI-1 enhanced migration and tumorigenicity [46]. The migratory phenotype was negated by simultaneously silencing matriptase, indicating that HAI-1 works, at least in part, to inhibit the oncogenic effects of matriptase in OSCC [46]. HAI-2 also plays a role in inhibition of the pro-oncogenic activity of matriptase in OSCC. In HAI-2 knock-out (KO) OSCC cells, levels of activated matriptase increased as well as the levels of prostasin [47]. RNA interference (RNAi)-mediated prostasin silencing reversed the reduced invasiveness of the HAI-2 KO cells, indicating that both HAI-2 and prostasin act as suppressors of cellular invasion. It was previously demonstrated that a matriptase–prostasin reciprocal zymogen activation complex exists [48], which could cause dysregulation of both matriptase and prostasin activity upon HAI-2 KO. The OSCC tumor microenvironment may also play a role in matriptase-mediated tumorigenicity. Conditioned media isolated from SAS cells, a tongue SCC cell line, enhanced migration of cancer-associated fibroblasts (CAFs), stromal cells that contribute to malignant progression [49]. HAI-1 silencing in SAS cells further enhanced the migratory CAF phenotype, due to an increase in active matriptase produced by the cancer cells [49]. The authors propose that CAF-expressed PAR-2 is activated by matriptase in a paracrine manner to stimulate cellular migration in OSCC [49]. IHC analysis of clinical samples of OSCC further supported a pro-oncogenic role of matriptase, as matriptase expression positively correlated with grade, stage, lymph node positivity, and metastasis to distant sites. Higher matriptase expression was also associated with poorer patient prognosis [50].
2.1.2 DESC1
The first published DESC1 (differentially expressed in squamous cell carcinoma 1) study concluded that DESC1 transcript is expressed in normal head and neck epithelial tissues, but that expression is dramatically decreased in HNSCC samples [51]. In some cases, DESC1 is undetectable in HNSCC, which suggests that loss of the protease may be important for the development of SCC [51]. In a subsequent study, it was demonstrated that DESC1 protein levels also decreased during cancer progression [52] and that DESC1 protein expression positively correlated with keratinocyte differentiation [52]. Thus, DESC1 is lost during the de-differentiation associated with malignant transformation in HNSCC, indicating a potential role as a tumor suppressor. To elucidate the mechanism by which DESC1 exerts its tumor-suppressive activity, apoptosis and proliferation assays were performed in DESC1-overexpressing cell lines derived from both well and poorly differentiated esophageal SCC (ESCC) primary tumors [53]. In both cell lines, the overexpression of DESC1 decreased cell viability by increasing apoptosis upon serum starvation (a cellular stress condition). This indicates that DESC1 may sensitize cells that are under stress to undergo apoptosis [53]. Furthermore, this study identified that DESC1 expression caused decreased Akt1 activation through modulation of EGFR signaling, which in turn led to a tumor-suppressive phenotype. Additionally, in an orthotopic grafting model, DESC1-overexpressing ESCC cells displayed decreased tumor growth in nude mice compared to control cells [53]. The effect of loss-of-function studies in vivo using DESC1 null mice would further shed light on its role in SCC progression. One possible mechanism by which DESC1 is downregulated in ESCC was recently described [54]. In ESCC tissues, the long noncoding RNA (lncRNA) TUSC7 is significantly downregulated compared to normal esophageal tissue, and TUSC7 negatively regulates the micro-RNA miR-224 expression under normal conditions. Importantly, miR-224 is overexpressed in ESCC, and DESC1 is a direct target for silencing by miR-224 [54].
2.1.3 HAT and HATL-5
IHC analysis demonstrated that human airway trypsin-like protease (HAT) protein is highly expressed in nonmalignant apical squamous epithelial cells, but protein levels significantly decrease as the grade of disease increases in both cervical and esophageal SCC [55]. Consequently, poorly differentiated carcinomas displayed little to no staining for HAT protein [55]. Much like DESC1, HAT levels appear to correlate with differentiated epithelium, and as cells undergo malignant transformation to SCC, HAT expression is gradually reduced with increasing progression and is in many cases lost entirely.
Human airway trypsin-like protease 5 (HATL-5) transcript and protein expression is reduced in carcinoma tissues of the cervix, esophagus, and head and neck, as compared to normal tissues [56]. Like HAT expression in SCC, HATL-5 protein levels decreased significantly with increasing grade of disease, where poorly differentiated high-grade carcinomas displayed weak or undetectable HATL-5 [56]. It remains to be determined whether HAT and HATL-5 are critical for suppression of malignant transformation and/or progression SCC in vivo.
2.1.4 TMPRSS3
TMPRSS3 transcript and protein expression is significantly upregulated in nasopharyngeal carcinoma (NPC) as compared to adjacent nonmalignant epithelium [57]. Similarly, NPC cell lines showed significantly increased TMPRSS3 expression compared to the NP69 nasopharyngeal epithelium cell line. Stable knock-down (KD) of TMPRSS3 in NPC cells reduced the proliferation and invasive phenotype of the cells in vitro by inhibition of the PI3K/Akt oncogenic signaling pathway. Subcutaneously implanted TMPRSS3-KD NPC cells displayed impaired tumor growth compared to control cells [57].
2.1.5 Prostasin
Prostasin (PRSS8) has been identified in the context of ESCC as a potential tumor suppressor [58]. Both transcript and protein levels of prostasin were significantly decreased in poorly differentiated ESCC tissues as compared to normal esophagus, carcinoma in situ, and well-differentiated ESCC samples. In ESCC tissues and cell lines with low or undetectable expression of prostasin, the CpG island-containing region of the PRSS8 gene promoter was hypermethylated [58]. This CpG island hypermethylation silenced prostasin expression and could be reversed by treatment of cells with the demethylating agent decitabine. A potential mechanism for the role of prostasin in ESCC tumor suppression is through the downregulation of cell cycle and epithelial-to-mesenchymal transition (EMT) proteins, including cyclin D1, Snail, and Twist [58].
2.2 Breast cancer
2.2.1 Matriptase
Matriptase was first described in 1993 as a new cancer-associated protease with gelatinolytic activity expressed by cultured human breast cancer cells [59]. In breast carcinomas, increased matriptase expression correlates with tumor grade and stage, and a high matriptase expression is predictive of poor survival [60,61,62,63]. The cognate matriptase inhibitors HAI-1 and HAI-2 are expressed at a significantly lower level in poorly differentiated breast tumors, and HAI-2 expression is inversely correlated with nodal involvement and tumor dissemination [64]. Interestingly, in a long-term survival study of node-negative breast cancer patients, 30-year survival data demonstrated that high expression of both matriptase and c-Met, the receptor for HGF, was significantly associated with poorer disease-free survival [61]. Using genetic mouse models, it was demonstrated that matriptase is critically involved in mammary carcinogenesis and that one of the molecular mechanisms through which matriptase exerts its pro-carcinogenic effects is activation of pro-HGF on the cancer cell surface, leading to initiation of the c-Met signaling pathway and elicitation of mitogenic and invasive responses in breast cancer [65]. Matriptase hypomorphic mice that displayed an approximate 75% reduction in matriptase protein levels in mammary glands were used [65]. When crossed into the mouse mammary tumor virus (MMTV) Polyomavirus middle T (PymT) antigen genetic mammary tumor model, matriptase hypomorphic mice displayed a significant delay in tumor onset, as well as a decreased tumor burden caused by abrogation of tumor cell proliferation [65]. For mechanistic studies, primary mammary carcinoma cells with genetic disruption of the matriptase encoding gene by tamoxifen-inducible Cre–loxP recombination were generated. Matriptase-null cells displayed an impaired ability to initiate activation of the c-Met signaling pathway in response to fibroblast-secreted pro-HGF [65]. The matriptase/c-Met signaling axis also mediates proliferation and invasion in human inflammatory breast cancer (IBC) cell lines and in non-IBC human triple-negative ductal carcinoma cell lines [65, 66].
Platelet-derived growth factor-C (PDGF-C) is another substrate of matriptase that contributes to breast cancer cell migration and survival in vitro [67]. MCF-7 luminal breast cancer cells engineered to overexpress PDGF-C produced proteases capable of cleaving PDGF-C to its active form. Increased PDGF-C expression enhanced cell proliferation, anchorage-independent cell growth, and tumor cell motility by autocrine signaling. Matriptase was identified as the major protease responsible for processing of PDGF-C in MCF-7 cells [67].
2.2.2 Matriptase-2
Matriptase-2 (TMPRSS6) is highly expressed in normal mammary tissue and mainly confined to the epithelial cells [68]. In breast carcinomas, matriptase-2 protein levels decreased with increasing tumor grade with very low matriptase-2 levels observed in undifferentiated ductal or lobular tumors. Reduced matriptase-2 levels in breast cancer tissues correlated with an overall poor prognosis [68, 69]. When matriptase-2 was stably expressed in the highly invasive breast cancer cell line MDA-MB-231, which does not endogenously express matriptase-2, reduced cell invasion and migration was observed in vitro. Furthermore, matriptase-2–expressing cells implanted subcutaneously into nude mice displayed significantly impaired tumor growth [68]. Thirteen single nucleotide polymorphisms (SNPs) in the TMPRSS6 gene were investigated in triple-negative breast cancer, and four variants were associated with reduced matriptase-2 expression and poor survival [69].
2.2.3 Hepsin
Hepsin is overexpressed in breast cancer tissues as compared to adjacent nonmalignant breast tissue [70, 71]. Hepsin expression also positively correlated with the tumor stage and lymph node metastases [70]. The overexpression of hepsin in mammary epithelial organoids was associated with a downregulation of HAI-1 and augmented HGF/c-Met signaling which caused deterioration of desmosomes and hemidesmosomes [70, 71]. Hepsin facilitates the invasive potential of breast cancer cells through remodeling of the basement membrane by cleavage of laminin-332, a component of the hemidesmosome at cell–cell junctions [72]. Decreasing hepsin activity with a selective inhibitor or its expression with siRNA-mediated silencing reduced desmosome cleavage and impaired the proliferation and invasiveness of cultured breast cancer cells [72, 73].
2.2.4 TMPRSS3
In an IHC study comparing breast cancer patient tissue samples to adjacent healthy breast tissue, a significantly higher expression of TMPRSS3 in cancerous tissue was demonstrated [74]. The expression level of TMPRSS3 also correlated with disease stage, lymph node positivity, and proliferation of the cancer cells. Consequently, high expression of TMPRSS3 led to lower disease-free and overall survival [74]. Additionally, TMPRSS3 was found to positively associate with distant organ metastasis in breast cancer [75]. In one study, the expression of TMPRSS3 in breast cancer samples was described to be low in poorly differentiated tumors, and low TMPRSS3 expression was significantly associated with reduced overall survival [76]. The expression levels and the clinical significance of TMPRSS3 in breast cancer therefore remain unclear.
2.2.5 Prostasin
In 2002, it was demonstrated that prostasin expression at both the transcript and protein level is undetectable in highly invasive and metastatic breast cancer cell lines, but the protease is expressed in normal breast epithelial cells and minimally invasive breast cancer cell lines [77]. Transgenic expression of prostasin in MDA-MB-231 and MDA-MB-435 breast cancer cell lines, which do not express endogenous prostasin, reduced the in vitro invasiveness of both cell lines. Prostasin expression may be lost in breast cancer cells lines due to methylation of its promoter region, as treatment of breast cancer cell lines with a DNA methyltransferase inhibitor was able to reactivate prostasin expression [77]. Matriptase and prostasin have also been shown to be co-expressed in breast cancer cell lines and human breast cancer tissue samples [78].
2.3 Colorectal cancer
2.3.1 Matriptase
A 2006 study demonstrated that the ratio of matriptase:HAI-1 mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals [79]. Additionally, a 2007 study that investigated the ratio of matriptase to HAI-1 via IHC analysis showed that the matriptase:HAI-1 ratio is higher in more differentiated colon adenocarcinoma and decreases in poorly and moderately differentiated cancers [80]. These studies indicate that the matriptase:HAI-1 ratio is important for CRC tumor development and that the ratio is dependent on the grade/differentiation of the tumor. Silencing of matriptase expression with siRNA or inhibition of matriptase activity using small molecule inhibitors in the CRC cell line DLD-1 led to decreased activation of pro-HGF and decreased cell invasion through an extracellular matrix in vitro [81].
Similar to matriptase-null mice, which have severe epidermal barrier defects [82], mice with matriptase ablation specifically in the intestinal epithelium (villin-Cre+/0;St14LoxP/−) display intestinal epithelial barrier defects due to decreased tight junction formation [83]. These mice also form colonic adenocarcinomas that resemble CRC that arise from inflammatory bowel disease. Loss of matriptase in the colon therefore leads to dysregulated epithelial barrier function, which allows for intestinal microbes and resident immune cells to cause chronic intestinal inflammation that eventually leads to adenocarcinoma formation [83]. Thus, inflammation-associated colon carcinogenesis can be initiated and promoted solely by an intrinsic intestinal permeability barrier perturbation, and in this context, matriptase acts as a tumor suppressor by supporting normal barrier function. The intestinal barrier defect in this matriptase loss-of-function model limits interpretation pertaining to the contribution of matriptase in CRC, since conclusions cannot be drawn regarding matriptase loss in the context of an intact intestinal barrier. Additional studies using alternative models, such as orthotopic xenografts assessing growth of matriptase-deficient CRC cells implanted in a normal intestinal background, would be informative.
2.3.2 TMPRSS4
TMPRSS4 mRNA and protein expression are significantly increased in CRC tissue samples compared to normal colon mucosa, and the expression of TMPRSS4 correlates with both the tumor grade as well as the presence of lymph node metastases [84, 85]. RNAi-mediated silencing of TMPRSS4 in the CRC cell line HCT116 demonstrated decreased cell proliferation, invasion, and migration, as well as a reduction in the cancer stem cell (CSC) markers CD44 and CD133 [84]. Thus, TMPRSS4 expression may be linked to the ability of CRC cells to self-renew.
2.3.3 Prostasin
Several studies have demonstrated that prostasin (PRSS8) is a tumor suppressor in CRC. Prostasin mRNA levels are modestly yet significantly decreased in CRC and dysplastic colon tissue as compared to matched normal tissue [86, 87]. Transcript levels of protease nexin-1 (PN-1), an endogenous inhibitor of prostasin, are significantly increased in dysplasia and CRC tissue, which may be a contributing factor to CRC progression due to a reduction in prostasin activity [86]. Furthermore, low prostasin protein expression is significantly associated with lower overall and disease-free survival of CRC patients [87]. The localization of prostasin in CRC tissues changes during cancer progression: in normal colon epithelium, prostasin is located on the apical plasma membrane, while HAI-1 is located on the basolateral plasma membrane [86]. In CRC tissue samples, prostasin co-localized with HAI-1 in IHC staining, suggesting a loss of cell polarity [86]. Prostasin has been shown to negatively associate with Sphk1, S1p, phosphorylated-Stat3, and phosphorylated-Akt levels in CRC cell lines [87]. This indicates that inflammatory signaling in the colon through the Sphk1/S1p/Stat3/Akt axis may be suppressed by prostasin under normal conditions, and this axis becomes dysregulated upon carcinogenesis. Subcutaneous xenografts of a prostasin-overexpressing HCT116 CRC cell line into nude mice showed that prostasin inhibited tumor growth and suppressed the Sphk1/S1p/Stat3/Akt axis in vivo [87]. Prostasin-overexpressing CRC cells displayed decreased growth and metastasis upon grafting into nude mice, as well as decreased invasion, migration, and colony and sphere formation in vitro compared to WT cells [88]. A conditional knock-out mouse that lacks prostasin expression specifically in the colon (Prss8fl/fl, p-Villin-Cre+) displays intestinal inflammation and develops spontaneous colitis at a young age, that eventually develops into proliferative, poorly differentiated intestinal tumors [88]. Mechanistic studies indicated that prostasin-dependent tumor suppression was mediated through targeting of the Wnt/β-catenin, EMT, and stem cell signaling pathways [88]. Since epidermal ablation of either prostasin or matriptase leads to identical epidermal barrier phenotypes [89, 90], it is plausible that the conditional prostasin KO mice have impaired intestinal barrier function which could promote inflammation and carcinogenesis as a secondary effect.
2.4 Ovarian cancer
2.4.1 Matriptase
Matriptase is highly expressed in ovarian carcinomas at the transcript and protein levels, while it is low to undetectable in normal ovaries [91,92,93]. The expression of matriptase in ovarian carcinomas decreases as the stage of disease increases [92, 94], with one study finding that 72% of stage I tumors expressed matriptase mRNA and protein, while less than 50% of stage II/III/IV tumors were positive for matriptase expression [92]. The reduction of matriptase expression with advancing disease stage may be a consequence of the cells acquiring a more mesenchymal phenotype [95]. Consequently, matriptase expression in ovarian cancer is significantly associated with better survival outcomes [92, 93]. However, it also appears that the ratio between the expression of matriptase and its cognate inhibitors HAI-1 and HAI-2 is an important factor in ovarian carcinogenesis. Both HAI-1 and HAI-2 protein levels are significantly decreased with increasing stage of disease, and lower expression of HAI-2 is associated with accumulation of ascites fluid and residual tumor diameter [96]. Disease-free and overall survival are also significantly decreased with low HAI-1 and HAI-2 expression [96]. In most advanced stage ovarian tumors that do express matriptase, HAI-1 was rarely co-expressed, while about 30% of lower stage tumors co-expressed matriptase and HAI-1 proteins [94]. Furthermore, when HAI-1 and HAI-2 were transiently transfected into the OVCAR-3 ovarian cancer cell line, matriptase protein level dramatically decreased and apoptosis increased [96]. Another study that specifically assessed the matriptase:HAI-1 ratio in HO-8910 ovarian cancer cells and in the highly metastatic HO-8910PM cells showed that in the latter, the ratio of matriptase to HAI-1 was significantly increased at both the transcript and protein levels compared to HO-8910 cells [97]. Cellular migration and invasion in these cells were significantly positively associated with the ratio of matriptase to HAI-1, and siRNA-mediated silencing of matriptase decreased the migratory and invasive ability of the HO-8910PM cells in vitro [97]. One proposed mechanism by which matriptase increases the invasiveness of ovarian cancer is through the activation of urokinase plasminogen activator (uPA) which is involved in degradation, via activation of plasminogen to plasmin, of the extracellular matrix surrounding tumor cells, allowing for their dissemination [98]. When matriptase was silenced in the ovarian cancer cell line HRA, pro-uPA conversion to active uPA was impaired, which may contribute to the observed abrogation of invasiveness through a reconstituted extracellular matrix in vitro [98].
2.4.2 TMPRSS3
TMPRSS3 (TAGD-12) overexpression in ovarian cancer was discovered nearly two decades ago [99], and TMPRSS3 was introduced as a potential therapeutic biomarker. TMPRSS3 is significantly increased at both the transcript and protein levels in epithelial ovarian cancer as compared to normal ovarian epithelium and ovarian tumors of low malignant potential (LMP) [99]. In a later study, genome-profiling was performed in epithelial ovarian cancer (EOC) cell lines following treatment with S-adenosyl-l-methionine (SAM, a compound that stimulates DNA methylation). Genes that were downregulated upon SAM treatment were considered hypomethylated in EOC [100]. The CpG island in the 5′ untranslated promoter region of the TMPRSS3 gene was hypomethylated in high-grade EOC tumors, causing increased expression of TMPRSS3 in EOC [100]. SAM treatment was also shown to decrease the protein expression of TMPRSS3 in the EOC cell lines SKOV3 and OVCAR3, indicating that increased DNA methylation of TMPRSS3 in EOC is capable of reducing its expression in vitro [100]. TMPRSS3 overexpression in the ovarian cancer cell line A2780 increased proliferation, invasion, and migration of the cells, and conversely, silencing of TMPRSS3 in HO8910 cells had the opposite effect [101] further suggesting a role of TMPRSS3 as a pro-oncogenic protease in ovarian cancer.
2.4.3 Testisin
Testisin is significantly upregulated at the transcript level in human ovarian clear-cell carcinoma and adenocarcinoma samples as compared to normal ovarian tissue [102]. This transcript increase was demonstrated to correlate with the stage of ovarian carcinoma, with testisin expression being significantly higher in advanced stage carcinomas than LMP tumors or adenomas [102]. In metastatic serous papillary ovarian tumors however, testisin gene expression decreases as compared to primary ovarian serous carcinomas [103], indicating that testisin may primarily be upregulated in primary, not metastatic, ovarian tumor cells. Testisin was detected at the mRNA and protein level in the ovarian cancer cell line CaOv3 and RNAi-mediated KD of testisin in these cells induced apoptosis by increasing the activity of caspase-3 and caspase-7 [38]. Testisin-overexpressing SKOV3 ovarian cancer cells grew significantly larger primary tumors upon subcutaneous implantation into mice, further implicating testisin as a pro-oncogenic protease [38]. The role of testisin in ovarian cancer metastasis was further investigated in vivo using an intraperitoneal xenograft model of late stage, metastatic tumorigenesis [104]. Xenografts of testisin-overexpressing clear-cell carcinoma cells displayed abrogated intraperitoneal tumor cell seeding and tumor metastasis. The mechanism for this inhibition of metastasis was proposed to involve proteolytic activation of PAR-2 by testisin, which antagonizes the pro-angiogenic angiopoietins ANG2 and ANGPTL4 to cause decreased ascites accumulation [104]. A novel experimental cancer therapeutic that utilizes modified anthrax toxin protective antigen to induce cancer cell killing upon testisin-mediated cleavage has shown efficacy in cell culture experiments and in vivo [105] (see Section 3.9).
2.5 Prostate cancer
2.5.1 Matriptase
Matriptase is significantly increased at both the mRNA and the protein level in prostate cancer patient tissues compared to normal prostate samples, and this increased expression also significantly correlates with tumor grade [106, 107]. Expression is restricted to the malignant epithelial prostate cells, as matriptase transcript is nearly undetectable in stromal cells [106]. A correlation between matriptase expression levels and prostate cancer aggressiveness was observed in the PC-3 and DU145 prostate cancer cell lines [108]. When matriptase expression was reduced in these cells using hammerhead ribozyme transgenes, the cells exhibited slower growth, reduced invasion and migration, and increased cell–cell adhesion. Furthermore, matriptase-deficient PC-3 cells caused significantly reduced tumor growth in a subcutaneous xenograft model [108].
As observed in many other cancers, the increased expression of matriptase in both prostate cancer cell lines and tissues was accompanied by a corresponding decrease in the expression of HAI-1, again showing the importance of matriptase:HAI-1 balance in malignant progression [107]. A later study demonstrated that HAI-1 protein level increases as well in all prostate proliferative diseases tested, including localized and aggressive cancer, benign prostate hyperplasia, and high-grade intraepithelial neoplasia [109]. The authors hypothesize that the reason for the discrepancy between studies investigating HAI-1 levels in prostate cancer may be due to different antibodies being used, different assays, as well as small sample sizes and different patient populations [109]. Studies using HAI-1-deficient PC-3 and DU145 prostate cancer cell lines have demonstrated that these cells displayed decreased invasiveness and slower growth compared to control cells in vitro [110]. This lends credence to the idea that dysregulation of HAI-1 expression, under some conditions, can lead to a more aggressive prostate cancer phenotype.
HAI-2 significantly decreases in malignant tissue compared to benign lesions and normal prostate at both the transcript and protein levels, and expression further decreases with increased Gleason score [106, 111]. The N1 and N2 prostate cancer cell lines were established through serial intraprostatic propagation of 103E human prostate cancer cells and isolation of the metastatic cells from nearby lymph nodes [112]. The invasion capability of these cells was revealed to gradually increase throughout the serial isolations (103E<N1<N2) and the expression of HAI-2, but not HAI-1 was significantly decreased throughout the progression in parallel with increased activation of matriptase [112]. Furthermore, shRNA-mediated silencing of HAI-2 in CWR22Rv1 prostate cancer cells demonstrated increased levels of active matriptase and increased prostate cancer invasiveness, while overexpression of HAI-2 in N2 cells showed co-localization with matriptase on the cell surface, reduced matriptase activity, and reduced invasive capability [112]. Silencing of matriptase reduced the invasive potential induced by HAI-2 silencing in CWR22Rv1 cells. In vivo, HAI-2 overexpression or matriptase silencing in N2 cells significantly decreased tumorigenicity and metastatic capability in orthotopically xenografted mice [112]. These results suggest that matriptase activity is primarily controlled by HAI-2 in prostate cancer and that an imbalance between HAI-2 and matriptase expression leads to matriptase-mediated cell migration, invasion, and metastasis [111,112,113]. The proform of HGF is a substrate for matriptase in prostate cancer. PC-3 cells, which express high levels of matriptase, treated with a matriptase inhibitor or matriptase siRNA, showed an abrogation of pro-HGF cleavage into its active form [114]. Matriptase may be activated in the prostate tumor microenvironment by extracellular acidification induced by platelet-derived growth factor-D (PDGF-D) [115]. PDGF-D signaling in a benign prostate epithelial cell line increased the nuclear localization of the transcription factor hypoxia-inducible factor-1α (HIF-1α), which increases expression of carbonic anhydrase IX (CAIX) and causes acidosis. This low pH induces the activation and shedding of matriptase, leading to a more aggressive prostate cancer phenotype, and potentially facilitates the invasion of cancer cells [115]. Matriptase is capable of cleaving the full-length PDGF-D dimer into a hemidimer and further into an active growth factor domain (GFD) dimer [116]. Matriptase further processes the GFD into a smaller, inactive GFD fragment, which is unable to activate the β-PDGF receptor [116]. Thus, the activity of PDGF-D is regulated by matriptase, and this regulation can also influence the binding of PDGF-D to the extracellular matrix (ECM). Matriptase also modulates the integrity of the ECM by cleaving laminin-332 (Ln-332), an ECM protein [117, 118]. A growth factor receptor which may play a role in matriptase activation in prostate cancer is ErbB-2 (also known as epidermal growth factor receptor 2 or HER2). Dysregulated ErbB-2 signaling is associated with cancer cell proliferation and invasion, and this signaling can be ligand-dependent as well as ligand-independent [119]. LnCaP C-33 cells, a prostate cancer cell line that is minimally invasive and has moderate levels of activated ErbB-2 and active matriptase, were stimulated with EGF to enhance the activation of ErbB-2, and this caused a corresponding increase in the levels of activated matriptase. Loss-of-function studies using the ErbB-2 inhibitor AG825 led to a reduction in active matriptase as well as significantly decreased migration and invasion of the prostate cancer cells. It was also demonstrated that the activation of matriptase and induction of an invasive phenotype by ErbB-2 stimulation occurs via the PI3K pathway [119].
Invasion can also be promoted by an androgen-dependent mechanism in prostate cancer. TMPRSS2 is commonly associated with prostate cancer progression and severity, and it was found that testosterone (DHT) increases TMPRSS2 expression in a dose-dependent manner [120]. Matriptase was identified as a TMPRSS2 substrate in prostate cancer cell lines, where TMPRSS2 can proteolytically activate matriptase and enhance its shedding. Thus, androgen signaling induces TMPRSS2 expression, which in turn increases matriptase activation to promote the invasion and migration of prostate cancer cells [120]. Matriptase activation and shedding is induced by high expression of the androgen receptor in PC-3 cells [118]. This matriptase activation mechanism is dependent on activation of Src tyrosine kinase by androgen binding to androgen receptor (AR), and silencing of matriptase with siRNA significantly decreases the AR-dependent invasiveness of the PC-3 cells [118]. Cyclooxygenase-2 (COX-2) also plays a role in the activation of matriptase and the invasive potential of PC-3 prostate cancer cells [121]. COX-2 silencing in PC-3 cells leads to a 90% decrease in levels of activated matriptase. COX-2 activity is induced by inflammation and generates prostaglandin E2 (PGE2) which acts through its receptors EP1/2 to activate matriptase to promote tumor growth in a PC-3 orthotopic xenograft model and cell invasion in vitro [121].
2.5.2 Hepsin
A 2001 study that investigated differentially expressed genes between normal and malignant human prostate samples identified hepsin as a significantly overexpressed protease in prostate cancer [122]. In situ hybridization showed low gene expression of hepsin in benign prostate epithelial cells and high hepsin expression in the carcinoma cells of cancerous samples and in prostatic intraepithelial neoplasia (PIN) cells. These results indicated that hepsin expression is associated with malignant transformation of prostate epithelium [122]. The gene expression results were confirmed by IHC analysis of hepsin protein expression in human tissue biopsies, as 100% of prostate carcinoma samples stained positive for hepsin, whereas about half of PIN samples were positive [123]. Only 11% of benign prostatic hyperplasia (BPN), and none of the normal prostate samples, were positive [123]. Several single SNPs have been identified in the hepsin gene, which differ significantly in frequencies between prostate cancer patients as compared to healthy controls. Furthermore, a major 11-locus haplotype was significantly associated with prostate cancer susceptibility and one of the SNPs correlated with Gleason score [124]. However, a later study indicated that these SNPs did not have significant associations with risk of developing prostate cancer, tumor aggressiveness, risk of recurrence, or risk of death due to prostate cancer [125]. The authors of the latter study suggest that discrepancies between the two studies may be due to differences in study populations, as well as differences in adjustments for age or other factors.
Hepsin on the surface of LNCaP prostate cancer cells has been shown to cleave pro-macrophage-stimulating protein (MSP) into its active form at lower concentrations than matriptase or hepatocyte growth factor activator (HGFA), indicating that the MSP/RON signaling pathway may be activated by hepsin in prostate cancer to promote prostate cancer progression [126].
Hepsin is also involved in promoting invasion of prostate cancer cells through the cleavage of Ln-332, an ECM protein [127]. Prostate cancer progression is accompanied by proteolytic processing of Ln-332, and LnCaP cells engineered to overexpress hepsin were significantly more invasive due to increased cleavage of Ln-332 [127]. Pro-HGF is another substrate of hepsin that has been strongly implicated in prostate epithelial transformation and prostate cancer malignancy [114, 128, 129]. As such, hepsin activity is also inhibited by the Kunitz-type inhibitors HAI-1 and HAI-2 [128]. Orthotopic xenografts were performed in mice using either LnCaP-17 cells that express low levels of hepsin, or LnCaP-34 cells that express high levels of hepsin. The LnCaP-34 xenografts grew at a significantly faster rate, were more invasive, and metastasized to lymph nodes [130]. Established LnCaP-34 tumors treated with a polyethylene glycol conjugated (PEGylated) form of HAI-1 Kunitz domain-1 (KD1) (see Section 3.6) showed significantly diminished invasion and metastasis, and prostate-specific antigen (PSA) levels were also significantly reduced over time [130]. Interestingly, treatment with KD1-PEG did not reduce the volume of the primary tumor that formed from the initial injection of cells into the left lobe of the prostate, indicating that hepsin may play a more significant role in promoting an invasive phenotype in prostate cancer [130]. This is in agreement with a previous study using transgenic overexpression of hepsin in the prostate of mice in combination with transgenic expression of a viral oncogene (see below), where proliferation in the primary tumor cells was not affected, but progression and metastasis to the liver, lung, and bone was observed [131]. Doxycycline-inducible overexpression of hepsin in PC-3 cells led to the inability to adhere to tissue culture plates which was associated with a corresponding reduction in Akt phosphorylation [132]. When the cells were grown on ECM produced by nontumorigenic prostate cells, Akt phosphorylation was restored [132] suggesting that the matrix surrounding the cells is important for mediating the effects of hepsin overexpression. The LPB-Tag/PB-Hepsin mouse is a double-transgenic model of metastatic prostate cancer where the oncogene SV40-large T antigen (Tag) is expressed under the control of the prostate-specific long probasin (LPB) promoter, and hepsin is expressed under the control of the probasin (PB) promoter [131, 133]. In this model, hepsin overexpression in the prostate epithelium leads to disorganization of the basement membrane and promotes primary prostate cancer progression and distant metastasis [131, 133]. A selective hepsin inhibitor (see Section 3.6) blocked the development of metastatic lesions in these mice, whereas the majority of mice in the control group developed metastatic lesions to the liver, lung, and bone [133]. In a different study, a mouse model was generated in which PB-Hepsin transgenic mice were crossed with mice in which adenomatous polyposis coli (APC, a tumor suppressor that inhibits Wnt signaling) was knocked out in the prostate (ApcPBKOHepsin mice) [134]. These mice developed significantly larger, more invasive, and hyperproliferative prostate tumors, compared to mice that only have APC knocked out, indicating that the Wnt/β-catenin pathway and hepsin act in concert to promote prostate cancer progression [134]. PB-Hepsin mice have also been crossed with transgenic mice overexpressing Myc in the prostate, resulting in PB-Hepsin/PB-Hi-myc mice [135]. These double-transgenic mice developed prostate adenocarcinomas more rapidly than PB-Hi-myc mice, indicating that the addition of hepsin overexpression caused accelerated malignant progression of the tumors. Interestingly, PB-Hi-myc tumors acquired hepsin expression over time as they progressed to higher grade tumors, suggesting an important role for hepsin in promoting aggressiveness [135].
2.5.3 TMPRSS2
Around 50% of prostate cancers harbor a gene rearrangement between TMPRSS2 and estrogen-regulated gene (ERG), a member of the erythroblast transformation-specific (ETS) family of transcription factors. This gene fusion causes constitutive activation of oncogenic ERG, which can lead to prostate cancer cell invasion and metastasis [136,137,138,139,140]. In these cases, the proteolytic activity of TMPRSS2 is not considered to be of importance. However, studies have shown a role for the function of TMPRSS2 itself in prostate cancer, without the fusion to ERG. TMPRSS2 is an androgen-regulated protease expressed in the prostate secretory epithelium under normal conditions [141]. In prostate hyperplasia, neoplasia, and in prostate cancer metastases, TMPRSS2 protein expression significantly increases, and the expression correlates significantly with Gleason score [141]. In a transgenic adenocarcinoma of the mouse prostate (TRAMP) mouse model in which TMPRSS2 was genetically ablated (Tmprss2−/−; TRAMP), mice developed prostate tumors at the same frequency as Tmprss2+/+ TRAMP mice; however, the sizes of tumors in the Tmprss2−/− TRAMP animals were significantly larger and tended to be more differentiated [142]. Importantly, only 7% of Tmprss2−/− TRAMP mice harbored macroscopic distant metastases, while 61% of Tmprss2+/+ TRAMP mice displayed metastases to the liver and lung. One possible mechanism for this promotion of prostate cancer metastasis by TMPRSS2 is through the HGF/c-Met signaling pathway, which has been shown to be involved in the EMT of cancer cells. TMPRSS2 can cleave pro-HGF into its active form, which then elicits an oncogenic and invasive phenotype by binding to its cognate receptor c-Met [142]. As mentioned above, TMPRSS2 is also able to activate matriptase in prostate cancer cell lines, which could also contribute to the increased invasion of prostate cancer cells [120].
2.5.4 Prostasin
Prostasin is expressed in normal prostate epithelial cells, but mRNA and protein expression decrease significantly in high-grade and invasive prostate cancer [143, 144]. In hormone-refractory metastatic prostate cancer, which progresses despite androgen deprivation therapy, transcript levels of prostasin are dramatically decreased compared to normal prostate or organ-confined prostate cancer [143]. Much like in ESCC, as mentioned above, the PRSS8 gene in PC-3 and DU-145 cell lines has a hypermethylated promoter region, which causes reduced prostasin gene expression [145]. In the invasive prostate cancer cell lines PC-3 and DU-145, restoration of prostasin expression by transfection with prostasin cDNA significantly reduced the invasiveness of both cell lines [144]. Prostasin exerts its anti-oncogenic properties in both protease-dependent and protease-independent manners [146]. Thus, the mRNA expression of uPA, uPAR, COX-2, and the inducible nitric oxide synthase (iNOS) was decreased by WT and active-site mutant prostasin in PC-3 cells [146]. Mutant prostasin reduced the protein level of EGFR upon treatment of the cells with EGF but did not affect the mRNA expression of EGFR or the expression of activated Erk2, a kinase downstream of EGFR signaling. WT prostasin reduced the protein and mRNA levels of EGFR and dramatically reduced activation of Erk2, indicating that expression of prostasin in PC-3 cells abrogates the activity of the EGFR/MAPK signaling pathway. It was further demonstrated that prostasin is involved in negatively regulating invasion of prostate cancer cells in that WT prostasin reduced the expression of Slug, an EMT protein. Furthermore, the mutant prostasin significantly upregulated the mRNA and protein expression of matriptase in PC-3 cells, indicating that both proteolytically active and inactive prostasin play a role in matriptase-mediated oncogenic signaling [146].
2.6 Endometrial and cervical cancer
2.6.1 Matriptase
Although matriptase is expressed in normal endometrium, the protein expression of matriptase is significantly higher in endometrial hyperplasia and adenocarcinoma samples [147]. Elevated matriptase levels also correlate significantly with many factors that determine disease severity, such as stage and grade of disease, invasion into the myometrium or cervix, lymph node metastases, and peritoneal cytology. Furthermore, high matriptase expression is significantly associated with poor progression-free and overall survival in patients with endometrial cancer [147].
Matriptase mRNA and protein expression were also detected in a small cohort of primary cervical adenocarcinoma and cervical SCC tumors, whereas normal cervical keratinocytes and normal cervical biopsies displayed no detectable matriptase expression [148]. Additionally, matriptase IHC staining intensity was significantly associated with the severity of cervical SCC, where low-grade squamous lesions had much lower matriptase protein expression than invasive SCC [149]. High HAI-1 and HAI-2 expression are significantly associated with better progression-free and overall survival in cervical cancer patients [150, 151]. These improved patient outcomes are likely due to HAI-1/HAI-2 inhibition of their oncogenic protease targets, including matriptase [150, 151].
2.6.2 Hepsin
Expression of hepsin is significantly increased in endometrial adenocarcinoma samples compared to endometrial hyperplasia and normal endometrium, as identified by IHC analysis [152]. This high expression also is significantly associated with a more severe disease burden, including factors such as disease stage and grade, invasion of the myometrium or cervix, and metastases to the lymph nodes and ovaries [152]. A later study contrasted some of the results of the previous study: while hepsin expression was significantly increased in endometrial cancer samples compared to endometrial hyperplasia, the expression of hepsin negatively correlated with disease grade, tumor size, and myometrial invasion [153]. The authors of the more recent study argue that hepsin may play an important role in the initial development and progression of endometrial tumors by cleavage of basement membrane proteins, but the expression decreases as the tumors become more aggressive and metastatic [153]. A possible mechanism for hepsin activity in endometrial cancer was described in a 2008 study where hepsin was stably transfected into endometrial cancer cell lines [154]. Hepsin-overexpressing cells exhibited cell cycle arrest, increased apoptosis, and abrogation of invasion in vitro, and in vivo hepsin-overexpressing tumors grew more slowly than tumors formed from WT cells [154]. El-Rebey et al. theorize that the expression of hepsin in different cancers, including endometrial, may play an oncogenic or tumor-suppressive role, depending on the phase of tumorigenesis [153]. The discrepancies in these small numbers of studies emphasize the importance of further studying the role of hepsin in the development and progression of endometrial cancer.
Hepsin is another target for inhibition by HAI-1 and HAI-2, and overexpression of HAI-1 and HAI-2 in HeLa and SiHa cervical cancer cell lines reduced hepsin protein expression and induced apoptosis [150, 151]. The idea that hepsin acts as a pro-oncogenic protease in cervical cancer is further supported by the investigation of hepsin expression in normal, paracancerous (tissue taken near the tumor), and cancerous cervical tissue. Thus, 90% of cervical tumors tested were positive for hepsin expression, while only 61% of paracancerous tissue and 10% of normal tissue stained positive for hepsin. Furthermore, the expression of hepsin positively associated with the severity of disease and negatively with patient prognosis [155].
2.6.3 HAT and HATL-5
As mentioned in Section 2.1, expression of HAT and HATL-5 is negatively associated with the development of cervical SCC, and well-differentiated tumors have significantly more HAT and HATL-5 expression than poorly differentiated tumors [55, 56]. Determining the role of these proteases in cancer awaits further investigation.
2.7 Pancreatic cancer
2.7.1 Matriptase
IHC staining of pancreatic ductal adenocarcinoma (PDAC) and normal pancreatic ductal tissue samples showed that matriptase expression is significantly increased in PDAC [156]. Active site serine protease inhibitors that selectively target matriptase reduced pro-uPA activation, c-Met phosphorylation, and invasiveness of pancreatic carcinoma cell lines, suggesting an important role of matriptase in PDAC oncogenesis [156]. HAI-1 expression has been shown to be critical for suppression of an invasive phenotype in pancreatic cancer. Thus, HAI-1 stable knock-down in the SUIT-2 pancreatic cancer cell line caused the cells to adopt an EMT-like phenotype, including reduced levels of the epithelial marker E-cadherin and increased expression of matrix metalloprotease-9 (MMP-9), a protease associated with ECM cleavage [157]. Stable knock-down of matriptase in HAI-1 knock-down SUIT-2 cells partially reversed the EMT-like phenotype of the cells, suggesting that matriptase may be acting alongside other HAI-1 targets like TMPRSS4 to enhance invasiveness of pancreatic cancer cells [157]. In vivo models using SUIT-2 and S2-CP8 pancreatic cancer cell xenografts showed that loss of HAI-1 in these cells increased the number of lung metastases, while forced overexpression of HAI-1 in S2-CP8 cells decreased the number of metastatic lesions [157, 158]. These studies demonstrate an important role for HAI-1 regulation of metastatic spreading in pancreatic cancer, mediated in part by dysregulation of matriptase expression.
2.7.2 TMPRSS4
TMPRSS4, previously known as TMPRSS3 [159], was identified in 2000 as a differentially expressed protease in pancreatic cancer samples compared to normal pancreas [160]. Northern blot analysis demonstrated that TMPRSS4 RNA was not detectable in normal pancreas tissue or in pancreatitis but was expressed in the majority of pancreatic carcinomas tested [160]. Additional studies in vitro and in vivo are needed to fully elucidate the role of TMPRSS4 in pancreatic cancer.
2.8 Testicular cancer
2.8.1 Testisin
Upon initial cloning of testisin, it was discovered that while present in normal testicular tissue, testisin expression was undetectable in testicular tumors [37]. This data was later confirmed using embryonal carcinoma cell lines from testicular tumors, as testisin transcript was not detected in any of the cell lines [161]. These initial studies led to further investigations into the potential for testisin acting as a tumor suppressor and the mechanisms by which the gene is silenced in testicular cancer. It was found that, in cell lines that do not express testisin like the embryonal carcinoma line Tera-2, the promoter CpG-rich region of the testisin gene is hypermethylated, leading to silencing of the gene [162]. Hypermethylation was also observed in primary testicular tumors, while adjacent normal testis tissue harbored mainly unmethylated testisin promoters. Stable transfection of testisin cDNA into the Tera-2 cell line suppressed tumor growth in vivo, and in vitro testisin-transfected cells formed significantly fewer colonies in a colony formation assay [162]. It was also demonstrated that testisin is capable of proteolytically cleaving PAR-2, causing downstream Ca2+ mobilization and activation of Erk1/2 and the NF-κB signaling pathway [163]. Interestingly, the cleavage of PAR-2 induces its internalization and intracellular signaling and may contribute to its degradation [163].
2.9 Hematological malignancies
2.9.1 Matriptase
Besides its important role in many different solid tumors, matriptase has also been implicated in hematological malignancies including lymphoma and leukemia. RT-PCR and western blot screening of various leukemia cell lines have shown that matriptase is expressed in B cell–derived cancer cells but was undetectable in T cell–derived leukemias [164]. In primary patient leukemia samples, matriptase protein and mRNA were detected in the majority of chronic lymphocytic leukemia (CLL) samples, whereas most acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML), and acute myeloid leukemia (AML) had low to undetectable matriptase levels [164]. Silencing of matriptase in B cell Burkitt’s lymphoma Namalwa cells caused a significant reduction in cell invasion through reconstituted basement membrane matrix, but the proliferation of the cells was unaffected [164]. Furthermore, inhibition of matriptase with a recombinant HAI-1 fragment significantly decreased invasion of Namalwa and Raji Burkitt’s lymphoma cell lines, as well as primary CLL cells. Importantly, the invasive phenotype of CML cells was unaffected by recombinant HAI-1 [164]. Matriptase expression in aggressive non-Hodgkin’s B cell lymphoma, frequently in the absence of HAI-1, was demonstrated [165]. Due to the lack of matriptase inhibition in these lymphomas, active matriptase is shed into the extracellular milieu rapidly and at high levels and can activate its oncogenic substrates pro-uPA and pro-HGF. Injection of shRNA-mediated matriptase KD cells into mice led to significant reductions in tumor growth accompanied by increased intratumoral apoptosis [165]. Although HAI-1 expression appears to be lost in most non-Hodgkin’s B cell lymphomas, it was discovered that HAI-2 is commonly co-expressed with matriptase in hematological malignancies, specifically in B cell–derived Burkitt’s lymphoma [166]. Importantly, although HAI-2 potently inhibits the catalytic activity of matriptase, a high ratio of HAI-2 to matriptase expression did not correlate with a reduction in matriptase shedding or activity in this cancer [166].
2.9.2 HATL-4
HATL-4 expression at the mRNA and protein levels was demonstrated in several AML- and CML-derived cell lines, but analysis of patient primary samples showed that HATL-4 was only expressed in AML cells, while negligible in CML, ALL, and CLL samples [167]. HATL-4 mRNA levels in AML significantly correlated with the percentage of minimal residual disease as well as poor prognosis. RNAi-mediated silencing of HATL-4 in the THP-1 AML cell line significantly reduced cell invasion, mediated by reduced activation of MMP-2 and ECM cleavage. These HATL-4 silenced cells were also inoculated into athymic nude mice, and the tumors grew significantly slower than tumors formed from WT THP-1 cells [167].
3 Targeting cell surface–anchored serine proteases in cancer
Our understanding of protease function is continuously advancing, and at the same time, protease-focused drug discovery is becoming increasingly sophisticated. Clinical trials using broad inhibition of matrix metalloprotease activity proved disappointing around the turn of the century [168]; however, new approaches that more specifically detect and/or modulate proteases hold promise to improve cancer management. For diagnostics, protease activity may be measured as a biomarker of cancer for early detection as well as monitoring therapeutic response. Many different technologies are being harnessed to develop therapeutics that can perturb or leverage protease activity for improved efficacy.
For the cell surface–anchored serine proteases, direct inhibition of protease activity by small molecules, peptides and peptidomimetics, modified macromolecular inhibitors, or antibodies is the most commonly described strategy for targeting these proteases in cancer. In more recent years, new approaches including protease-activated cellular toxins and antibody–toxin conjugates are emerging as alternatives to direct proteolytic targeting.
3.1 Matriptase inhibitors—earliest studies
Over the past 20 years, a wide variety of matriptase inhibitors have been developed and tested in cell-free systems, cell culture models, and mouse models. In 1999, macromolecular inhibitors of serine proteases of the chymotrypsin fold, ecotin and ecotin variants, were described as subnanomolar inhibitors of matriptase [169]. An additional ecotin variant (MT-6) with differential inhibitory activity toward matriptase compared to FXa, FXIIa, and plasma kallikrein was subsequently generated [170]. In 2001, bis-benzamidines were described as novel small molecule inhibitors of matriptase [171]. The lead bis-benzamidine compound inhibited matriptase and thrombin, whereas a different analog displayed a 13-fold selectivity toward matriptase [171]. The same year, the sunflower-derived trypsin inhibitor (SFTI-1) was described as a potent inhibitor of matriptase [172]. This 14-amino acid long bicyclic peptide, previously isolated from sunflower seed, is a potent natural peptide trypsin inhibitor that displayed comparable potency against matriptase (Ki = 0.92 nM) [172]. In 2004, the small molecule matriptase inhibitor CVS-3983 was used for the treatment of androgen-independent human prostate cancer xenograft models [173]. CVS-3983 contains an argininealdehyde at the P1 position, a glycine at P2, and a quaternary center at P3. Kinetic analysis revealed specific inhibition of matriptase (compared to factor Xa1, plasmin, trypsin, tPA, and uPA) with an IC50 value of 3.3 nM [173]. Xenografts in mice were established subcutaneously and CVS-3983 was administered i.p. twice daily for 7 days. A significant reduction in final mean tumor volume was observed in CVS-3983–treated compared to vehicle-treated mice. Additionally, in a cell culture assay, CVS-3983 reduced the invasion of prostate cancer cell lines through reconstituted ECM [173]. In an orthotopic xenograft model of prostate cancer, the effects of two small molecule matriptase inhibitors, analogs 8 and 59, on primary tumor growth and metastasis were determined. Mice with established PC-3 tumors received daily i.p. injection for 4 weeks [26]. The inhibitors are bis-basic secondary amides of sulfonylated 3-amidino-phenylalanine that have a high affinity for matriptase (Ki = 46 and 6.7 nM for analogs 8 and 59, respectively) and low affinity toward related trypsin-like serine proteases (factor Xa1, plasmin, trypsin, tPA, and uPA) [174]. The inhibitors significantly reduced primary tumor burden and abdominal/thoracic metastasis formation. In cell culture studies, analog 8 significantly reduced pro-HGF–induced invasion of prostate cancer and colorectal cancer cells at micromolar concentrations [81, 174]. Although CVS-3983 and analogs 8 and 59 are highly selective toward matriptase when compared to the tested secreted serine proteases, it cannot be ruled out that the inhibition of additional proteases including TTSP family members plays a role in the cell culture and in vivo models used. The selectivity of the earliest inhibitors toward matriptase compared to other TTSP family members was not assessed because the family was just emerging, and studies characterizing biochemical properties and the role of family members in normal physiology and in cancer progression were still in their early stages [7].
3.2 Matriptase activity inhibitory antibodies and toxin-conjugated antibodies
A significant challenge associated with the development of selective and potent inhibitors of serine proteases, including TTSPs, is that the target protease is frequently co-expressed with other similar proteases that differ only slightly in sequence, structure, and substrate specificity. Efforts have been made using a variety of technologies to address these challenges. One promising strategy is the generation of monoclonal antibodies (MAbs) and single-chain antibodies (single-chain variable fragment, scFv) because (1) the cell surface localization of the target protease renders it accessible to antibodies and (2) as therapeutic reagents, antibodies can be less toxic and more selective than small molecule inhibitors and other macromolecular inhibitors. Selective and potent human scFvs that directly inhibit matriptase proteolytic activity were identified by screening a phage-displayed library against matriptase [175,176,177]. The apparent Ki of the scFvs ranged from 50 pM to 129 nM. Two of the scFvs had approximately 800- and 1500-fold selectivity when tested against the most homologous serine protease, the mouse ortholog of matriptase (epithin) [178], that exhibits 86.6% sequence identity. One scFv was shown to detect denatured matriptase by western blot analysis with no cross-reactivity to other family members in HeLa or PC-3 cell lysates [177]. In follow-up studies, scFvs selective for the active form of matriptase were fluorescently labeled and used as probes for matriptase activity in cell culture and in vivo [179]. A proteolytic activity assay with several matriptase-expressing human cancer cell lines and matriptase negative control cells was used to verify that matriptase scFvs bound to and inhibited the full-length endogenous protease. Fluorescence microscopy confirmed the cell surface localization of labeled scFvs bound to matriptase. For in vivo imaging, the lead anti-matriptase antibody fragment was converted into the larger and more potent IgG with a Ki of 35 pM, and no detectable cross-reactivity with matriptase zymogen, HAI-1–bound matriptase, or epithin was detected [179]. Fluorescently labeled anti-matriptase IgG was administered via tail vein injection to mice bearing xenograft tumors that were positive or negative for matriptase. Antibodies localized to matriptase-positive tumors with minimal staining in matriptase-negative tumors or other tissues [179]. The preclinical utility of anti-matriptase IgG for the detection and quantitation of active matriptase in vivo was further investigated using the nuclear imaging modality single-photon emission computed tomography (SPECT) [180]. The radiolabeled matriptase probe showed uptake in both xenografted cell lines and patient-derived xenografts (PDX) using SPECT/X-ray computed tomography (SPECT/CT) imaging [180]. The significance of active matriptase at the protein level in cancer was further documented by IHC using anti-matriptase IgG on paraffin-embedded human tissue samples and corresponding normal tissue. Active matriptase was detected in colon adenocarcinomas of every stage in contrast to colon samples from healthy donors where active matriptase was undetectable [180].
In 2019, two antibody-based competitive inhibitors that target both the zymogen and activated forms of matriptase were generated and characterized [25]. MAbs were generated in mice immunized with a mutated variant of human matriptase in which the serine protease domain is locked in the zymogen conformation (R614A). The detectable activity of this zymogen-locked mutant was equivalent to the zymogen preparation having ∼ 3% of the activity of an activated matriptase preparation [25]. The MAbs displayed Ki values of 21 and 9 nM, respectively, and did not inhibit activity of uPA, hepatocyte growth factor activator (HGFA), or the TTSP hepsin at concentrations 20-fold higher than the inhibitory constant toward matriptase. Furthermore, no inhibition was observed toward epithin. It was proposed that these antibodies may provide an efficient way to regulate matriptase activity in preclinical applications by targeting the protease both before zymogen activation, which presumably would inhibit zymogen-mediated transactivation of other matriptase zymogens (auto-activation), and after its activation [25]. The in vivo anti-tumor activity of these inhibitory antibodies remains to be documented.
Using an alternative strategy to direct inhibition of matriptase activity, a novel matriptase antibody drug conjugate (ADC) was generated [181]. The ADC was synthesized by using the potent anti-tubulin toxin, monomethyl auristatin-E (MMAE), linked to the activated matriptase-specific monoclonal antibody (M69) via a lysosomal protease-cleavable dipeptide linker. M69 has previously been shown to bind active matriptase and matriptase in complex with HAI-1 with no cross-activity with epithin [182,183,184]. M69-MMAE displayed IC50 equivalent values ranging from 30 to 322 pM in cell lines expressing high levels of matriptase versus 758–838 pM in cell lines with low expression [181]. Anti-tumor activity of M69-MMAE was tested against human triple-negative breast cancer (TNBC) xenografts in immunocompromised mice. MDA-MB-468 and MDA-MB-231 TNBC cells were implanted subcutaneously and treatment started once tumors were palpable using twice weekly i.p. injection of M69-MMAE. The effect of the ADC was compared to treatment with the unconjugated antibody and the ADC displayed significant potent anti-tumor activity by decreasing tumor size, while the unconjugated antibody had no effect [181]. There was no evidence of toxicity as measured by mouse body weight. Anti-tumor activity of M69-MMAE against TNBC-PDX tumors was also observed. Furthermore, the ADC enhanced the anti-tumor response of cisplatin in MDA-MB-468 xenografts, which suggests that targeted therapy against matriptase in combination with conventional chemotherapy may provide treatment benefits for breast cancer patients [181]. In a different study by the same group, the M69-MMAE was tested in four matriptase-positive mantle cell lymphoma cell lines (MCL) [185]. The ADC was cytotoxic to all four cell lines and also showed a dose-dependent anti-tumor effect by reducing tumor size of an MCL cell line (JeKo-1) in subcutaneous xenografts in mice [185].
An important limitation of testing antibodies in vivo that specifically recognize the human protease and not the endogenous mouse orthologue including human/mouse matriptase in mouse models is that potential toxicity caused by matriptase inhibition or ADC-induced cell death in nontumor tissue cannot be assessed [180, 181]. It is worth noting that acute ablation of endogenous matriptase in adult mice led to severe gastrointestinal defects and their demise within 1 week [186]. In humans with mutations in the matriptase encoding ST14 gene rendering the protease inactive or retaining very low activity, no gastrointestinal symptoms were reported [187,188,189,190,191]. Therefore, it still remains of high priority to assess the safety of matriptase targeting in clinical settings.
3.3 Peptidomimetic and protein matriptase inhibitors
In 2012, a slow, tight-binding (Ki = 0.011 nM) benzothiazole-containing RQAR-peptidomimetic inhibitor of matriptase, which mimics the P1–P4 substrate recognition sequence of the enzyme, was characterized (Inhibitor 1, IN-1) [192]. IN-1 was demonstrated to be a potent matriptase inhibitor that demonstrated selectivity when compared to secreted trypsin-like proteases and the TTSPs matriptase-2, hepsin, and HAT [192]. Subsequently, it was demonstrated that IN-1 efficiently inhibits matriptase-mediated pro-HGF/c-Met activation in breast cancer cells [65, 66]. A limitation for the use of peptide-based inhibitors in vivo is that they often display short half-lives. Studies of peptidomimetics/semipeptidic TTSP inhibitors revealed that removal of the N-terminal amino group (desamino) yielded more biologically stable compounds [193]. Additionally, the replacement of natural amino acids with unnatural amino acids did not only profoundly increase their plasma stability but did also yield more potent and selective compounds [193].
Two groups have reported that the cyclic microprotein squash Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II) is an efficient matriptase inhibitor [194, 195]. Quimbar et al. used structure–activity relationships for SFTI-1 and MCoTI-II to design inhibitors with enhanced inhibitory activity. It was demonstrated that MCoTI-II is a significantly more potent matriptase inhibitor than SFTI-1 and that all alanine mutants of both peptides, generated using positional scanning mutagenesis, had decreased trypsin affinity, whereas several mutations resulted in enhanced matriptase inhibitory activity—the most potent with a Ki of 290 pM [195]. Gray et al. demonstrated that MCoTI-II is a high-affinity (Ki 9 nM) and highly selective inhibitor with a greater than 1000-fold differential in Ki values between matriptase and hepsin [194]. In cell culture assays, MCoTI-II efficiently inhibited the proteolytic activation of pro-HGF by matriptase but not by hepsin and inhibited pro-HGF–induced cell scattering and invasion of prostate cancer cells [194]. The authors point out that since matriptase and hepsin are closely related proteases with overlapping protein substrate specificity, the ability of MCoTI-II to discriminate between matriptase and hepsin is an important feature of its inhibitory activity. The use of disulfide-rich, cyclic peptides as scaffolds is emerging as a powerful approach for the design of novel drug candidates, and modified naturally occurring cyclic peptides have displayed long half-life in serum [196] as well as oral activity [197, 198]. These properties suggest that selective inhibitors of matriptase and other cell surface serine proteases, based on cyclic peptide scaffolds, might hold significant promise for the treatment of cancer. Anti-tumor properties of peptidomimetic and cyclic peptide matriptase inhibitors in vivo have to our knowledge not yet been reported.
Macromolecular matriptase inhibitors include ecotin and variants (described above in Section 3.1), eglin C, and KD1-KD2/1-Fc (engineered HAI-1) [169, 170, 199, 200]. In order to identify potent matriptase inhibitors, a cDNA library expressing variants of the protease inhibitor eglin C was screened [199]. The most potent of these, R1K4′-eglin, which had the wild-type Pro45 (P1 position) and Tyr49 (P4′ position) residues replaced with arginine and lysine, respectively, led to the production of a selective, high affinity (Ki of 4 nM), and proteolytically stable inhibitor of matriptase [199]. Combinatorial approaches based on the previous findings that the first Kunitz domain (KD1) of HAI-1 acts as a minimal matriptase-binding and inhibiting domain, whereas the second Kunitz domain (KD2) is inactive (Fig. 1b) [201, 202], were used to engineer a potent and stable matriptase inhibitor [200]. The KD2 domain was replaced with an engineered chimeric variant of KD2/KD1 domains which was fused with an antibody Fc domain to increase valency and circulating serum half-life. The final protein variant (KD1-KD2/1-Fc) contains four stoichiometric binding sites that effectively inhibit matriptase with a Ki of 70 pM and a relative selectivity for matriptase at >1000-fold over trypsin 3 and uPA, 110-fold over kallikrein 4, and 20-fold over hepsin. Furthermore, cell culture assays demonstrated efficient inhibition of pro-HGF–induced scattering of MDCK cells as well as invasion of lung and breast cell lines [200].
3.4 Triplex inhibitors of matriptase, hepsin, and HGFA
Structure–activity relationship studies based on the tetrapeptide inhibitors Ac-KQLR-ketobenzothiazole (kbt) and Ac-SKLR-kbt corresponding to the N-terminal portion of the pro-HGF and pro-MSP substrate cleavage sites for HGFA, matriptase, and hepsin were used to develop substrate-based covalent P1′ α-ketobenzothiazole mechanism-based peptide triplex inhibitors of all three proteases [203,204,205]. HGFA is a secreted protease present at high levels in the blood, and like matriptase and hepsin, HGFA efficiently activates pro-HGF and pro-MSP [206]. It was demonstrated in cell culture studies that these inhibitors caused a dose-dependent decrease of c-Met signaling in breast cancer cells and blocked pro-HGF–induced migration of prostate cancer cells [203,204,205]. Small molecule nonpeptide tetrahydropyrimidin-2(1H)-one triplex inhibitors of matriptase, hepsin, and HGFA were also developed [205, 207,208,209,210]. The SRI31215 small molecule inhibitor impaired pro-HGF–induced migration and c-Met activation in prostate cancer cells. Additionally, SRI31215 reduced primary resistance to cetuximab and gefitinib in pro-HGF–producing colon cancer cells, blocked crosstalk between c-MET–amplified nonsmall cell lung cancer (NSCLC) cells and pro-HGF–secreting fibroblasts, and perturbed fibroblast-mediated resistance to c-Met kinase inhibition in NSCLC cells [208, 209]. The authors argue that cancer cells commonly overexpress a combination of pro-HGF–activating proteases which makes triplex inhibitors efficient in blocking activation of pro-HGF in cancer cells that display expression/activation of multiple proteases. However, they recently also developed new, smaller molecular weight dipeptides that are selective inhibitors of matriptase or hepsin, in order study their individual roles in cell biology [73] (see below in Section 3.5). Studies on in vivo stability and anti-tumor efficacy of triplex protease inhibitors have not yet been published.
3.5 Hepsin inhibitors
In addition to the triplex matriptase, hepsin, and HGFA inhibitors described above, selective inhibitors of hepsin and matriptase which contain a P1 arginine (R) kbt warhead were recently developed and used to study protease function in cell junction integrity in breast epithelial cells [73, 205]. Damalanka et al. demonstrated that the selective hepsin inhibitor 6k inhibited hepsin-mediated desmosome degradation and loss of the desmosomal cadherin desmoglein 2 in an immortalized mammary epithelial cell line with transgenic overexpression of hepsin. The matriptase selective inhibitor had no effect on desmosome integrity in this cellular model [73]. Based on studies by Han et al. describing the tetrapeptide hepsin inhibitor Ac-KQLR-ketothiazole (kt) [204], the minimal structural requirements for hepsin inhibitory activity by truncating amino acids at the N terminus were further investigated by Kwon et al. [211]. It was demonstrated that the dipeptide analog Ac-LR-kbt (15) exhibited strong inhibition of hepsin activity with Ki values of 3.38 and 2.91 nM for the (S)-epimer and the (R)-epimer, respectively. Furthermore, both 15S and 15R showed selectivity for hepsin over matriptase by > 60-fold [211]. In a 2019 follow-up paper, the dipeptide-derived hepsin inhibitors were conjugated to near-infrared (NIR) optical dyes in order to generate imaging probes [212]. The fluorophores were separated from the hepsin-binding moiety by a polyethyleneglycol (PEG) linker, to decrease steric hindrance. The Leu-Arg dipeptides attached to borondipyrromethene (BODIPY) or SulfoCy7 exhibited strong hepsin binding affinities with Ki values of 21 and 22 nM, respectively, and high selectivity over matriptase [212]. In cell uptake studies, SulfoCy7-conjugated inhibitor exhibited selective targeting and retention in hepsin-overexpressing prostate cancer cells [212].
3.6 Hepsin inhibitor in vivo studies
In vivo prostate cancer treatment studies were performed using the small molecule hepsin inhibitor HepIn-13 [133]. The IC50 value of HepIn-13 was determined to be 0.33 μM in a chromogenic peptide assay. HepIn-13 is orally available and could be detected in serum after the mice were exposed to soft rodent chow containing HepIn-13 at concentrations that were significantly higher than its IC50 [133]. During 5 weeks of treatment, mice did not show detectable liver toxicity or any other signs of adverse effects. To determine the effect of HepIn-13 on prostate cancer progression in vivo, animal studies using the LPB-Tag/PB-Hepsin mice (described above in Section 2.5), which consistently show bone metastasis, were performed. Starting at 10 weeks of age, when the animals presented with low-grade prostate tumors, mice received HepIn-13 in the chow for 3 weeks. At this time, HepIn-13–treated mice displayed no significant reduction in primary tumor size; however, a significant reduction in bone, liver, and lung metastasis compared to control mice was observed. The authors cautioned that while HepIn-13 is a poor inhibitor of matriptase, potential inhibitory effects on other proteases cannot be ruled out [133].
A HAI-1 protein-based hepsin inhibitor was also tested in vivo in an orthotopic prostate cancer model using LnCap-34 cells with stable hepsin overexpression [130]. Recombinant HAI-1 KD1 domain was used because it by itself is a significantly better hepsin inhibitor than the entire HAI-1 extracellular domain [213]. The small molecular mass of the KD1 domain (∼ 6 versus 58 kDa for HAI-1) confers a short circulating half-life of 20 min, which limits its therapeutic efficacy. Chemical conjugation of KD1 to PEG showed significant extension in serum half-life (up to 96 h) [130]. Daily i.p. administration of KD1-PEG to mice with established tumors for up to 14 weeks significantly decreased contralateral prostate invasion and lymph node metastasis. Moreover, serum PSA level remained reduced during the entire treatment period [130]. A caveat with using KD1-PEG as hepsin inhibitor in tumor models is that KD1 targets several other serine proteases including matriptase and HGFA.
3.7 Hepsin antibody inhibitors
In addition to small molecule and HAI-1 KD1 inhibitors, the development of several antibodies inhibiting hepsin activity has been reported. In 2006, MAbs against proteolytically active hepsin were generated in hepsin knock-out mice [214]. Three MAbs were shown to inhibit hepsin proteolytic activity and to impair invasion of prostate and ovarian cancer cells in vitro [214]. In 2012, allosteric anti-hepsin antibodies were reported by two different groups [215, 216]. The anti-hepsin antibody Fab25 was identified by Ganesan et al. by screening of a Fab phage display library where hepsin with its S1 pocket occupied by 3,4-dichloro-isocoumarin was used as the “bait” [215]. Fab25 inhibited human and mouse hepsin enzymatic activity with IC50 values of 4.1 and 329 nM, respectively, and was highly specific with no inhibitory effect toward nine different serine proteases including the cell surface–anchored proteases matriptase and prostasin. Biochemical and enzymatic studies with synthetic substrates of variable length suggested that Fab25 acts as an allosteric inhibitor based on noncompetitive inhibition kinetics. In cell culture assays, Fab25 blocked DU145 prostate cancer cell migration in response to hepsin-mediated cleavage of Ln-332 [215]. In another study published the same year by Koschubs et al., a monoclonal humanized antibody hH35 that selectively inhibited hepsin activity (over matriptase, HAT, endopeptidase, and trypsin) was generated [216]. Kinetic characterization revealed nonlinear, slow, and tight-binding inhibition. This correlated with the crystal structure obtained for the human hepsin-hH35 antibody Fab fragment complex, which showed that the antibody binds hepsin around α3-helix, located far from the active center [216]. The authors state that endogenous levels of hepsin in tumor cell lines were below the detection level in flow cytometry or immunocytochemistry analysis; however, cell surface hepsin was detected by hH35 in HEK293 cells stably overexpressing full-length hepsin [216]. The anti-tumorigenic effects of hH35 in cultured cancer cells or in vivo were not reported.
3.8 Prostasin inhibitors
In 2008, structure-based design was utilized to guide the early stage optimization of a substrate-like inhibitor to afford potent peptidomimetic inhibitors of prostasin [217]. The first X-ray crystal structures of prostasin with small molecule inhibitors bound to the active site were also reported. Guided by the X-ray structure, the peptidomimetic scaffold was optimized to generate potent, reversible, and low nanomolar Ki inhibitors of prostasin [217]. Reports on prostasin inhibitors used in cancer cell culture studies or in vivo are not available.
3.9 Testisin-mediated proteolytic activation of bacterial toxin
Modified bacterial toxins are biologically inspired technologies that are being leveraged for novel tumor treatment strategies. Anthrax toxin is a three-component toxin of which the protective antigen (PrAg) component is cleaved at the cell surface by furin-like proteases, and this cleavage is absolutely required for the subsequent steps in toxin action [218]. The unique requirement that PrAg be activated on the target cell surface provides an opportunity to engineer this protein to make its activation dependent on specific tumor cell surface proteases. It was first demonstrated that PrAg can be made specific for MMP or uPA-expressing cells by replacing the furin site with MMP or uPA preferred sites [219, 220]. Later, a dual MMP/uPA-activated toxin was generated which was highly effective in vivo with low toxicity in a preclinical model of melanoma [221, 222]. This technology was also utilized to develop a toxin targeting testisin-expressing tumor cells [105]. The native activation sequence of PrAG was mutated to a sequence derived from protein C inhibitor (PCI), that can be cleaved by membrane-anchored serine proteases, to generate the mutant protein PrAg-PCIS. PrAg-PCIS was cytotoxic to multiple human tumor cell lines when combined with FP59, a chimeric anthrax toxin lethal factor-Pseudomonas exotoxin fusion protein [105]. PrAg-PCIS toxin was activated by both recombinant mouse testisin and endogenous cell surface human testisin, and the toxin inhibited the growth of the testisin-expressing cancer cells in culture. Furthermore, in a subcutaneous xenograft model using HeLa cells, PrAg-PCIS treatment led to a significant reduction in primary tumor size and was well-tolerated by the mice with no obvious adverse effects [105]. It should be noted that while the engineered cleavage site in PrAg-PCIS showed a preference for activation cleavage by testisin in a cell-free assay, it also was cleaved by the recombinant catalytic domains of hepsin and matriptase. PrAg-PCIS was also cleaved by hepsin on the cell surface of HeLa cells transfected with full-length WT hepsin and not by a catalytically inactive mutant of hepsin. In the same model, matriptase was ineffective at activating PrAg-PCIS toxin when expressed as a full-length protein on the cell surface. Taken together, it is possible that testisin-activated toxin may be activated by additional pericellular serine proteases in vivo. The low toxicity which could be assessed in the mice because endogenous proteases cleave PrAg-PCIS makes cell surface serine protease–activated toxins a potentially promising strategy for targeted cancer therapy.
4 Conclusions
Cell surface–anchored serine proteases represent promising targets for cancer diagnostic imaging and therapeutic intervention. Although no specific inhibitors are yet in clinical trials, multiple studies on the development of novel activity probes and inhibitors are currently ongoing in laboratories across the world. It is the hope that the combination of basic science and drug discovery will provide a stepping stone for future clinical applications and ultimately lead to better outcomes for cancer patients.
Abbreviations
- ADC :
-
antibody drug conjugate
- ALL :
-
acute lymphocytic leukemia
- AML :
-
acute myeloid leukemia
- APC :
-
adenomatous polyposis coli
- AR :
-
androgen receptor
- ARIH :
-
autosomal recessive icthyosis with hypotrichosis
- BODIPY :
-
borondipyrromethene
- CAF :
-
cancer-associated fibroblast
- CAP1 :
-
channel-activating protease-1
- CLL :
-
chronic lymphocytic leukemia
- CML :
-
chronic myeloid leukemia
- COX-2 :
-
cyclooxygenase-2
- CRC :
-
colorectal cancer
- CT :
-
computed tomography
- DESC :
-
differentially expressed in squamous cell carcinoma
- DMBA :
-
dimethylbenzanthracene
- DHT :
-
dihydrotestosterone
- ECM :
-
extracellular matrix
- ECRG :
-
esophageal cancer-related gene
- EGFR :
-
epidermal growth factor receptor
- EMT :
-
epithelial to mesenchymal transition
- EOC :
-
epithelial ovarian cancer
- ERG :
-
estrogen-regulated gene
- ERK :
-
extracellular signal-regulated kinase
- ESCC :
-
esophageal squamous cell carcinoma
- ETS :
-
erythroblast transformation specific
- FAP :
-
fibroblast activation protein
- GnT-V :
-
beta1,6-n-acetylglucosaminyltransferase-V
- GPI :
-
glycosyl-phosphatidylinositol
- HAI-1 :
-
hepatocyte growth factor inhibitor-1
- HAI-2 :
-
hepatocyte growth factor inhibitor-2
- HAT :
-
human airway trypsin-like
- HGF :
-
hepatocyte growth factor
- HGFA :
-
hepatocyte growth factor activator
- HIF-1a :
-
hypoxia-inducible factor 1-alpha
- HNSCC :
-
head and neck squamous cell carcinoma
- IHC :
-
immunohistochemistry
- IN-1 :
-
matriptase inhibitor-1
- K5 :
-
keratin-5
- K14 :
-
keratin-14
- kbt :
-
ketobenzothiazole
- KD :
-
knock-down
- KD1-PEG :
-
PEGylated form of the Kunitz domain-1
- KO :
-
knock-out
- kt :
-
ketothiazole
- Ln-332 :
-
laminin-332
- LPB :
-
large probasin
- LMP :
-
low malignant potential
- Mab :
-
monoclonal antibody
- MAPK :
-
mitogen-activated protein kinase
- MCL :
-
mantle cell lymphoma
- MCOTI-II :
-
Momordica cochinchinensis trypsin inhibitor-II
- MMAE :
-
monomethyl auristatin-E
- MMP :
-
matrix metalloprotease
- MMTV :
-
mouse mammary tumor virus
- MSP :
-
macrophage stimulating protein
- mTor :
-
mammalian/mechanistic target of rapamycin
- NF :
-
nuclear factor
- NIR :
-
near-infrared
- NPC :
-
nasopharyngeal carcinoma
- NSCLC :
-
nonsmall cell lung cancer
- OSCC :
-
oral squamous cell carcinoma
- PAR-2 :
-
proteinase-activated receptor-2
- PB :
-
probasin
- PCI :
-
protein C inhibitor
- PDAC :
-
pancreatic ductal adenocarcinoma
- PDX :
-
patient-derived xenograft
- PDGF :
-
platelet-derived growth factor
- PDGFR :
-
platelet-derived growth factor receptor
- PEG :
-
polyethylene glycol
- PGE 2 :
-
prostaglandin E2
- PI3K :
-
Phosphoinositide 3-kinase
- PIN :
-
prostatic intraepithelial neoplasia
- PN-1 :
-
protease nextin-1
- PrAg :
-
protective antigen
- PSA :
-
prostate-specific antigen
- PymT :
-
polyomavirus middle T
- RON :
-
Recepteur d’Origine Nantais
- S1p :
-
sphingosine-1-phosphate
- SAM :
-
S-adenosyl-l-methionine
- SAR :
-
structure–activity relationship
- SCC :
-
squamous cell carcinoma
- SCID :
-
severe combined immunodeficiency
- scFv :
-
single-chain variable fragment
- SFTI-1 :
-
unflower-derived trypsin inhibitor
- SNP :
-
single nucleotide polymorphism
- SPECT :
-
single-photon emission computed tomography
- Sphk1 :
-
sphingosine kinase 1
- STAT3 :
-
signal transducer and activator of transcription 3
- Tag :
-
T antigen
- TMPRSS :
-
transmembrane protease, serine
- TNBC :
-
triple-negative breast cancer
- TRAMP :
-
transgenic adenocarcinoma of the mouse prostate
- TTSP :
-
type II transmembrane serine protease
- UPA :
-
urokinase type plasminogen activator
- uPAR :
-
urokinase-type plasminogen activator receptor
- WT :
-
wild-type
References
Puente, X. S., Sanchez, L. M., Gutierrez-Fernandez, A., Velasco, G., & Lopez-Otin, C. (2005). A genomic view of the complexity of mammalian proteolytic systems. Biochemical Society Transactions, 33(Pt 2), 331–334. https://doi.org/10.1042/bst0330331.
Caughey, G. H. (2007). Mast cell tryptases and chymases in inflammation and host defense. Immunological Reviews, 217, 141–154. https://doi.org/10.1111/j.1600-065X.2007.00509.x.
Netzel-Arnett, S., Hooper, J. D., Szabo, R., Madison, E. L., Quigley, J. P., Bugge, T. H., et al. (2003). Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Reviews, 22(2–3), 237–258.
Busek, P., Mateu, R., Zubal, M., Kotackova, L., & Sedo, A. (2018). Targeting fibroblast activation protein in cancer—prospects and caveats. Frontiers in Bioscience (Landmark Ed), 23, 1933–1968.
Zi, F., He, J., He, D., Li, Y., Yang, L., & Cai, Z. (2015). Fibroblast activation protein alpha in tumor microenvironment: recent progression and implications (review). Molecular Medicine Reports, 11(5), 3203–3211. https://doi.org/10.3892/mmr.2015.3197.
Antalis, T. M., Buzza, M. S., Hodge, K. M., Hooper, J. D., & Netzel-Arnett, S. (2010). The cutting edge: membrane-anchored serine protease activities in the pericellular microenvironment. The Biochemical Journal, 428(3), 325–346. https://doi.org/10.1042/BJ20100046.
Hooper, J. D., Clements, J. A., Quigley, J. P., & Antalis, T. M. (2001). Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. The Journal of Biological Chemistry, 276(2), 857–860. https://doi.org/10.1074/jbc.R000020200.
Bugge, T. H., Antalis, T. M., & Wu, Q. (2009). Type II transmembrane serine proteases. The Journal of Biological Chemistry, 284(35), 23177–23181. https://doi.org/10.1074/jbc.R109.021006.
Bugge, T. H., List, K., & Szabo, R. (2007). Matriptase-dependent cell surface proteolysis in epithelial development and pathogenesis. Frontiers in Bioscience, 12, 5060–5070.
Szabo, R., & Bugge, T. H. (2008). Type II transmembrane serine proteases in development and disease. The International Journal of Biochemistry & Cell Biology, 40(6–7), 1297–1316. https://doi.org/10.1016/j.biocel.2007.11.013.
Szabo, R., & Bugge, T. H. (2011). Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annual Review of Cell and Developmental Biology, 27, 213–235. https://doi.org/10.1146/annurev-cellbio-092910-154247.
Murray, A. S., Varela, F. A., & List, K. (2016). Type II transmembrane serine proteases as potential targets for cancer therapy. Biological Chemistry, 397(9), 815–826. https://doi.org/10.1515/hsz-2016-0131.
Tanabe, L. M., & List, K. (2017). The role of type II transmembrane serine protease-mediated signaling in cancer. The FEBS Journal, 284(10), 1421–1436. https://doi.org/10.1111/febs.13971.
Antalis, T. M., Bugge, T. H., & Wu, Q. (2011). Membrane-anchored serine proteases in health and disease. Progress in Molecular Biology and Translational Science, 99, 1–50. https://doi.org/10.1016/b978-0-12-385504-6.00001-4.
Takeuchi, T., Harris, J. L., Huang, W., Yan, K. W., Coughlin, S. R., & Craik, C. S. (2000). Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. The Journal of Biological Chemistry, 275(34), 26333–26342. https://doi.org/10.1074/jbc.M002941200.
Jiang, J., Yang, J., Feng, P., Zuo, B., Dong, N., Wu, Q., et al. (2014). N-glycosylation is required for matriptase-2 autoactivation and ectodomain shedding. The Journal of Biological Chemistry, 289(28), 19500–19507. https://doi.org/10.1074/jbc.M114.555110.
Qiu, D., Owen, K., Gray, K., Bass, R., & Ellis, V. (2007). Roles and regulation of membrane-associated serine proteases. Biochemical Society Transactions, 35(Pt 3), 583–587. https://doi.org/10.1042/bst0350583.
Afar, D. E., Vivanco, I., Hubert, R. S., Kuo, J., Chen, E., Saffran, D. C., et al. (2001). Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Research, 61(4), 1686–1692.
Guipponi, M., Vuagniaux, G., Wattenhofer, M., Shibuya, K., Vazquez, M., Dougherty, L., et al. (2002). The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Human Molecular Genetics, 11(23), 2829–2836. https://doi.org/10.1093/hmg/11.23.2829.
Andreasen, D., Vuagniaux, G., Fowler-Jaeger, N., Hummler, E., & Rossier, B. C. (2006). Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in Xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. Journal of the American Society of Nephrology, 17(4), 968–976. https://doi.org/10.1681/asn.2005060637.
Murray, A. S., Varela, F. A., Hyland, T. E., Schoenbeck, A. J., White, J. M., Tanabe, L. M., et al. (2017). Phosphorylation of the type II transmembrane serine protease, TMPRSS13, in hepatocyte growth factor activator inhibitor-1 and -2-mediated cell-surface localization. The Journal of Biological Chemistry, 292(36), 14867–14884. https://doi.org/10.1074/jbc.M117.775999.
Oberst, M. D., Williams, C. A., Dickson, R. B., Johnson, M. D., & Lin, C. Y. (2003). The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. The Journal of Biological Chemistry, 278(29), 26773–26779. https://doi.org/10.1074/jbc.M304282200.
Tseng, I. C., Xu, H., Chou, F. P., Li, G., Vazzano, A. P., Kao, J. P., et al. (2010). Matriptase activation, an early cellular response to acidosis. The Journal of Biological Chemistry, 285(5), 3261–3270. https://doi.org/10.1074/jbc.M109.055640.
Godiksen, S., Soendergaard, C., Friis, S., Jensen, J. K., Bornholdt, J., Sales, K. U., et al. (2013). Detection of active matriptase using a biotinylated chloromethyl ketone peptide. PLoS One, 8(10), e77146. https://doi.org/10.1371/journal.pone.0077146.
Tamberg, T., Hong, Z., De Schepper, D., Skovbjerg, S., Dupont, D. M., Vitved, L., et al. (2019). Blocking the proteolytic activity of zymogen matriptase with antibody-based inhibitors. The Journal of Biological Chemistry, 294(1), 314–326. https://doi.org/10.1074/jbc.RA118.004126.
Inouye, K., Yasumoto, M., Tsuzuki, S., Mochida, S., & Fushiki, T. (2010). The optimal activity of a pseudozymogen form of recombinant matriptase under the mildly acidic pH and low ionic strength conditions. Journal of Biochemistry, 147(4), 485–492. https://doi.org/10.1093/jb/mvp190.
Friis, S., Tadeo, D., Le-Gall, S. M., Jurgensen, H. J., Sales, K. U., Camerer, E., et al. (2017). Matriptase zymogen supports epithelial development, homeostasis and regeneration. BMC Biology, 15(1), 46. https://doi.org/10.1186/s12915-017-0384-4.
Kawaguchi, T., Qin, L., Shimomura, T., Kondo, J., Matsumoto, K., Denda, K., et al. (1997). Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. The Journal of Biological Chemistry, 272(44), 27558–27564. https://doi.org/10.1074/jbc.272.44.27558.
Shimomura, T., Denda, K., Kitamura, A., Kawaguchi, T., Kito, M., Kondo, J., et al. (1997). Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. The Journal of Biological Chemistry, 272(10), 6370–6376.
Kataoka, H., Kawaguchi, M., Fukushima, T., & Shimomura, T. (2018). Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): emerging key players in epithelial integrity and cancer. Pathology International, 68(3), 145–158. https://doi.org/10.1111/pin.12647.
Yu, J. X., Chao, L., & Chao, J. (1994). Prostasin is a novel human serine proteinase from seminal fluid. Purification, tissue distribution, and localization in prostate gland. The Journal of Biological Chemistry, 269(29), 18843–18848.
Chen, L. M., Skinner, M. L., Kauffman, S. W., Chao, J., Chao, L., Thaler, C. D., et al. (2001). Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. The Journal of Biological Chemistry, 276(24), 21434–21442. https://doi.org/10.1074/jbc.M011423200.
Szabo, R., Hobson, J. P., List, K., Molinolo, A., Lin, C. Y., & Bugge, T. H. (2008). Potent inhibition and global co-localization implicate the transmembrane Kunitz-type serine protease inhibitor hepatocyte growth factor activator inhibitor-2 in the regulation of epithelial matriptase activity. The Journal of Biological Chemistry, 283(43), 29495–29504. https://doi.org/10.1074/jbc.M801970200.
Chen, Y. W., Wang, J. K., Chou, F. P., Chen, C. Y., Rorke, E. A., Chen, L. M., et al. (2010). Regulation of the matriptase-prostasin cell surface proteolytic cascade by hepatocyte growth factor activator inhibitor-1 during epidermal differentiation. The Journal of Biological Chemistry, 285(41), 31755–31762. https://doi.org/10.1074/jbc.M110.150367.
Fan, B., Wu, T. D., Li, W., & Kirchhofer, D. (2005). Identification of hepatocyte growth factor activator inhibitor-1B as a potential physiological inhibitor of prostasin. The Journal of Biological Chemistry, 280(41), 34513–34520. https://doi.org/10.1074/jbc.M502119200.
Inoue, M., Kanbe, N., Kurosawa, M., & Kido, H. (1998). Cloning and tissue distribution of a novel serine protease esp-1 from human eosinophils. Biochemical and Biophysical Research Communications, 252(2), 307–312. https://doi.org/10.1006/bbrc.1998.9645.
Hooper, J. D., Nicol, D. L., Dickinson, J. L., Eyre, H. J., Scarman, A. L., Normyle, J. F., et al. (1999). Testisin, a new human serine proteinase expressed by premeiotic testicular germ cells and lost in testicular germ cell tumors. Cancer Research, 59(13), 3199–3205.
Tang, T., Kmet, M., Corral, L., Vartanian, S., Tobler, A., & Papkoff, J. (2005). Testisin, a glycosyl-phosphatidylinositol-linked serine protease, promotes malignant transformation in vitro and in vivo. Cancer Research, 65(3), 868–878.
Kempkensteffen, C., Christoph, F., Weikert, S., Krause, H., Kollermann, J., Schostak, M., et al. (2006). Epigenetic silencing of the putative tumor suppressor gene testisin in testicular germ cell tumors. Journal of Cancer Research and Clinical Oncology, 132(12), 765–770. https://doi.org/10.1007/s00432-006-0124-6.
Manton, K. J., Douglas, M. L., Netzel-Arnett, S., Fitzpatrick, D. R., Nicol, D. L., Boyd, A. W., et al. (2015). Hypermethylation of the 5’ CpG island of the gene encoding the serine protease Testisin promotes its loss in testicular tumorigenesis. British Journal of Cancer, 113(11), 1640. https://doi.org/10.1038/bjc.2015.384.
List, K., Szabo, R., Molinolo, A., Sriuranpong, V., Redeye, V., Murdock, T., et al. (2005). Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes & Development, 19(16), 1934–1950. https://doi.org/10.1101/gad.1300705.
Bocheva, G., Rattenholl, A., Kempkes, C., Goerge, T., Lin, C. Y., D'Andrea, M. R., et al. (2009). Role of matriptase and proteinase-activated receptor-2 in nonmelanoma skin cancer. The Journal of Investigative Dermatology, 129(7), 1816–1823. https://doi.org/10.1038/jid.2008.449.
Sales, K. U., Friis, S., Abusleme, L., Moutsopoulos, N. M., & Bugge, T. H. (2015). Matriptase promotes inflammatory cell accumulation and progression of established epidermal tumors. Oncogene, 34(35), 4664–4672. https://doi.org/10.1038/onc.2014.391.
Sales, K. U., Friis, S., Konkel, J. E., Godiksen, S., Hatakeyama, M., Hansen, K. K., et al. (2015). Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene, 34(3), 346–356. https://doi.org/10.1038/onc.2013.563.
Szabo, R., Rasmussen, A. L., Moyer, A. B., Kosa, P., Schafer, J. M., Molinolo, A. A., et al. (2011). c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene, 30(17), 2003–2016. https://doi.org/10.1038/onc.2010.586.
Baba, T., Kawaguchi, M., Fukushima, T., Sato, Y., Orikawa, H., Yorita, K., et al. (2012). Loss of membrane-bound serine protease inhibitor HAI-1 induces oral squamous cell carcinoma cells’ invasiveness. The Journal of Pathology, 228(2), 181–192. https://doi.org/10.1002/path.3993.
Yamamoto, K., Kawaguchi, M., Shimomura, T., Izumi, A., Konari, K., Honda, A., et al. (2018). Hepatocyte growth factor activator inhibitor type-2 (HAI-2)/SPINT2 contributes to invasive growth of oral squamous cell carcinoma cells. Oncotarget, 9(14), 11691–11706. https://doi.org/10.18632/oncotarget.24450.
Friis, S., Uzzun Sales, K., Godiksen, S., Peters, D. E., Lin, C. Y., Vogel, L. K., et al. (2013). A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. The Journal of Biological Chemistry, 288(26), 19028–19039. https://doi.org/10.1074/jbc.M113.469932.
Kanemaru, A., Yamamoto, K., Kawaguchi, M., Fukushima, T., Lin, C. Y., Johnson, M. D., et al. (2017). Deregulated matriptase activity in oral squamous cell carcinoma promotes the infiltration of cancer-associated fibroblasts by paracrine activation of protease-activated receptor 2. International Journal of Cancer, 140(1), 130–141. https://doi.org/10.1002/ijc.30426.
Cheng, M. F., Huang, M. S., Lin, C. S., Lin, L. H., Lee, H. S., Jiang, J. C., et al. (2014). Expression of matriptase correlates with tumour progression and clinical prognosis in oral squamous cell carcinoma. Histopathology, 65(1), 24–34. https://doi.org/10.1111/his.12361.
Lang, J. C., & Schuller, D. E. (2001). Differential expression of a novel serine protease homologue in squamous cell carcinoma of the head and neck. British Journal of Cancer, 84(2), 237–243. https://doi.org/10.1054/bjoc.2000.1586.
Sedghizadeh, P. P., Mallery, S. R., Thompson, S. J., Kresty, L., Beck, F. M., Parkinson, E. K., et al. (2006). Expression of the serine protease DESC1 correlates directly with normal keratinocyte differentiation and inversely with head and neck squamous cell carcinoma progression. Head & Neck, 28(5), 432–440. https://doi.org/10.1002/hed.20346.
Ng, H. Y., Ko, J. M., Yu, V. Z., Ip, J. C., Dai, W., Cal, S., et al. (2016). DESC1, a novel tumor suppressor, sensitizes cells to apoptosis by downregulating the EGFR/AKT pathway in esophageal squamous cell carcinoma. International Journal of Cancer, 138(12), 2940–2951. https://doi.org/10.1002/ijc.30034.
Chang, Z.-W., Jia, Y.-X., Zhang, W.-J., Song, L.-J., Gao, M., Li, M.-J., et al. (2018). LncRNA-TUSC7/miR-224 affected chemotherapy resistance of esophageal squamous cell carcinoma by competitively regulating DESC1. Journal of Experimental & Clinical Cancer Research: CR, 37(1), 56–56. https://doi.org/10.1186/s13046-018-0724-4.
Duhaime, M. J., Page, K. O., Varela, F. A., Murray, A. S., Silverman, M. E., Zoratti, G. L., et al. (2016). Cell surface human airway trypsin-like protease is lost during squamous cell carcinogenesis. Journal of Cellular Physiology, 231(7), 1476–1483. https://doi.org/10.1002/jcp.25173.
Miller, G. S., Zoratti, G. L., Murray, A. S., Bergum, C., Tanabe, L. M., & List, K. (2014). HATL5: a cell surface serine protease differentially expressed in epithelial cancers. PLoS One, 9(2), e87675. https://doi.org/10.1371/journal.pone.0087675.
Wang, J. Y., Jin, X., & Li, X. F. (2018). Knockdown of TMPRSS3, a transmembrane serine protease, inhibits proliferation, migration, and invasion in human nasopharyngeal carcinoma cells. Oncology Research, 26(1), 95–101. https://doi.org/10.3727/096504017x14920318811695.
Bao, Y., Wang, Q., Guo, Y., Chen, Z., Li, K., Yang, Y., et al. (2016). PRSS8 methylation and its significance in esophageal squamous cell carcinoma. Oncotarget, 7(19), 28540–28555. https://doi.org/10.18632/oncotarget.8677.
Lin, C. Y., Wang, J. K., Torri, J., Dou, L., Sang, Q. A., & Dickson, R. B. (1997). Characterization of a novel, membrane-bound, 80-kDa matrix-degrading protease from human breast cancer cells. Monoclonal antibody production, isolation, and localization. The Journal of Biological Chemistry, 272(14), 9147–9152.
Jin, J. S., Cheng, T. F., Tsai, W. C., Sheu, L. F., Chiang, H., & Yu, C. P. (2007). Expression of the serine protease, matriptase, in breast ductal carcinoma of Chinese women: correlation with clinicopathological parameters. Histology and Histopathology, 22(3), 305–309.
Kang, J. Y., Dolled-Filhart, M., Ocal, I. T., Singh, B., Lin, C. Y., Dickson, R. B., et al. (2003). Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Research, 63(5), 1101–1105.
Oberst, M., Anders, J., Xie, B., Singh, B., Ossandon, M., Johnson, M., et al. (2001). Matriptase and HAI-1 are expressed by normal and malignant epithelial cells in vitro and in vivo. The American Journal of Pathology, 158(4), 1301–1311. https://doi.org/10.1016/S0002-9440(10)64081-3.
Siddiqui, S. F., Pawelek, J., Handerson, T., Lin, C. Y., Dickson, R. B., Rimm, D. L., et al. (2005). Coexpression of beta1,6-N-acetylglucosaminyltransferase V glycoprotein substrates defines aggressive breast cancers with poor outcome. Cancer Epidemiology, Biomarkers & Prevention, 14(11 Pt 1), 2517–2523. https://doi.org/10.1158/1055-9965.epi-05-0464.
Parr, C., Watkins, G., Mansel, R. E., & Jiang, W. G. (2004). The hepatocyte growth factor regulatory factors in human breast cancer. Clinical Cancer Research, 10(1 Pt 1), 202–211.
Zoratti, G. L., Tanabe, L. M., Varela, F. A., Murray, A. S., Bergum, C., Colombo, E., et al. (2015). Targeting matriptase in breast cancer abrogates tumour progression via impairment of stromal-epithelial growth factor signalling. Nature Communications, 6, 6776. https://doi.org/10.1038/ncomms7776.
Zoratti, G. L., Tanabe, L. M., Hyland, T. E., Duhaime, M. J., Colombo, E., Leduc, R., et al. (2016). Matriptase regulates c-Met mediated proliferation and invasion in inflammatory breast cancer. Oncotarget, 7(36), 58162–58173. https://doi.org/10.18632/oncotarget.11262.
Hurst, N. J., Jr., Najy, A. J., Ustach, C. V., Movilla, L., & Kim, H. R. (2012). Platelet-derived growth factor-C (PDGF-C) activation by serine proteases: implications for breast cancer progression. The Biochemical Journal, 441(3), 909–918. https://doi.org/10.1042/bj20111020.
Parr, C., Sanders, A. J., Davies, G., Martin, T., Lane, J., Mason, M. D., et al. (2007). Matriptase-2 inhibits breast tumor growth and invasion and correlates with favorable prognosis for breast cancer patients. Clinical Cancer Research, 13(12), 3568–3576. https://doi.org/10.1158/1078-0432.ccr-06-2357.
Tuhkanen, H., Hartikainen, J. M., Soini, Y., Velasco, G., Sironen, R., Nykopp, T. K., et al. (2013). Matriptase-2 gene (TMPRSS6) variants associate with breast cancer survival, and reduced expression is related to triple-negative breast cancer. International Journal of Cancer, 133(10), 2334–2340. https://doi.org/10.1002/ijc.28254.
Xing, P., Li, J. G., Jin, F., Zhao, T. T., Liu, Q., Dong, H. T., et al. (2011). Clinical and biological significance of hepsin overexpression in breast cancer. Journal of Investigative Medicine, 59(5), 803–810. https://doi.org/10.2310/JIM.0b013e31821451a1.
Tervonen, T. A., Belitškin, D., Pant, S. M., Englund, J. I., Marques, E., Ala-Hongisto, H., et al. (2016). Deregulated hepsin protease activity confers oncogenicity by concomitantly augmenting HGF/MET signalling and disrupting epithelial cohesion. Oncogene, 35(14), 1832–1846. https://doi.org/10.1038/onc.2015.248.
Pant, S. M., Belitskin, D., Ala-Hongisto, H., Klefstrom, J., & Tervonen, T. A. (2018). Analyzing the type II transmembrane serine protease hepsin-dependent basement membrane remodeling in 3D cell culture. Methods in Molecular Biology, 1731, 169–178. https://doi.org/10.1007/978-1-4939-7595-2_16.
Damalanka, V. C., Han, Z., Karmakar, P., O'Donoghue, A. J., La Greca, F., Kim, T., et al. (2019). Discovery of selective matriptase and hepsin serine protease inhibitors: useful chemical tools for cancer cell biology. Journal of Medicinal Chemistry, 62(2), 480–490. https://doi.org/10.1021/acs.jmedchem.8b01536.
Rui, X., Li, Y., Jin, F., & Li, F. (2015). TMPRSS3 is a novel poor prognostic factor for breast cancer. International Journal of Clinical and Experimental Pathology, 8(5), 5435–5442.
Luo, P., Lu, G., Fan, L. L., Zhong, X., Yang, H., Xie, R., et al. (2017). Dysregulation of TMPRSS3 and TNFRSF11B correlates with tumorigenesis and poor prognosis in patients with breast cancer. Oncology Reports, 37(4), 2057–2062. https://doi.org/10.3892/or.2017.5449.
Pelkonen, M., Luostari, K., Tengström, M., Ahonen, H., Berdel, B., Kataja, V., et al. (2015). Low expression levels of hepsin and TMPRSS3 are associated with poor breast cancer survival. BMC Cancer, 15, 431–431. https://doi.org/10.1186/s12885-015-1440-5.
Chen, L. M., & Chai, K. X. (2002). Prostasin serine protease inhibits breast cancer invasiveness and is transcriptionally regulated by promoter DNA methylation. International Journal of Cancer, 97(3), 323–329.
Bergum, C., Zoratti, G., Boerner, J., & List, K. (2012). Strong expression association between matriptase and its substrate prostasin in breast cancer. Journal of Cellular Physiology, 227(4), 1604–1609. https://doi.org/10.1002/jcp.22877.
Vogel, L. K., Saebo, M., Skjelbred, C. F., Abell, K., Pedersen, E. D., Vogel, U., et al. (2006). The ratio of matriptase/HAI-1 mRNA is higher in colorectal cancer adenomas and carcinomas than corresponding tissue from control individuals. BMC Cancer, 6, 176. https://doi.org/10.1186/1471-2407-6-176.
Tsai, W. C., Sheu, L. F., Chao, Y. C., Chen, A., Chiang, H., & Jin, J. S. (2007). Decreased matriptase/HAI-1 ratio in advanced colorectal adenocarcinoma of Chinese patients. The Chinese Journal of Physiology, 50(5), 225–231.
Forbs, D., Thiel, S., Stella, M. C., Sturzebecher, A., Schweinitz, A., Steinmetzer, T., et al. (2005). In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. International Journal of Oncology, 27(4), 1061–1070.
List, K., Haudenschild, C. C., Szabo, R., Chen, W., Wahl, S. M., Swaim, W., et al. (2002). Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene, 21(23), 3765–3779. https://doi.org/10.1038/sj.onc.1205502.
Kosa, P., Szabo, R., Molinolo, A. A., & Bugge, T. H. (2012). Suppression of tumorigenicity-14, encoding matriptase, is a critical suppressor of colitis and colitis-associated colon carcinogenesis. Oncogene, 31(32), 3679–3695. https://doi.org/10.1038/onc.2011.545.
Huang, A., Zhou, H., Zhao, H., Quan, Y., Feng, B., & Zheng, M. (2014). TMPRSS4 correlates with colorectal cancer pathological stage and regulates cell proliferation and self-renewal ability. Cancer Biology & Therapy, 15(3), 297–304. https://doi.org/10.4161/cbt.27308.
Kim, S., Kang, H. Y., Nam, E. H., Choi, M. S., Zhao, X. F., Hong, C. S., et al. (2010). TMPRSS4 induces invasion and epithelial-mesenchymal transition through upregulation of integrin alpha5 and its signaling pathways. Carcinogenesis, 31(4), 597–606. https://doi.org/10.1093/carcin/bgq024.
Selzer-Plon, J., Bornholdt, J., Friis, S., Bisgaard, H. C., Lothe, I. M., Tveit, K. M., et al. (2009). Expression of prostasin and its inhibitors during colorectal cancer carcinogenesis. BMC Cancer, 9, 201. https://doi.org/10.1186/1471-2407-9-201.
Bao, Y., Li, K., Guo, Y., Wang, Q., Li, Z., Yang, Y., et al. (2016). Tumor suppressor PRSS8 targets Sphk1/S1P/Stat3/Akt signaling in colorectal cancer. Oncotarget, 7(18), 26780–26792. https://doi.org/10.18632/oncotarget.8511.
Bao, Y., Guo, Y., Yang, Y., Wei, X., Zhang, S., Zhang, Y., et al. (2019). PRSS8 suppresses colorectal carcinogenesis and metastasis. Oncogene.
List, K., Szabo, R., Wertz, P. W., Segre, J., Haudenschild, C. C., Kim, S. Y., et al. (2003). Loss of proteolytically processed filaggrin caused by epidermal deletion of matriptase/MT-SP1. The Journal of Cell Biology, 163(4), 901–910. https://doi.org/10.1083/jcb.200304161.
Leyvraz, C., Charles, R. P., Rubera, I., Guitard, M., Rotman, S., Breiden, B., et al. (2005). The epidermal barrier function is dependent on the serine protease CAP1/Prss8. The Journal of Cell Biology, 170(3), 487–496. https://doi.org/10.1083/jcb.200501038.
Tanimoto, H., Underwood, L. J., Wang, Y., Clarke, J., & O’Brien, T. J. Cloning and expression of TADG-15, a novel serine protease expressed in ovarian cancer. 1998 Proc. Am. Assoc. Cancer Res., 39, 648.
Tanimoto, H., Shigemasa, K., Tian, X., Gu, L., Beard, J. B., Sawasaki, T., et al. (2005). Transmembrane serine protease TADG-15 (ST14/matriptase/MT-SP1): expression and prognostic value in ovarian cancer. British Journal of Cancer, 92(2), 278–283. https://doi.org/10.1038/sj.bjc.6602320.
Ji, M., Li, S., Xie, Y., Zhao, Z., Chang, W., Li, Y., et al. (2017). Expression and prognostic value of matriptase in ovarian serous adenocarcinoma. Oncology Letters, 13(3), 1741–1744. https://doi.org/10.3892/ol.2017.5600.
Oberst, M. D., Johnson, M. D., Dickson, R. B., Lin, C. Y., Singh, B., Stewart, M., et al. (2002). Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical outcome and tumor clinicopathological parameters. Clinical Cancer Research, 8(4), 1101–1107.
Johnson, M. D., Oberst, M. D., Lin, C. Y., & Dickson, R. B. (2003). Possible role of matriptase in the diagnosis of ovarian cancer. Expert Review of Molecular Diagnostics, 3(3), 331–338. https://doi.org/10.1586/14737159.3.3.331.
Nakamura, K., Abarzua, F., Kodama, J., Hongo, A., Nasu, Y., Kumon, H., et al. (2009). Expression of hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2) in ovarian cancer. International Journal of Oncology, 34(2), 345–353.
Sun, P., Jiang, Z., Chen, X., Xue, L., Mao, X., Ruan, G., et al. (2016). Decreasing the ratio of matriptase/HAI-1 by downregulation of matriptase as a potential adjuvant therapy in ovarian cancer. Molecular Medicine Reports, 14(2), 1465–1474. https://doi.org/10.3892/mmr.2016.5435.
Suzuki, M., Kobayashi, H., Kanayama, N., Saga, Y., Lin, C. Y., Dickson, R. B., et al. (2004). Inhibition of tumor invasion by genomic down-regulation of matriptase through suppression of activation of receptor-bound pro-urokinase. The Journal of Biological Chemistry, 279(15), 14899–14908. https://doi.org/10.1074/jbc.M313130200.
Underwood, L. J., Shigemasa, K., Tanimoto, H., Beard, J. B., Schneider, E. N., Wang, Y., et al. (2000). Ovarian tumor cells express a novel multi-domain cell surface serine protease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1502(3), 337–350. https://doi.org/10.1016/S0925-4439(00)00058-2.
Guerrero, K., Wang, Z., Bachvarova, M., Gregoire, J., Renaud, M.-C., Plante, M., et al. (2012). A novel genome-based approach correlates TMPRSS3 overexpression in ovarian cancer with DNA hypomethylation. Gynecologic Oncology, 125(3), 720–726. https://doi.org/10.1016/j.ygyno.2012.03.026.
Zhang, D., Qiu, S., Wang, Q., & Zheng, J. (2016). TMPRSS3 modulates ovarian cancer cell proliferation, invasion and metastasis. Oncology Reports, 35(1), 81–88. https://doi.org/10.3892/or.2015.4356.
Shigemasa, K., Underwood, L. J., Beard, J., Tanimoto, H., Ohama, K., Parmley, T. H., et al. (2000). Overexpression of testisin, a serine protease expressed by testicular germ cells, in epithelial ovarian tumor cells. Journal of the Society for Gynecologic Investigation, 7(6), 358–362.
Bignotti, E., Tassi, R. A., Calza, S., Ravaggi, A., Bandiera, E., Rossi, E., et al. (2007). Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. American Journal of Obstetrics and Gynecology, 196(3), 245.e241–245.e211. https://doi.org/10.1016/j.ajog.2006.10.874.
Conway, G. D., Buzza, M. S., Martin, E. W., Duru, N., Johnson, T. A., Peroutka, R. J., et al. (2019). PRSS21/testisin inhibits ovarian tumor metastasis and antagonizes proangiogenic angiopoietins ANG2 and ANGPTL4. Journal of Molecular Medicine, 97(5), 691–709. https://doi.org/10.1007/s00109-019-01763-3.
Martin, E. W., Buzza, M. S., Driesbaugh, K. H., Liu, S., Fortenberry, Y. M., Leppla, S. H., et al. (2015). Targeting the membrane-anchored serine protease testisin with a novel engineered anthrax toxin prodrug to kill tumor cells and reduce tumor burden. Oncotarget, 6(32), 33534–33553. https://doi.org/10.18632/oncotarget.5214.
Riddick, A. C., Shukla, C. J., Pennington, C. J., Bass, R., Nuttall, R. K., Hogan, A., et al. (2005). Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. British Journal of Cancer, 92(12), 2171–2180. https://doi.org/10.1038/sj.bjc.6602630.
Saleem, M., Adhami, V. M., Zhong, W., Longley, B. J., Lin, C. Y., Dickson, R. B., et al. (2006). A novel biomarker for staging human prostate adenocarcinoma: overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiology, Biomarkers & Prevention, 15(2), 217–227. https://doi.org/10.1158/1055-9965.EPI-05-0737.
Sanders, A. J., Parr, C., Davies, G., Martin, T. A., Lane, J., Mason, M. D., et al. (2006). Genetic reduction of matriptase-1 expression is associated with a reduction in the aggressive phenotype of prostate cancer cells in vitro and in vivo. Journal of Experimental Therapeutics & Oncology, 6(1), 39–48.
Warren, M., Twohig, M., Pier, T., Eickhoff, J., Lin, C. Y., Jarrard, D., et al. (2009). Protein expression of matriptase and its cognate inhibitor HAI-1 in human prostate cancer: a tissue microarray and automated quantitative analysis. Applied Immunohistochemistry & Molecular Morphology, 17(1), 23–30. https://doi.org/10.1097/PAI.0b013e31817c3334.
Sanders, A. J., Parr, C., Mason, M. D., & Jiang, W. G. (2007). Suppression of hepatocyte growth factor activator inhibitor-1 leads to a more aggressive phenotype of prostate cancer cells in vitro. International Journal of Molecular Medicine, 20(4), 613–619.
Bergum, C., & List, K. (2010). Loss of the matriptase inhibitor HAI-2 during prostate cancer progression. Prostate, 70(13), 1422–1428. https://doi.org/10.1002/pros.21177.
Tsai, C. H., Teng, C. H., Tu, Y. T., Cheng, T. S., Wu, S. R., Ko, C. J., et al. (2014). HAI-2 suppresses the invasive growth and metastasis of prostate cancer through regulation of matriptase. Oncogene, 33(38), 4643–4652. https://doi.org/10.1038/onc.2013.412.
Wu, S. R., Teng, C. H., Tu, Y. T., Ko, C. J., Cheng, T. S., Lan, S. W., et al. (2017). The Kunitz domain I of hepatocyte growth factor activator inhibitor-2 inhibits matriptase activity and invasive ability of human prostate cancer cells. Scientific Reports, 7(1), 15101. https://doi.org/10.1038/s41598-017-15415-4.
Owen, K. A., Qiu, D., Alves, J., Schumacher, A. M., Kilpatrick, L. M., Li, J., et al. (2010). Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. The Biochemical Journal, 426(2), 219–228. https://doi.org/10.1042/bj20091448.
Najy, A. J., Dyson, G., Jena, B. P., Lin, C. Y., & Kim, H. R. (2016). Matriptase activation and shedding through PDGF-D-mediated extracellular acidosis. American Journal of Physiology. Cell Physiology, 310(4), C293–C304. https://doi.org/10.1152/ajpcell.00043.2015.
Huang, W., & Kim, H. R. (2015). Dynamic regulation of platelet-derived growth factor D (PDGF-D) activity and extracellular spatial distribution by matriptase-mediated proteolysis. The Journal of Biological Chemistry, 290(14), 9162–9170. https://doi.org/10.1074/jbc.M114.610865.
Tripathi, M., Potdar, A. A., Yamashita, H., Weidow, B., Cummings, P. T., Kirchhofer, D., et al. (2011). Laminin-332 cleavage by matriptase alters motility parameters of prostate cancer cells. Prostate, 71(2), 184–196. https://doi.org/10.1002/pros.21233.
Zarif, J. C., Lamb, L. E., Schulz, V. V., Nollet, E. A., & Miranti, C. K. (2015). Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget, 6(9), 6862–6876. https://doi.org/10.18632/oncotarget.3119.
Wu, S. R., Cheng, T. S., Chen, W. C., Shyu, H. Y., Ko, C. J., Huang, H. P., et al. (2010). Matriptase is involved in ErbB-2-induced prostate cancer cell invasion. The American Journal of Pathology, 177(6), 3145–3158. https://doi.org/10.2353/ajpath.2010.100228.
Ko, C. J., Huang, C. C., Lin, H. Y., Juan, C. P., Lan, S. W., Shyu, H. Y., et al. (2015). Androgen-induced TMPRSS2 activates matriptase and promotes extracellular matrix degradation, prostate cancer cell invasion, tumor growth, and metastasis. Cancer Research, 75(14), 2949–2960. https://doi.org/10.1158/0008-5472.CAN-14-3297.
Ko, C. J., Lan, S. W., Lu, Y. C., Cheng, T. S., Lai, P. F., Tsai, C. H., et al. (2017). Inhibition of cyclooxygenase-2-mediated matriptase activation contributes to the suppression of prostate cancer cell motility and metastasis. Oncogene, 36(32), 4597–4609. https://doi.org/10.1038/onc.2017.82.
Magee, J. A., Araki, T., Patil, S., Ehrig, T., True, L., Humphrey, P. A., et al. (2001). Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Research, 61(15), 5692–5696.
Goel, M. M., Agrawal, D., Natu, S. M., & Goel, A. (2011). Hepsin immunohistochemical expression in prostate cancer in relation to Gleason’s grade and serum prostate specific antigen. Indian Journal of Pathology & Microbiology, 54(3), 476–481. https://doi.org/10.4103/0377-4929.85078.
Pal, P., Xi, H., Kaushal, R., Sun, G., Jin, C. H., Jin, L., et al. (2006). Variants in the HEPSIN gene are associated with prostate cancer in men of European origin. Human Genetics, 120(2), 187–192. https://doi.org/10.1007/s00439-006-0204-3.
Holt, S. K., Kwon, E. M., Lin, D. W., Ostrander, E. A., & Stanford, J. L. (2010). Association of hepsin gene variants with prostate cancer risk and prognosis. Prostate, 70(9), 1012–1019. https://doi.org/10.1002/pros.21135.
Ganesan, R., Kolumam, G. A., Lin, S. J., Xie, M. H., Santell, L., Wu, T. D., et al. (2011). Proteolytic activation of pro-macrophage-stimulating protein by hepsin. Molecular Cancer Research, 9(9), 1175–1186. https://doi.org/10.1158/1541-7786.MCR-11-0004.
Tripathi, M., Nandana, S., Yamashita, H., Ganesan, R., Kirchhofer, D., & Quaranta, V. (2008). Laminin-332 is a substrate for hepsin, a protease associated with prostate cancer progression. The Journal of Biological Chemistry, 283(45), 30576–30584. https://doi.org/10.1074/jbc.M802312200.
Kirchhofer, D., Peek, M., Lipari, M. T., Billeci, K., Fan, B., & Moran, P. (2005). Hepsin activates pro-hepatocyte growth factor and is inhibited by hepatocyte growth factor activator inhibitor-1B (HAI-1B) and HAI-2. FEBS Letters, 579(9), 1945–1950. https://doi.org/10.1016/j.febslet.2005.01.085.
Mi, J., Hooker, E., Balog, S., Zeng, H., Johnson, D. T., He, Y., et al. (2018). Activation of hepatocyte growth factor/MET signaling initiates oncogenic transformation and enhances tumor aggressiveness in the murine prostate. The Journal of Biological Chemistry, 293(52), 20123–20136. https://doi.org/10.1074/jbc.RA118.005395.
Li, W., Wang, B. E., Moran, P., Lipari, T., Ganesan, R., Corpuz, R., et al. (2009). Pegylated kunitz domain inhibitor suppresses hepsin-mediated invasive tumor growth and metastasis. Cancer Research, 69(21), 8395–8402. https://doi.org/10.1158/0008-5472.CAN-09-1995.
Klezovitch, O., Chevillet, J., Mirosevich, J., Roberts, R. L., Matusik, R. J., & Vasioukhin, V. (2004). Hepsin promotes prostate cancer progression and metastasis. Cancer Cell, 6(2), 185–195. https://doi.org/10.1016/j.ccr.2004.07.008.
Wittig-Blaich, S. M., Kacprzyk, L. A., Eismann, T., Bewerunge-Hudler, M., Kruse, P., Winkler, E., et al. (2011). Matrix-dependent regulation of AKT in Hepsin-overexpressing PC3 prostate cancer cells. Neoplasia, 13(7), 579–589. https://doi.org/10.1593/neo.11294.
Tang, X., Mahajan, S. S., Nguyen, L. T., Beliveau, F., Leduc, R., Simon, J. A., et al. (2014). Targeted inhibition of cell-surface serine protease Hepsin blocks prostate cancer bone metastasis. Oncotarget, 5(5), 1352–1362. https://doi.org/10.18632/oncotarget.1817.
Valkenburg, K. C., Hostetter, G., & Williams, B. O. (2015). Concurrent Hepsin overexpression and adenomatous polyposis coli deletion causes invasive prostate carcinoma in mice. Prostate, 75(14), 1579–1585. https://doi.org/10.1002/pros.23032.
Nandana, S., Ellwood-Yen, K., Sawyers, C., Wills, M., Weidow, B., Case, T., et al. (2010). Hepsin cooperates with MYC in the progression of adenocarcinoma in a prostate cancer mouse model. Prostate, 70(6), 591–600. https://doi.org/10.1002/pros.21093.
Demichelis, F., Fall, K., Perner, S., Andren, O., Schmidt, F., Setlur, S. R., et al. (2007). TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene, 26(31), 4596–4599. https://doi.org/10.1038/sj.onc.1210237.
Tomlins, S. A., Laxman, B., Dhanasekaran, S. M., Helgeson, B. E., Cao, X., Morris, D. S., et al. (2007). Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature, 448(7153), 595–599. https://doi.org/10.1038/nature06024.
Tomlins, S. A., Rhodes, D. R., Perner, S., Dhanasekaran, S. M., Mehra, R., Sun, X. W., et al. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science, 310(5748), 644–648. https://doi.org/10.1126/science.1117679.
Vaarala, M. H., Porvari, K., Kyllonen, A., Lukkarinen, O., & Vihko, P. (2001). The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: detection of mutated TMPRSS2 form in a case of aggressive disease. International Journal of Cancer, 94(5), 705–710.
Wang, Z., Wang, Y., Zhang, J., Hu, Q., Zhi, F., Zhang, S., et al. (2017). Significance of the TMPRSS2:ERG gene fusion in prostate cancer. Molecular Medicine Reports, 16(4), 5450–5458. https://doi.org/10.3892/mmr.2017.7281.
Lucas, J. M., True, L., Hawley, S., Matsumura, M., Morrissey, C., Vessella, R., et al. (2008). The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. The Journal of Pathology, 215(2), 118–125. https://doi.org/10.1002/path.2330.
Lucas, J. M., Heinlein, C., Kim, T., Hernandez, S. A., Malik, M. S., True, L. D., et al. (2014). The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discovery, 4(11), 1310–1325. https://doi.org/10.1158/2159-8290.CD-13-1010.
Takahashi, S., Suzuki, S., Inaguma, S., Ikeda, Y., Cho, Y. M., Hayashi, N., et al. (2003). Down-regulated expression of prostasin in high-grade or hormone-refractory human prostate cancers. Prostate, 54(3), 187–193. https://doi.org/10.1002/pros.10178.
Chen, L. M., Hodge, G. B., Guarda, L. A., Welch, J. L., Greenberg, N. M., & Chai, K. X. (2001). Down-regulation of prostasin serine protease: a potential invasion suppressor in prostate cancer. Prostate, 48(2), 93–103. https://doi.org/10.1002/pros.1085.
Chen, L. M., Zhang, X., & Chai, K. X. (2004). Regulation of prostasin expression and function in the prostate. Prostate, 59(1), 1–12. https://doi.org/10.1002/pros.10346.
Chen, M., Fu, Y. Y., Lin, C. Y., Chen, L. M., & Chai, K. X. (2007). Prostasin induces protease-dependent and independent molecular changes in the human prostate carcinoma cell line PC-3. Biochimica et Biophysica Acta, 1773(7), 1133–1140. https://doi.org/10.1016/j.bbamcr.2007.04.013.
Nakamura, K., Hongo, A., Kodama, J., Abarzua, F., Nasu, Y., Kumon, H., et al. (2009). Expression of matriptase and clinical outcome of human endometrial cancer. Anticancer Research, 29(5), 1685–1690.
Santin, A. D., Cane, S., Bellone, S., Bignotti, E., Palmieri, M., De Las Casas, L. E., et al. (2003). The novel serine protease tumor-associated differentially expressed gene-15 (matriptase/MT-SP1) is highly overexpressed in cervical carcinoma. Cancer, 98(9), 1898–1904. https://doi.org/10.1002/cncr.11753.
Lee, J. W., Yong Song, S., Choi, J. J., Lee, S. J., Kim, B. G., Park, C. S., et al. (2005). Increased expression of matriptase is associated with histopathologic grades of cervical neoplasia. Human Pathology, 36(6), 626–633. https://doi.org/10.1016/j.humpath.2005.03.003.
Nakamura, K., Abarzua, F., Hongo, A., Kodama, J., Nasu, Y., Kumon, H., et al. (2009). Hepatocyte growth factor activator inhibitor-2 (HAI-2) is a favorable prognosis marker and inhibits cell growth through the apoptotic pathway in cervical cancer. Annals of Oncology, 20(1), 63–70. https://doi.org/10.1093/annonc/mdn556.
Nakamura, K., Abarzua, F., Hongo, A., Kodama, J., Nasu, Y., Kumon, H., et al. (2009). The role of hepatocyte growth factor activator inhibitor-1 (HAI-1) as a prognostic indicator in cervical cancer. International Journal of Oncology, 35(2), 239–248.
Matsuo, T., Nakamura, K., Takamoto, N., Kodama, J., Hongo, A., Abrzua, F., et al. (2008). Expression of the serine protease hepsin and clinical outcome of human endometrial cancer. Anticancer Research, 28(1a), 159–164.
El-Rebey, H. S., Kandil, M. A., Samaka, R. M., Al-Sharaky, D. R., & El Deeb, K. (2017). The role of hepsin in endometrial carcinoma. Applied Immunohistochemistry & Molecular Morphology, 25(9), 624–631. https://doi.org/10.1097/pai.0000000000000352.
Nakamura, K., Takamoto, N., Abarzua, F., Hongo, A., Kodama, J., Nasu, Y., et al. (2008). Hepsin inhibits the cell growth of endometrial cancer. International Journal of Molecular Medicine, 22(3), 389–397.
Cheng, H., Wang, W., Zhang, Y., Zhang, B., Cheng, J., Teng, P., et al. (2017). Expression levels and clinical significance of hepsin and HMGB1 proteins in cervical carcinoma. Oncology Letters, 14(1), 159–164. https://doi.org/10.3892/ol.2017.6116.
Uhland, K., Siphos, B., Arkona, C., Schuster, M., Petri, B., Steinmetzer, P., et al. (2009). Use of IHC and newly designed matriptase inhibitors to elucidate the role of matriptase in pancreatic ductal adenocarcinoma. International Journal of Oncology, 35(2), 347–357.
Cheng, H., Fukushima, T., Takahashi, N., Tanaka, H., & Kataoka, H. (2009). Hepatocyte growth factor activator inhibitor type 1 regulates epithelial to mesenchymal transition through membrane-bound serine proteinases. Cancer Research, 69(5), 1828–1835. https://doi.org/10.1158/0008-5472.CAN-08-3728.
Ye, J., Kawaguchi, M., Haruyama, Y., Kanemaru, A., Fukushima, T., Yamamoto, K., et al. (2014). Loss of hepatocyte growth factor activator inhibitor type 1 participates in metastatic spreading of human pancreatic cancer cells in a mouse orthotopic transplantation model. Cancer Science, 105(1), 44–51. https://doi.org/10.1111/cas.12306.
Keppner, A., Andreasen, D., Merillat, A. M., Bapst, J., Ansermet, C., Wang, Q., et al. (2015). Epithelial sodium channel-mediated sodium transport is not dependent on the membrane-bound serine protease CAP2/Tmprss4. PLoS One, 10(8), e0135224. https://doi.org/10.1371/journal.pone.0135224.
Wallrapp, C., Hahnel, S., Muller-Pillasch, F., Burghardt, B., Iwamura, T., Ruthenburger, M., et al. (2000). A novel transmembrane serine protease (TMPRSS3) overexpressed in pancreatic cancer. Cancer Research, 60(10), 2602–2606.
Hooper, J. D., Bowen, N., Marshall, H., Cullen, L. M., Sood, R., Daniels, R., et al. (2000). Localization, expression and genomic structure of the gene encoding the human serine protease testisin. Biochimica et Biophysica Acta, 1492(1), 63–71. https://doi.org/10.1016/s0167-4781(00)00071-3.
Manton, K. J., Douglas, M. L., Netzel-Arnett, S., Fitzpatrick, D. R., Nicol, D. L., Boyd, A. W., et al. (2005). Hypermethylation of the 5’ CpG island of the gene encoding the serine protease Testisin promotes its loss in testicular tumorigenesis. British Journal of Cancer, 92(4), 760–769. https://doi.org/10.1038/sj.bjc.6602373.
Driesbaugh, K. H., Buzza, M. S., Martin, E. W., Conway, G. D., Kao, J. P., & Antalis, T. M. (2015). Proteolytic activation of the protease-activated receptor (PAR)-2 by the glycosylphosphatidylinositol-anchored serine protease testisin. The Journal of Biological Chemistry, 290(6), 3529–3541. https://doi.org/10.1074/jbc.M114.628560.
Gao, L., Liu, M., Dong, N., Jiang, Y., Lin, C. Y., Huang, M., et al. (2013). Matriptase is highly upregulated in chronic lymphocytic leukemia and promotes cancer cell invasion. Leukemia, 27(5), 1191–1194. https://doi.org/10.1038/leu.2012.289.
Chou, F. P., Chen, Y. W., Zhao, X. F., Xu-Monette, Z. Y., Young, K. H., Gartenhaus, R. B., et al. (2013). Imbalanced matriptase pericellular proteolysis contributes to the pathogenesis of malignant B cell lymphomas. The American Journal of Pathology, 183(4), 1306–1317. https://doi.org/10.1016/j.ajpath.2013.06.024.
Chiu, Y.-L., Wu, Y.-Y., Barndt, R. B., Yeo, Y. H., Lin, Y.-W., Sytwo, H.-P., et al. (2019). Aberrant regulation favours matriptase proteolysis in neoplastic B cells that co-express HAI-2. Journal of Enzyme Inhibition and Medicinal Chemistry, 34(1), 692–702. https://doi.org/10.1080/14756366.2019.1577831.
Yan, R., Liu, M., Hu, Y., Wang, L., Wang, C., Jiang, Y., et al. (2019). Ectopic expression of human airway trypsin-like protease 4 in acute myeloid leukemia promotes cancer cell invasion and tumor growth. Cancer Medicine, 8(5), 2348–2359. https://doi.org/10.1002/cam4.2074.
Coussens, L. M., Fingleton, B., & Matrisian, L. M. (2002). Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science, 295(5564), 2387–2392. https://doi.org/10.1126/science.1067100.
Takeuchi, T., Shuman, M. A., & Craik, C. S. (1999). Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proceedings of the National Academy of Sciences of the United States of America, 96(20), 11054–11061.
Stoop, A. A., & Craik, C. S. (2003). Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nature Biotechnology, 21(9), 1063–1068. https://doi.org/10.1038/nbt860.
Enyedy, I. J., Lee, S. L., Kuo, A. H., Dickson, R. B., Lin, C. Y., & Wang, S. (2001). Structure-based approach for the discovery of bis-benzamidines as novel inhibitors of matriptase. Journal of Medicinal Chemistry, 44(9), 1349–1355.
Long, Y. Q., Lee, S. L., Lin, C. Y., Enyedy, I. J., Wang, S., Li, P., et al. (2001). Synthesis and evaluation of the sunflower derived trypsin inhibitor as a potent inhibitor of the type II transmembrane serine protease, matriptase. Bioorganic & Medicinal Chemistry Letters, 11(18), 2515–2519.
Galkin, A. V., Mullen, L., Fox, W. D., Brown, J., Duncan, D., Moreno, O., et al. (2004). CVS-3983, a selective matriptase inhibitor, suppresses the growth of androgen independent prostate tumor xenografts. Prostate, 61(3), 228–235. https://doi.org/10.1002/pros.20094.
Steinmetzer, T., Schweinitz, A., Sturzebecher, A., Donnecke, D., Uhland, K., Schuster, O., et al. (2006). Secondary amides of sulfonylated 3-amidinophenylalanine. New potent and selective inhibitors of matriptase. Journal of Medicinal Chemistry, 49(14), 4116–4126. https://doi.org/10.1021/jm051272l.
Farady, C. J., Sun, J., Darragh, M. R., Miller, S. M., & Craik, C. S. (2007). The mechanism of inhibition of antibody-based inhibitors of membrane-type serine protease 1 (MT-SP1). Journal of Molecular Biology, 369(4), 1041–1051. https://doi.org/10.1016/j.jmb.2007.03.078.
Schneider, E. L., Lee, M. S., Baharuddin, A., Goetz, D. H., Farady, C. J., Ward, M., et al. (2012). A reverse binding motif that contributes to specific protease inhibition by antibodies. Journal of Molecular Biology, 415(4), 699–715. https://doi.org/10.1016/j.jmb.2011.11.036.
Sun, J., Pons, J., & Craik, C. S. (2003). Potent and selective inhibition of membrane-type serine protease 1 by human single-chain antibodies. Biochemistry, 42(4), 892–900. https://doi.org/10.1021/bi026878f.
Kim, M. G., Chen, C., Lyu, M. S., Cho, E. G., Park, D., Kozak, C., et al. (1999). Cloning and chromosomal mapping of a gene isolated from thymic stromal cells encoding a new mouse type II membrane serine protease, epithin, containing four LDL receptor modules and two CUB domains. Immunogenetics, 49(5), 420–428.
Darragh, M. R., Schneider, E. L., Lou, J., Phojanakong, P. J., Farady, C. J., Marks, J. D., et al. (2010). Tumor detection by imaging proteolytic activity. Cancer Research, 70(4), 1505–1512. https://doi.org/10.1158/0008-5472.can-09-1640.
LeBeau, A. M., Lee, M., Murphy, S. T., Hann, B. C., Warren, R. S., Delos Santos, R., et al. (2013). Imaging a functional tumorigenic biomarker in the transformed epithelium. Proceedings of the National Academy of Sciences of the United States of America, 110(1), 93–98. https://doi.org/10.1073/pnas.1218694110.
Rather, G. M., Lin, S. Y., Lin, H., Banach-Petrosky, W., Hirshfield, K. M., Lin, C. Y., et al. (2018). Activated matriptase as a target to treat breast cancer with a drug conjugate. Oncotarget, 9(40), 25983–25992. https://doi.org/10.18632/oncotarget.25414.
Lin, C. Y., Anders, J., Johnson, M., & Dickson, R. B. (1999). Purification and characterization of a complex containing matriptase and a Kunitz-type serine protease inhibitor from human milk. The Journal of Biological Chemistry, 274(26), 18237–18242.
Benaud, C., Dickson, R. B., & Lin, C. Y. (2001). Regulation of the activity of matriptase on epithelial cell surfaces by a blood-derived factor. European Journal of Biochemistry, 268(5), 1439–1447.
Benaud, C., Oberst, M., Hobson, J. P., Spiegel, S., Dickson, R. B., & Lin, C. Y. (2002). Sphingosine 1-phosphate, present in serum-derived lipoproteins, activates matriptase. The Journal of Biological Chemistry, 277(12), 10539–10546. https://doi.org/10.1074/jbc.M109064200.
Rather, G. M., Lin, S. Y., Lin, H., Szekely, Z., & Bertino, J. R. (2019). A novel antibody-toxin conjugate to treat mantle cell lymphoma. Frontiers in Oncology, 9, 258. https://doi.org/10.3389/fonc.2019.00258.
List, K., Kosa, P., Szabo, R., Bey, A. L., Wang, C. B., Molinolo, A., et al. (2009). Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. The American Journal of Pathology, 175(4), 1453–1463. https://doi.org/10.2353/ajpath.2009.090240.
List, K., Currie, B., Scharschmidt, T. C., Szabo, R., Shireman, J., Molinolo, A., et al. (2007). Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. The Journal of Biological Chemistry, 282(50), 36714–36723. https://doi.org/10.1074/jbc.M705521200.
Alef, T., Torres, S., Hausser, I., Metze, D., Tursen, U., Lestringant, G. G., et al. (2009). Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. The Journal of Investigative Dermatology, 129(4), 862–869. https://doi.org/10.1038/jid.2008.311.
Avrahami, L., Maas, S., Pasmanik-Chor, M., Rainshtein, L., Magal, N., Smitt, J., et al. (2008). Autosomal recessive ichthyosis with hypotrichosis syndrome: further delineation of the phenotype. Clinical Genetics, 74(1), 47–53. https://doi.org/10.1111/j.1399-0004.2008.01006.x.
Basel-Vanagaite, L., Attia, R., Ishida-Yamamoto, A., Rainshtein, L., Ben Amitai, D., Lurie, R., et al. (2007). Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. American Journal of Human Genetics, 80(3), 467–477. https://doi.org/10.1086/512487.
Desilets, A., Beliveau, F., Vandal, G., McDuff, F. O., Lavigne, P., & Leduc, R. (2008). Mutation G827R in matriptase causing autosomal recessive ichthyosis with hypotrichosis yields an inactive protease. The Journal of Biological Chemistry, 283(16), 10535–10542. https://doi.org/10.1074/jbc.M707012200.
Colombo, E., Desilets, A., Duchene, D., Chagnon, F., Najmanovich, R., Leduc, R., et al. (2012). Design and synthesis of potent, selective inhibitors of matriptase. ACS Medicinal Chemistry Letters, 3(7), 530–534. https://doi.org/10.1021/ml3000534.
St-Georges, C., Desilets, A., Beliveau, F., Ghinet, M., Dion, S. P., Colombo, E., et al. (2017). Modulating the selectivity of matriptase-2 inhibitors with unnatural amino acids. European Journal of Medicinal Chemistry, 129, 110–123. https://doi.org/10.1016/j.ejmech.2017.02.006.
Gray, K., Elghadban, S., Thongyoo, P., Owen, K. A., Szabo, R., Bugge, T. H., et al. (2014). Potent and specific inhibition of the biological activity of the type-II transmembrane serine protease matriptase by the cyclic microprotein MCoTI-II. Thrombosis and Haemostasis, 112(2), 402–411. https://doi.org/10.1160/TH13-11-0895.
Quimbar, P., Malik, U., Sommerhoff, C. P., Kaas, Q., Chan, L. Y., Huang, Y. H., et al. (2013). High-affinity cyclic peptide matriptase inhibitors. The Journal of Biological Chemistry, 288(19), 13885–13896. https://doi.org/10.1074/jbc.M113.460030.
Swedberg, J. E., Nigon, L. V., Reid, J. C., de Veer, S. J., Walpole, C. M., Stephens, C. R., et al. (2009). Substrate-guided design of a potent and selective kallikrein-related peptidase inhibitor for kallikrein 4. Chemistry & Biology, 16(6), 633–643. https://doi.org/10.1016/j.chembiol.2009.05.008.
Clark, R. J., Jensen, J., Nevin, S. T., Callaghan, B. P., Adams, D. J., & Craik, D. J. (2010). The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angewandte Chemie (International Ed. in English), 49(37), 6545–6548. https://doi.org/10.1002/anie.201000620.
Wong, C. T., Rowlands, D. K., Wong, C. H., Lo, T. W., Nguyen, G. K., Li, H. Y., et al. (2012). Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angewandte Chemie (International Ed. in English), 51(23), 5620–5624. https://doi.org/10.1002/anie.201200984.
Desilets, A., Longpre, J. M., Beaulieu, M. E., & Leduc, R. (2006). Inhibition of human matriptase by eglin c variants. FEBS Letters, 580(9), 2227–2232. https://doi.org/10.1016/j.febslet.2006.03.030.
Mitchell, A. C., Kannan, D., Hunter, S. A., Parra Sperberg, R. A., Chang, C. H., & Cochran, J. R. (2018). Engineering a potent inhibitor of matriptase from the natural hepatocyte growth factor activator inhibitor type-1 (HAI-1) protein. The Journal of Biological Chemistry, 293(14), 4969–4980. https://doi.org/10.1074/jbc.M117.815142.
Kojima, K., Tsuzuki, S., Fushiki, T., & Inouye, K. (2008). Roles of functional and structural domains of hepatocyte growth factor activator inhibitor type 1 in the inhibition of matriptase. The Journal of Biological Chemistry, 283(5), 2478–2487. https://doi.org/10.1074/jbc.M709073200.
Zhao, B., Yuan, C., Li, R., Qu, D., Huang, M., & Ngo, J. C. (2013). Crystal structures of matriptase in complex with its inhibitor hepatocyte growth factor activator inhibitor-1. The Journal of Biological Chemistry, 288(16), 11155–11164. https://doi.org/10.1074/jbc.M113.454611.
Han, Z., Harris, P. K., Karmakar, P., Kim, T., Owusu, B. Y., Wildman, S. A., et al. (2016). alpha-Ketobenzothiazole serine protease inhibitors of aberrant HGF/c-MET and MSP/RON kinase pathway signaling in cancer. ChemMedChem, 11(6), 585–599. https://doi.org/10.1002/cmdc.201500600.
Han, Z., Harris, P. K., Jones, D. E., Chugani, R., Kim, T., Agarwal, M., et al. (2014). Inhibitors of HGFA, matriptase, and hepsin serine proteases: a nonkinase strategy to block cell signaling in cancer. ACS Medicinal Chemistry Letters, 5(11), 1219–1224. https://doi.org/10.1021/ml500254r.
Damalanka, V. C., & Janetka, J. W. (2019). Recent progress on inhibitors of the type II transmembrane serine proteases, hepsin, matriptase and matriptase-2. Future Medicinal Chemistry, 11(7), 743–769. https://doi.org/10.4155/fmc-2018-0446.
Miyazawa, K., Shimomura, T., Kitamura, A., Kondo, J., Morimoto, Y., & Kitamura, N. (1993). Molecular cloning and sequence analysis of the cDNA for a human serine protease reponsible for activation of hepatocyte growth factor. Structural similarity of the protease precursor to blood coagulation factor XII. The Journal of Biological Chemistry, 268(14), 10024–10028.
Franco, F. M., Jones, D. E., Harris, P. K., Han, Z., Wildman, S. A., Jarvis, C. M., et al. (2015). Structure-based discovery of small molecule hepsin and HGFA protease inhibitors: evaluation of potency and selectivity derived from distinct binding pockets. Bioorganic & Medicinal Chemistry, 23(10), 2328–2343. https://doi.org/10.1016/j.bmc.2015.03.072.
Owusu, B. Y., Bansal, N., Venukadasula, P. K., Ross, L. J., Messick, T. E., Goel, S., et al. (2016). Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling. Oncotarget, 7(20), 29492–29506. https://doi.org/10.18632/oncotarget.8785.
Owusu, B. Y., Thomas, S., Venukadasula, P., Han, Z., Janetka, J. W., Galemmo, R. A., Jr., et al. (2017). Targeting the tumor-promoting microenvironment in MET-amplified NSCLC cells with a novel inhibitor of pro-HGF activation. Oncotarget, 8(38), 63014–63025. https://doi.org/10.18632/oncotarget.18260.
Venukadasula, P. K., Owusu, B. Y., Bansal, N., Ross, L. J., Hobrath, J. V., Bao, D., et al. (2016). Design and synthesis of nonpeptide inhibitors of hepatocyte growth factor activation. ACS Medicinal Chemistry Letters, 7(2), 177–181. https://doi.org/10.1021/acsmedchemlett.5b00357.
Kwon, H., Kim, Y., Park, K., Choi, S. A., Son, S. H., & Byun, Y. (2016). Structure-based design, synthesis, and biological evaluation of Leu-Arg dipeptide analogs as novel hepsin inhibitors. Bioorganic & Medicinal Chemistry Letters, 26(2), 310–314. https://doi.org/10.1016/j.bmcl.2015.12.023.
Kim, K., Kwon, H., Choi, D., Lim, T., Minn, I., Son, S. H., et al. (2019). Design and synthesis of dye-conjugated hepsin inhibitors. Bioorganic Chemistry, 89, 102990. https://doi.org/10.1016/j.bioorg.2019.102990.
Shia, S., Stamos, J., Kirchhofer, D., Fan, B., Wu, J., Corpuz, R. T., et al. (2005). Conformational lability in serine protease active sites: structures of hepatocyte growth factor activator (HGFA) alone and with the inhibitory domain from HGFA inhibitor-1B. Journal of Molecular Biology, 346(5), 1335–1349. https://doi.org/10.1016/j.jmb.2004.12.048.
Xuan, J. A., Schneider, D., Toy, P., Lin, R., Newton, A., Zhu, Y., et al. (2006). Antibodies neutralizing hepsin protease activity do not impact cell growth but inhibit invasion of prostate and ovarian tumor cells in culture. Cancer Research, 66(7), 3611–3619. https://doi.org/10.1158/0008-5472.CAN-05-2983.
Ganesan, R., Zhang, Y., Landgraf, K. E., Lin, S. J., Moran, P., & Kirchhofer, D. (2012). An allosteric anti-hepsin antibody derived from a constrained phage display library. Protein Engineering, Design & Selection, 25(3), 127–133. https://doi.org/10.1093/protein/gzr067.
Koschubs, T., Dengl, S., Durr, H., Kaluza, K., Georges, G., Hartl, C., et al. (2012). Allosteric antibody inhibition of human hepsin protease. The Biochemical Journal, 442(3), 483–494. https://doi.org/10.1042/BJ20111317.
Tully, D. C., Vidal, A., Chatterjee, A. K., Williams, J. A., Roberts, M. J., Petrassi, H. M., et al. (2008). Discovery of inhibitors of the channel-activating protease prostasin (CAP1/PRSS8) utilizing structure-based design. Bioorganic & Medicinal Chemistry Letters, 18(22), 5895–5899. https://doi.org/10.1016/j.bmcl.2008.08.029.
Klimpel, K. R., Molloy, S. S., Thomas, G., & Leppla, S. H. (1992). Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proceedings of the National Academy of Sciences of the United States of America, 89(21), 10277–10281. https://doi.org/10.1073/pnas.89.21.10277.
Liu, S., Netzel-Arnett, S., Birkedal-Hansen, H., & Leppla, S. H. (2000). Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Research, 60(21), 6061–6067.
Liu, S., Bugge, T. H., & Leppla, S. H. (2001). Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. The Journal of Biological Chemistry, 276(21), 17976–17984. https://doi.org/10.1074/jbc.M011085200.
Peters, D. E., Hoover, B., Cloud, L. G., Liu, S., Molinolo, A. A., Leppla, S. H., et al. (2014). Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities. Toxicology and Applied Pharmacology, 279(2), 220–229. https://doi.org/10.1016/j.taap.2014.06.010.
Liu, S., Redeye, V., Kuremsky, J. G., Kuhnen, M., Molinolo, A., Bugge, T. H., et al. (2005). Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nature Biotechnology, 23(6), 725–730. https://doi.org/10.1038/nbt1091.
Funding
This work was supported by NIH/NCI R01CA222359A (KL), Susan G. Komen IBC17511329 (KL) and IBC18511329 (K.L), and an NIH/NCI Training grant (CEM) awarded to Wayne State University Cancer Biology Graduate Program (Ruth L. Kirschstein National Research Service Award T32-CA009531).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Martin, C.E., List, K. Cell surface–anchored serine proteases in cancer progression and metastasis. Cancer Metastasis Rev 38, 357–387 (2019). https://doi.org/10.1007/s10555-019-09811-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-019-09811-7