
Abstract
Changing the characteristics of cells from epithelial states to mesenchymal properties is a key process involved in developmental and physiological processes as well as in many diseases with cancer as the most prominent example. Nowadays, a great deal of work and literature concerns the understanding of the process of epithelial-to-mesenchymal transition (EMT) in terms of its molecular regulation and its implications for cancer. Similar statements can certainly be made regarding the investigation of the more than 500 proteases typically encoded by a mammalian genome. Specifically, the impact of proteases on tumor biology has been a long-standing topic of interest. However, although EMT actively regulates expression of many proteases and proteolytic enzymes are clearly involved in survival, division, differentiation, and movements of cells, information on the diverse roles of proteases in EMT has been rarely compiled. Here we aim to conceptually connect the scientific areas of “EMT” and “protease” research by describing how several important classes of proteolytic enzymes are regulated by EMT and how they are involved in initiation and execution of the EMT program. To do so, we briefly introduce the evolving key features of EMT and its regulation followed by discussion of protease involvement in this process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction to key characteristics of EMT
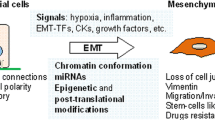
Epithelial-to-mesenchymal transition (EMT) is an evolutionarily conserved program of cellular plasticity that allows polarized, immotile epithelial cells to loosen their cell-cell adhesion, detach from neighboring cells, and to convert into motile mesenchymal cells (Fig. 1). As the term transition implies, EMT is not a binary switch but a gradual transformation through numerous hybrid and intermediate E/M-states in a reversible manner.

Cellular characteristics during EMT. A) Changes in cellular morphology and extracellular matrix organization during EMT. Immotile epithelial cells loosen their cell–cell contacts (desmosomes, gap-, tight-, adherens junctions), detach from neighboring cells and convert into motile mesenchymal cells. Concomitantly, remodeling of extracellular matrix to a state of radially aligned fibers supports cell motility. Intracellular (such as DUBs, SENPs, cathepsins), membrane bound (e.g., ADAMs, MT-MMPs, TTSPs), and extracellular proteases (MMPs) contribute to these processes by influencing cell signaling, breakdown of the basement membrane, and remodeling of the extracellular matrix. B) Molecular processes underlying the cellular plasticity during EMT. Cues from the microenvironment (gray box) converge and lead to induction of EMT-transcription factors. The following gradual loss of epithelial characteristics is accompanied by a re-organization of the actin cytoskeleton leading to an elongated cell shape with loss of cell–cell adhesion, gain of front-rear polarity, and increased invasive and migratory abilities. Molecular markers characteristic of different EMT-states, like E-cadherin and Epcam as well as N-cadherin and Vimentin, are shown. In general, enhanced proteolysis is characteristic for cells that underwent EMT. Abbreviations: ADAM—a disintegrin and metalloprotease; DUB—deubiquinating enzyme; MMP—matrix metalloprotease; MT-MMP—membrane type matrix metalloprotease; ROS—reactive oxygen species; TTSP—type II transmembrane serine protease family; SENP—sentrin specific protease
Historically, EMT has first been described in embryogenesis, but already in the early 1980s, Greenburg and Hay were able to observe EMT in adult tissues [1]. EMT processes are physiologically activated during tissue and organ development such as gastrulation, heart formation, neural crest migration, somitogenesis, and palate formation [2]. In the adult organism, EMT is transiently activated as a response to injury for example during wound healing in the skin or during the postovulatory ovarian remodeling process [3, 4]. In contrast, pathologically sustained EMT is a key mechanism of tissue fibrosis in multiple organs and plays a role in the pathophysiology of skin, renal, and liver fibrosis leading to tissue degeneration [5,6,7]. Importantly, pathological activation of EMT is also frequently detected in tumorigenesis [8].
Induction of EMT is mediated by cues from the microenvironment, such as TGF-β and Wnt-ligands as well as cell-intrinsic signals such as metabolic stress or oncogenic mutations in the RAS-Raf-Erk pathway (Fig. 1) [9,10,11]. Both intrinsic and extrinsic signals converge to provoke the expression of EMT-inducing transcription factors (EMT-TFs) for signal transmission. A small number of transcription factors function as core EMT-TFs. They belong to the Snail (SNAIL, SLUG), basic helix-loop-helix (TWIST1), and ZEB (ZEB1, ZEB2) families. These families are not related, and the members differ in structure and size, stability, spatiotemporal expression patterns, and target gene profile [12]. EMT-TFs control the expression of proteins implicated in cell–cell junctions (Claudins, Desmoplakin, Placophilins, Occludin, ZO-3) [13,14,15], cell polarity (Crumbs3, LGL2, PATJ) [16], cytoskeletal structure (reviewed in [17]), and extracellular matrix degradation (MMP-1, MMP-2, MMP-7, MMP-9, MMP-14, MMP-15) [18,19,20,21,22]. The common denominator of all core EMT-TFs is the direct or indirect repression of the key epithelial gene E-cadherin. This dynamic regulation of adherens junctions triggers signaling pathways and alterations in the organization of the actin cytoskeleton that are involved in cell motility. The complex signaling network that regulates EMT is active on epigenetic, transcriptional, and post-transcriptional level and includes signaling pathways, transcription factors, non-coding RNAs, and also proteases [11, 23].
2 EMT in cancer progression and metastasis
EMT in cancer is associated with tumor initiation and progression, stemness, survival, and therapy resistance. Classically, EMT has been thought to be critical for local invasion and cancer cell dissemination in the body. Subsequently, the reversal of EMT, termed mesenchymal-to-epithelial transition (MET), is thought to allow disseminated tumor cells to colonize the foreign soil at distant organ sites to form macrometastases that are often dismal signs for the prognosis of cancer patients [11, 24,25,26].
However, EMT plays an active role not only in metastasis but also in tumor initiation. In pre-malignant lesions, oncogene-induced senescence, and apoptosis limit tumor growth through the activation of the p19Arf-p53 and p16Ink4A-RB (retinoblastoma protein) oncosuppressive pathways. These fail-safe barriers prevent conversion from pre-malignant lesions to invasive carcinomas. EMT-TF expression in pre-malignant tumor states promotes escape from oncogene-induced senescence and provides survival advantages under different stress conditions even before tumor cell dissemination occurs. In this respect, TWIST, ZEB, and SNAIL proteins regulate p53 and RB signaling by repressing the transcription of upstream activators (like p16Ink4A and p15Ink4B) of either pathway as well as by directly downregulating p53 levels [2, 11]. Hence, EMT-TFs actively weaken tumor-suppressive signaling pathways that maintain epithelial cell characteristics, thereby allowing progress toward malignancy.
Carcinomas show a substantial intratumoral heterogeneity of cancer cells, to which EMT contributes considerably [27,28,29]. While a fraction of epithelial cells is likely to undergo a full EMT, meaning transition from a fully epithelial into a fully mesenchymal state, other cells only partially transform and reside in a hybrid E/M state with simultaneous expression of epithelial and mesenchymal markers. These intermediate E/M states are associated with increased tumor-initiating abilities and aggressive cancer progression [30,31,32,33]. In a comprehensive study, Pastushenko and colleagues identified seven distinct transition states along the epithelial to mesenchymal axis based on the expression of only six surface markers: Epcam, Keratin-14, Vimentin, CD51/αv-integrin, CD61/β3-integrin, and CD106/Vcam1 [32]. The authors further demonstrate that these transition states differ in the degree of stemness, plasticity, and metastatic ability. In addition, the tumor cells show distinct transcriptional and chromatin landscapes and localize within different niches of a primary tumor. Interestingly, according to the cell’s transitory state, it expresses different chemokines, pro-inflammatory, and pro-angiogenic molecules. Thereby tumor cells attract and regulate specific stromal cells, such as macrophages and fibroblasts, to shape a defined tumor microenvironment. All these findings point to the fact that cells undergoing a partial EMT express epithelial as well as mesenchymal markers, show high tumor-initiating capacity, are prone to metastatic dissemination, and are resistant to chemotherapy.
3 Proteases in cancer progression, invasion, and metastasis
Proteolysis contributes to tumorigenesis when conducted by tumor cells or by proteases originating from cells of the tumor microenvironment. Proteases have been shown to play various roles at different steps of tumorigenesis and invasion—both pro-tumorigenic as well as anti-tumorigenic—depending on the type and grade of cancer. The contribution of proteases to cancer progression and metastasis was long thought to be limited to extracellular matrix (ECM) and basement membrane breakdown as well as ECM-remodeling, thus enabling cells to invade adjacent tissue. More recently, it became obvious that proteases contribute to carcinogenesis not only by paving the way for tumor cells but also by influencing cell signaling in mitosis, apoptosis, autophagy, and inflammation [34,35,36,37]. In the clinics, most protease inhibitors failed due to the redundancy and multifunctionality of the enzymes, which led to considerable toxicity or even adverse effects [38, 39]. On the other hand, proteases possess great value as prognostic markers, prodrug activators as well as tools for image-guided surgery [40,41,42]. We focus this review on the role of proteases in signaling processes leading to EMT and thus to invasion and metastasis because the general functions of proteases in tumor biology, and their possible roles as drug targets have been extensively discussed in recent reviews [42,43,44,45,46,47,48,49].
Protease involvement in EMT occurs in three distinct steps: first, EMT leads to differential expression of a variety of proteases. Second, proteases function as EMT executors by promoting cell motility and invasion by degrading basement membrane and ECM. Third, proteases upregulated in tumor cells can themselves act as EMT initiators. Those processes are linked because proteolytic liberation of cytokines and growth factors from the ECM results in induction of protease expression thereby establishing a positive feedback loop. Of note, EMT is not mandatory for cells to invade adjacent tissue. It has been shown that there are various ways for cells to move and invade, from amoeboid and mesenchymal single-cell motility to multicellular streaming and collective migration of which the amoeboid invasion is independent of protease activity [50, 51]. In the following sections, we will review the ability of some major protease classes to induce the EMT process during cancer invasion and metastasis.
4 Metalloproteases and serine proteases as extracellular EMT triggers
Cells secrete a plethora of proteases to the extracellular space and express a large number of transmembrane proteases functioning near the extracellular surface of the plasma membrane. We will discuss three major classes of extracellular proteases: matrix metalloproteases (MMP), ADAMs (a disintegrin and metalloprotease), and extracellularly acting serine proteases.
In embryogenesis and physiological tissue repair, extracellular proteolysis is an integral part of an organized morphogenetic program. In the complex cancer environment, however, this physiological program is hijacked, and high extracellular protease levels are induced. Notably an increased expression or activity of MMPs, ADAMs, and transmembrane serine proteases has been demonstrated for almost every type of human cancer. This correlates with advanced tumor stage, increased invasion, and metastasis as well as decreased survival [52,53,54]. Extracellular protease contribution to cancer progression and metastasis is mainly due to three mechanisms: (1) proteases are often upregulated in invasive cancer cells undergoing an EMT and degrade or modify ECM and cell-cell contacts. (2) Proteases can activate stromal cells, which in turn produce more proteolytic enzymes thus driving cancer progression. (3) Proteases in the microenvironment can directly induce EMT in epithelial cells.
The protease substrates comprise cell adhesion molecules such as E-cadherin, CD44, or αv-integrin [55,56,57]. Several metalloproteases, including MMP-3, MMP-7, MMP-9, ADAM10, and MT1-MMP, were shown to be involved in E-cadherin cleavage [55, 58,59,60,61]. Further, growth factors and their receptors with links to the EMT program such as FGF receptor 1, members of the EGF receptor family, or the HGF receptor c-met are extensively influenced by extracellular proteolysis [62,63,64,65]. The phenomenon that pericellular proteolysis can directly induce EMT has been shown in several epithelial cell types including the lung, kidney, prostate, and mammary cells [59, 66,67,68,69,70,71]. In fact, it is often very difficult to discriminate whether increased motility and invasiveness is due to EMT-induced protease expression or protease-induced EMT.
4.1 Typical matrix metalloproteinases
For MMP-3, also known as stromelysin-1, it was shown already in the late 1990s that overexpression of the secreted protease reduced the levels of keratins, induced the expression of Vimentin, and led to the loss of E-cadherin and β-catenin from cell–cell contacts [59]. In 3D culture, these cells lost their differentiation and gained invasiveness, clearly indicating that these cells converted from an epithelial to a mesenchymal phenotype. Some years later, Radisky et al. established that the observed changes are due to MMP-3 dependent stimulation of Rac1b expression, a constitutively activated splice variant of Rac1 [72]. Subsequently, Rac1b induced EMT in mouse mammary epithelial cells by rising levels of cellular reactive oxygen species. This led on the one hand to the expression of the EMT activator SNAIL and, on the other hand, to oxidative damage to the DNA, genomic instability and thus malignant transformation. Interestingly, the EMT induction by MMP-3 seemed to depend on protein composition and stiffness of the ECM [73, 74]. The intertwining roles of MMP-3 and other MMPs on EMT induction and ECM remodeling highlight these enzymes as important promotors of tumor progression and metastasis.
MMP-19 expression was linked to poor prognosis of non-small cell lung cancer (NSCLC), and its overexpression promoted EMT, migration, and invasion in multiple NSCLC cell lines independently of its enzymatic activity [75]. The mechanism by which MMP-19 promotes EMT remains to be elucidated. Of interest, Pastushenko et al. linked MMP19 expression to a late stage of EMT proposing it as prognostic marker for full EMT rather than early or intermediate stages [32, 76].
The transmembrane protease MMP-14, also known as MT1-MMP, has been shown to be differentially expressed during partial EMT of tumor-initiating murine breast cancer cells [77]. It is upregulated and correlates with clinical stage in squamous cell carcinomas of the upper digestive tract [78, 79]. Esophageal or oral squamous cancer cells with high MMP-14 levels underwent morphological changes from an epithelial morphology to a fibroblast-like appearance, increased cell motility and invasion. MMP-14 activity was associated with SNAIL-mediated transcriptional repression of E-cadherin and increased expression of mesenchymal markers [78, 79]. MMP-14 expression in prostate adenocarcinoma cells activated TGF-β signaling, one of the key signaling pathways to trigger EMT in cancer cells [80]. This induced CUX1 and subsequently Wnt5a leading to EMT. Mesenchymal LNCaP cells reverted to an epithelial phenotype upon inhibition of MMP-14 or TGF-β. MMP-14 catalytically increased the availability of active TGF-β without affecting total TGF-β levels, which caused the induction of EMT in adjacent cells. Thus, MMP-14 increases tumor invasiveness through activation of TGF-β signaling and subsequent EMT in a paracrine and autocrine fashion, which highlights the importance of the microenvironment. TGF-β signaling is also implicated in EMT induction by other MMPs, such as MMP-8. MMP-8 and TGF-β1 accumulate in highly invasive hepatocellular carcinoma (HCC) cell lines and in HCC specimens from patients [81]. MMP-8 was able to restore TGF-β1 expression in TGF-β1-depleted HCC cells via the activation of PI3K/Akt/Rac1 pathway. On the other hand, MMP-8 expression was restored in MMP-8 depleted cells by TGF-β1 treatment indicating a reciprocal activation of MMP-8 and TGF-β1. This positive feedback loop was associated with poor patient outcome. A similar mechanism where MMP activation and TGF-β1 activation are linked has been shown for MMP-9 in esophageal squamous cell carcinoma [82]. Here TGF-β1 induced MMP-9, which in turn mediated the downregulation of E-cadherin leading to EMT. In A549 lung adenocarcinoma cells, MMP-28 was induced by TGF-β signaling, leading to loss of cell surface E-cadherin and the proteolytic liberation of active TGF-β from latent TGF-β complexes [66]. Again, MMP inhibition or neutralizing antibodies against TGF-β were able to abolish EMT. Interestingly, an increasing number of studies demonstrate that MMPs, such as MMP-2, MMP-9, and MMP-14, can stimulate TGF-β1 activity by cleaving latent TGF-β-binding protein-1. This happens in a complex interplay between MMPs, TGF-β, and urokinase-type plasminogen activator (uPA) [83, 84].
4.2 Transmembrane metalloproteases interacting with integrins
ADAMs (a disintegrin and metalloprotease) are transmembrane proteins implicated in multiple biological processes such as cell adhesion, proliferation, migration, and proteolysis. Ectodomains of other transmembrane proteins are their main substrates. Thus, ADAMs participate in the processing of growth factor and cytokine precursors and receptors as wells as other adhesion proteins. With regard to their substrates, it is not surprising that dysregulation of ADAM activity has been shown to be implicated in cancer development and progression [53, 85, 86]. The disintegrin domain of ADAMs has been shown to interact with various integrins thereby further multiplying and diversifying their functions [87].
In HCC, ADAM9 was shown to be upregulated by IL-6 through the JNK signaling pathway. This led to NADPH oxidase 1-induced ROS generation and EMT, which could be blocked by ADAM9 knockdown [88]. A positive correlation of ADAM9 expression with IL-6 or SNAIL expression in human HCC tissue samples demonstrated the clinical significance of these findings. ADAM10 and ADAM17 are critical sheddases for a variety of cell surface receptors [89]. ADAM10 cleaves E-cadherin in its ectodomain, which modulates cell–cell adhesion and migration as well as subcellular localization of β-catenin and thus its downstream signaling and EMT [60]. Atapattu et al. found an active form of ADAM10 being predominant in cancers compared with healthy tissue, and that this isoform is linked to an active Notch signaling, chemoresistance, and stemness [90]. Increased ADAM17 expression has been linked to lung adenocarcinoma [91]. In this study, the authors further showed that ADAM17 regulates E-cadherin and Vimentin expression in lung adenocarcinoma cell lines toward a mesenchymal phenotype and affected cell invasion. They suggest expression control of ADAM17 by miR-326, which is known to be involved in invasion and EMT. However, the molecular mechanism how ADAM17 regulates EMT in lung cancer needs further investigation.
ADAM12 plays a role in the context of TGF-β-induced EMT [92]. In this paper, a correlation was shown between ADAM12 expression and EMT markers in human breast cancer cell lines and tissue. TGF-β-induced EMT increased the expression of the membrane-anchored long form (ADAM12L) and expression of ADAM12L alone was sufficient to induce EMT and chemoresistance in MCF10A cells as a function of the cytoplasmic tail of ADAM12L. The authors observed changes in phosphorylation of SMAD3, Akt and Erk suggesting a contribution of ADAM12 to TGF-β-induced EMT through TGF-β receptor-dependent and independent pathways. In pituitary adenomas, a correlation between ADAM12 expression and EMT was observed as well [93]. Silencing of ADAM12 led to significant inhibition of the process. The proposed mechanism of this study is the ectodomain shedding of EGFR ligands and thus enhanced EGFR/Erk signaling by ADAM12. Erk pathway signaling was also shown to be affected by ADAM15 expression in breast carcinoma [94]. Najy et al. verified the ability of ADAM15 to cleave E-cadherin in response to growth factor deprivation in breast cancer cells. Interestingly they found the cleaved fragment in complex with HER2 and HER3 receptors in these cells, apparently stabilizing HER2/HER3 heterodimerization. This led to enhanced receptor activation and Erk signaling.
4.3 Transmembrane (TMPRSSs) and secreted serine proteases
The hepsin/TMPRSS (transmembrane protease/serine) subfamily of the type 2 transmembrane serine protease (TTSP) family has seven members: hepsin, TMPRSS2, TMPRSS3, TMPRSS4, TMPRSS5/spinesin, MSPL (mosaic serine protease large-form), and enteropeptidase [95]. Due to their common transmembrane domain, TTSPs are membrane-bound proteases and their activity is typically confined to the cell surface mainly regulating cell–cell and cell–matrix interactions. Aberrant expression of TMPRSS has been observed in several cancers [96]. TMPRSS4 was found overexpressed in lung cancer tissues and in a variety of human cancer cell lines [97]. Knockdown of TMPRSS4 in lung and colon cancer cell lines decreased invasion and cell-matrix adhesion whereas overexpression in SW480 colorectal cancer cells resulted in ZEB2 induction, E-cadherin loss, and an EMT phenotype [97]. The role of TMPRSS4 in EMT induction has also been shown in HCC [98]. In this work, TMPRSS4 stimulated the of Raf/MEK/Erk1/2 pathway leading to activation of the EMT-TFs SNAIL and SLUG. Recently, a role for TMPRSS3 in EMT induction in gastric cancer was attributed to activation of Erk1/2 and PI3K/Akt signaling [99]. However, information on the direct molecular targets of the transmembrane serine proteases remained sparse. In contrast, TMPRSS2 activation of pro-HGF to the mature HGF was identified as critical step for downstream signaling of the HGF-receptor c-MET leading to EMT and motility of prostate cancer cells [100]. TMPRSS2 was strongly expressed in high-grade prostate cancers as well as in most prostate cancer metastases, and a TMPRSS2 inhibitor suppressed metastasis in a mouse model of prostate cancer.
Another recently emerging role of membrane-anchored serine proteases in cancer is their ability to cleave and activate protease-activated receptors (PARs). PARs (PAR1-PAR4) are transmembrane G protein–coupled receptors activated by proteolytic cleavage of their extracellular domain. When the N-terminal part is cleaved, the resulting fragment serves as a ligand activating the receptor and subsequently starting the G protein-dependent signaling cascade. A variety of proteases is able to cleave PARs, among them proteases from the coagulation cascade, the digestive tract, and inflammatory cells. PAR2 in particular has been shown to be overexpressed in a variety of advanced-stage tumors. TMPRSS control the timing and strength of PAR signaling by activating these receptors. Furthermore, proteases adjacent to PAR2 tether PAR2 signaling to local membrane microdomains thereby providing topically restricted input of PAR2 on cellular signaling networks [101].
In contrast to the transmembrane serine proteases, the secreted serine protease HTRA1 dampens cancer progression. High HTRA1 levels were associated with favorable outcome in breast cancer and other cancer entities [102, 103]. Epigenetic silencing mechanisms downregulate HTRA1 in premalignant lesions, invasive breast carcinoma, and breast cancer cell lines but not in normal mammary epithelium [104]. HTRA1 knockdown in non-tumorigenic MCF10A mammary cells triggered an EMT with all its molecular and phenotypic hallmarks. Furthermore, HTRA1 knockdown resulted in DNA damage response activation while HTRA1 overexpression prevented it. Consequently, the authors suggested a tumor-suppressive and drug-sensitizing role for HTRA1, although the molecular cues explaining these interesting findings remain obscure to date.
5 Regulating EMT by removal of protein modifications in the cytoplasm: deubiquitination and desumoylation
Ubiquitination is a fate-determining event for proteins. Modifying a target protein with the small proteinaceous ubiquitin tag defines its activity, intracellular localization, and stability, the latter by enabling the degradation of poly-ubiquitinated proteins by the proteasome [105, 106]. Ubiquitin ligases execute ubiquitination, which is frequently reverted by deubiquitinating enzymes (DUBs) [107, 108]. The human genome encodes for 102 putative DUB genes comprising two main classes: cysteine proteases and metalloproteases. The interplay of ubiquitin ligases and DUBs results in highly dynamic post-translational modifications with impact on many cellular signaling pathways. The ubiquitin-specific proteases (USP) are the largest family of DUBs and have lately come into focus as possible anticancer drug targets [109].
Interestingly, the ubiquitin-system regulates core EMT-inducing transcriptional factors: USPs take an active part in the induction of EMT by stabilizing EMT inducers like SLUG and SNAIL and interfering with the main EMT inducing pathways like Wnt/β-catenin and hedgehog signaling. For example it was shown that hypoxia-induced EMT in colorectal cancer cells is mediated through USP47-dependent stabilization of SNAIL [110]. USP47 is often upregulated in colorectal cancer tissues as well as cell lines in which hypoxia leads to activation of Sox9, a transcription factor that has been shown to regulate EMT genes like E-cadherin, N-cadherin, and Vimentin. Choi et al. could show that Sox9 also activates USP47 transcription, and the subsequent stabilization of SNAIL enabled this transcription factor to translocate to the nucleus and regulate the expression of further EMT genes. Along this line, USP10 and USP5 have been described to deubiquitinate the EMT transcription factor SLUG thereby preventing its degradation by the proteasome and promoting EMT [111, 112].
In addition to their role in stabilizing EMT-TFs, USPs regulate the pathways inducing the key EMT-TFs. For example, in osteosarcoma, USP7 induces EMT by direct binding to β-catenin, thus the Wnt/β-catenin signaling pathway is activated and EMT achieved [113]. Much work focused on the role of USPs in TGF-β signaling [114]. USPs, such as USP4, USP15, and UCH37 deubiquitinate the receptors of the TGF-β superfamily [115,116,117]. This affects trafficking and/or stability of the receptors. Furthermore, USPs are also involved in regulating the signal mediators of TGF-β, namely the SMAD proteins. USP9x as well as the DUBs OTUB1 and CYLD deubiquitinate SMAD4, SMAD2/3, and SMAD7, respectively [118,119,120].
The small ubiquitin-like modifier (SUMO) is a posttranslational modification similar to ubiquitin. The sentrin-specific protease (SENP) family of endopeptidases cleaves the isopeptide bond connecting SUMO with its target protein. However, SENPs are also important for maturing pro-SUMO to the form that can be ligated to its targets [121]. In principle, SENPs can interfere with EMT again at the level of the core EMT-TFs and with the intracellular signal transduction activating those. Compared with DUBs considerably fewer investigations addressed SENPs in EMT. SENP2 has been shown to interact with SMAD4 and desumoylate it [122]. In consequence, cells gained motility and other EMT features. An interesting feature is the interconnection of the ubiquitin- and sumoylation-systems. For example, SENP1 and SENP 2 can regulate the Ubiquitin ligase Snurf2, which is involved in TGF-β signaling [123].
6 Lysosomal proteases: EMT amplifiers and mediators of cell motility
The proteases of the endolysosomal cell compartment, often referred to as “the cathepsins”, have a high impact on cancer progression in various cancer entities [46, 47]. Located inside the acidic vesicles of cells, cathepsins have long been thought to merely digest the proteins shuttled to the lysosome. In recent years, however, it became clear that cathepsins have multiple roles in many physiological as well as pathophysiological processes related to neurodegenerative disorders, autophagy and aging [124, 125], antigen presentation [126], and cancer and metastasis [36, 48, 127]. This extended range of pathological involvement is due to the fact that cathepsins can be secreted from cells and can be active in the extracellular space. Slightly acidified conditions, as found in inflammation and cancer, support cathepsin stability and activity. Extracellularly, cathepsins can degrade ECM proteins but are also able to execute more selective membrane protein cleavages near the cell surface [128]. Intracellularly, cathepsins can escape the lysosome by a so-far ill-defined process called lysosomal membrane permeabilization and perform specific cleavages in the cytosol and nucleus [129, 130]. Nucleocytosolic cathepsins are frequently associated with cell death; however, as discussed below, also, more regulatory functions have been ascribed to them.
Similar to the other proteases discussed in this review, cathepsins interact with the TGF-β pathway. Kern et al. demonstrated that lysosomal protease activity during EMT of normal and malignant mammary epithelial cells was increased [131]..While broad-spectrum cysteine cathepsin inhibition had no effect on the TGF-β1-induced EMT signaling and did not affect migration of normal mammary epithelial cells, it did reduce the invasion of murine breast cancer cells. Interestingly, overexpressed SNAIL promoted Cathepsin B expression and secretion in colorectal cancer cells [132]. High levels of Cathepsins B and Z promoted EMT in various tumor cell lines and were associated with a more mesenchymal phenotype [133]. Simultaneous knockdown or inhibition of both cathepsins reverted the cells to a less aggressive, epithelial-like phenotype that could represent a mesenchymal-to-epithelial transition (MET). In HCC, a frequently observed genetic alteration is amplification of Cathepsin Z at 20q13.3 [134]. In line with the involvement of this protease in EMT, increased motility, upregulation of mesenchymal markers, and downregulation of epithelial markers were found in Cathepsin Z overexpressing cells as well as an upregulation of MMP-2,MMP-3, and MMP-9.
A complex involvement of cathepsins in the EMT program was suggested for Cathepsin L: Burton et al. found a positive feedback loop, where SNAIL promotes Cathepsin L activity with subsequent cleavage-mediated activation of the transcription factor CUX1 that induced again the transcription of SNAIL [135]. It had been shown before that CUX1 is able to stimulate migration and invasion by inducing the expression of SLUG and SNAIL and thus leading to transcriptional repression of E-cadherin and Occludin [136]. Hence, Cathepsin L appears to ensure efficient execution of EMT by activating CUX1. Cathepsin L also has a role in EMT-mediated drug resistance of lung cancer cells [137]. Cells resistant to DNA-damaging drugs had a typical mesenchymal appearance and EMT marker profile as well as high Cathepsin L levels. Silencing of Cathepsin L reverted the drug resistance in those cells. Similar observations were made for non-small cell lung cancers, in which irradiation caused p53 mutations, Cathepsin L overexpression, and EMT [138]. Furthermore, in MCF-7 breast and A549 lung carcinoma cells, knockdown of Cathepsin L suppressed the TGF-β-mediated activation of PI3K/Akt and Wnt signaling pathways, thereby abrogating SNAIL expression and EMT [139].
7 Synopsis: EMT, proteases, and cancer
Our extensive literature survey revealed that multiple protease classes are tightly linked to initiation, regulation, and execution of EMT as a key oncogenic mechanism (Table 1). Of note, researchers studied the involvement of extracellular acting proteases, i.e., MMPs, ADAMs, and serine proteases, in EMT more extensively than that of intracellular proteases. Extracellular proteases often liberate and activate latent EMT-inducing factors. A prominent example is the proteolytic release of TGF-β from latent TGF-β complexes by several MMPs [83, 84]. Extracellular proteases also cleave and disable cell adhesion molecules, as for example ADAM15 cleaving E-cadherin, thereby ensuring effective EMT progression [94]. Finally, extracellular proteases also cleave ECM for remodeling the tumor stroma as prerequisite for cancer invasion [140, 141]. Although there are clear-cut examples for any of the steps of extracellular protease involvement in EMT, the truth is probably that a given protease contributes to several of the EMT stages, e.g., by liberating EMT-inducing growth factors and degrading ECM. Furthermore, it is conceivable that several extracellular proteolytic systems cooperate in those tasks; however, those matters have not been addressed comprehensively yet and are subject to further exploration by systems biology approaches [142].
In this review, we have been focusing on the deubiquitinating and desumoylating proteases as major representatives of intracellular cytosolic proteases. These proteases function in all intracellular EMT pathways with most experimental work devoted to TGF-β signaling. In addition, DUBs directly enhance EMT by stabilizing key EMT-TFs by removal of degradation marks. However, most DUBs and SENPs have multiple targets and several members of the protease families can remove ubiquitin or SUMO from a given target protein. As slightly different results have been obtained for different cancer types and/or different cancer cell lines, one should probably be aware of a great context specificity of the observations made. On the other hand, this situation offers the chance for therapeutic windows in which “EMT-proteases” critical for a cancer type could be selectively targeted without unacceptable general toxicity.
Surprisingly, also lysosomal proteases show strong association with EMT progression. Part of the explanation for those findings might be that cancer cells, and also fibroblasts and immune cells of the tumor microenvironment, frequently secrete lysosomal cathepsins. Subsequently cathepsins act as extracellular proteases as discussed above. There has been much work on the action of cytosolic and nuclear cathepsins, especially Cathepsin L, on EMT regulation under conditions not inducing apoptotic or necrotic lysosomal cell death. Remarkably, there is little evidence for interference of cytosolic cathepsins with typical EMT signaling, i.e., TGF-β and Wnt pathways. Instead, proteolytic activation of the transcription factor CUX1 results in subsequent transcriptional upregulation of core EMT-TFs. Since EMT and EMT-TFs also induce cathepsins, nucleocytosolic cathepsins appear to function in positive feedback loops as EMT amplifiers. To date, cathepsin involvement in EMT has not been much studied in the context of the primary localization of these proteases—the lysosome. This is surprising, because research of the past decade identified the lysosome as central hub for cell signaling [143]. It might be worthwhile to pursue along those avenues.
A final note is that many of the reports cited in this review showed positive associations of protease expression, EMT, and poor patient prognosis. However, studies scrutinizing whether EMT protease expressions are indeed independent prognostic factors are missing. In addition, one should not stick to the dogma of proteases being poor prognostic factors and EMT drivers. There is a longstanding notion of proteases having tumor-suppressive functions [144, 145]. In this review, one example for a potentially EMT-suppressive protease is the serine protease HTRA1. We are convinced that further exploration of this issues will result in both—exciting science and benefit for cancer patients.
Abbreviations
- A549:
-
lung carcinoma cell line
- ADAM:
-
a disintegrin and metalloprotease
- c-MET:
-
MET proto-oncogene
- CRC:
-
colorectal cancer
- CUX1:
-
cut like homeobox 1
- CYLD:
-
CYLD lysine 63 deubiquitinase
- DUB:
-
deubiquitinating enzymes
- e.g. :
-
exempli gratia
- E/M:
-
epithelial/mesenchymal
- ECM:
-
extracellular matrix
- EGFR:
-
epidermal growth factor receptor
- EMT:
-
epithelial-to-mesenchymal transition
- EMT-TF:
-
epithelial-to-mesenchymal transition—transcription factor
- Epcam, Vcam:
-
epithelial cell adhesion molecule, vascular cell adhesion molecule
- ERK:
-
extracellular signal-regulated kinase (also known as mitogen-activated protein kinase 1—MAPK1)
- ESCC:
-
esophageal squamous cell carcinoma
- FGF:
-
fibroblast growth factor
- HCC:
-
hepatocellular carcinoma
- HER:
-
human epidermal growth factor receptor
- HGF:
-
hepatocyte growth factor
- HTRA1:
-
HtrA serine peptidase 1
- i.e. :
-
id est
- IL-6:
-
interleukin-6
- JNK:
-
c-Jun N-terminal kinase
- LA:
-
lung adenocarcinoma
- LGL:
-
lethal giant larvae protein
- LNCaP cells:
-
prostate adenocarcinoma cell from lymph node metastasis
- MCF10A:
-
non-tumorigenic mammary cell line (human origin)
- MCF-7:
-
breast carcinoma cell line
- MET:
-
mesenchymal-to-epithelial transition
- MMP:
-
matrix metalloprotease
- MSPL:
-
mosaic serine protease large-form
- MT-MMP:
-
membrane type matrix metalloprotease
- NMuMG:
-
a non-transformed mouse mammary gland epithelial cell line
- NSCLC:
-
non-small cell lung cancer
- OTUB:
-
OTU deubiquitinase
- PAR:
-
protease-activated receptor
- PATJ:
-
PALS1-associated tight junction protein
- PI3K/Akt/Rac1:
-
phosphatidylinositol-4,5-bisphosphate 3-kinase/AKT serine/threonine kinase 1/Rac1
- Rac1:
-
Ras-related C3 botulinum toxin substrate 1 (rho family, small GTP binding protein Rac1)
- RB:
-
retinoblastoma protein
- ROS:
-
reactive oxygen species
- SENP:
-
sentrin specific protease (family of endopeptidases)
- SMAD:
-
from “MAD—mothers against decapentaplegic” (gene in Drosophila melanogaster) and “Sma—small body size” (protein in Caenorhabditis elegans)
- SW480:
-
human colon cancer cell line
- TGF-β:
-
transforming growth factor-beta
- TMPRSS:
-
transmembrane protease/serine subfamily of the type II transmembrane serine protease (TTSP) family
- TTSP:
-
type II transmembrane serine protease family
- uPA:
-
urokinase-type plasminogen activator
- USP:
-
ubiquitin specific proteases
- Wnt:
-
from “Wg—wingless” (gene in Drosophila melanogaster) and Int-1 (gene in Mus musculus)
- ZEB:
-
zinc finger E-box binding homeobox
- ZO-3:
-
zonula occludens 3, a tight junction protein
References
Greenburg, G., & Hay, E. D. (1982). Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. Journal of Cell Biology, 95(1), 333–339. https://doi.org/10.1083/jcb.95.1.333.
Thiery, J. P., Acloque, H., Huang, R. Y. J., & Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890. https://doi.org/10.1016/j.cell.2009.11.007.
Ahmed, N., Maines-Bandiera, S., Quinn, M. A., Unger, W. G., Dedhar, S., & Auersperg, N. (2006). Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. American Journal of Physiology-Cell Physiology, 290(6), C1532–C1542. https://doi.org/10.1152/ajpcell.00478.2005.
Arnoux, V., Nassour, M., L’Helgoualc’h, A., Hipskind, R. A., & Savagner, P. (2008). Erk5 controls slug expression and keratinocyte activation during wound healing. Molecular Biology of the Cell, 19(11), 4738–4749. https://doi.org/10.1091/mbc.e07-10-1078.
Iwano, M., Plieth, D., Danoff, T. M., Xue, C., Okada, H., & Neilson, E. G. (2002). Evidence that fibroblasts derive from epithelium during tissue fibrosis. The Journal of Clinical Investigation, 110(3), 341–350. https://doi.org/10.1172/JCI15518.
Stone, R. C., Pastar, I., Ojeh, N., Chen, V., Liu, S., Garzon, K. I., & Tomic-Canic, M. (2016). Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell and Tissue Research. NIH Public Access. https://doi.org/10.1007/s00441-016-2464-0.
Grande, M. T., Sánchez-Laorden, B., López-Blau, C., De Frutos, C. A., Boutet, A., Arévalo, M., … Nieto, M. A. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nature Medicine, 21(9), 989–997. doi:https://doi.org/10.1038/nm.3901.
Peinado, H., Olmeda, D., & Cano, A. (2007, March). Snail, ZEB and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews. Cancer. https://doi.org/10.1038/nrc2131.
Shin, S., Dimitri, C. A., Yoon, S. O., Dowdle, W., & Blenis, J. (2010). ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Molecular Cell, 38(1), 114–127. https://doi.org/10.1016/j.molcel.2010.02.020.
Janda, E., Lehmann, K., Killisch, I., Jechlinger, M., Herzig, M., Downward, J., … Grünert, S. (2002). Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis. The Journal of Cell Biology, 156(2), 299–314. doi:https://doi.org/10.1083/jcb.200109037.
Puisieux, A., Brabletz, T., & Caramel, J. (2014). Oncogenic roles of EMT-inducing transcription factors. Nature Cell Biology. https://doi.org/10.1038/ncb2976.
Stemmler, M. P., Eccles, R. L., Brabletz, S., & Brabletz, T. (2019). Non-redundant functions of EMT transcription factors. Nature Cell Biology. https://doi.org/10.1038/s41556-018-0196-y.
Savagner, P., Yamada, K. M., & Thiery, J. P. (1997). The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor–induced epithelial–mesenchymal transition. The Journal of Cell Biology, 137(6), 1403–1419. https://doi.org/10.1083/JCB.137.6.1403.
Vandewalle, C., Comijn, J., De Craene, B., Vermassen, P., Bruyneel, E., Andersen, H., … Berx, G. (2005). SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Research, 33(20), 6566–78. doi:https://doi.org/10.1093/nar/gki965.
Aigner, K., Dampier, B., Descovich, L., Mikula, M., Sultan, A., Schreiber, M., … Eger, A. (2007). The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene, 26(49), 6979–6988. doi:https://doi.org/10.1038/sj.onc.1210508.
Spaderna, S., Schmalhofer, O., Wahlbuhl, M., Dimmler, A., Bauer, K., Sultan, A., … Brabletz, T. (2008). The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Research, 68(2), 537–544. doi:https://doi.org/10.1158/0008-5472.CAN-07-5682.
Yilmaz, M., & Christofori, G. (2009). EMT, the cytoskeleton, and cancer cell invasion. Cancer and Metastasis Reviews, 28(1–2), 15–33. https://doi.org/10.1007/s10555-008-9169-0.
Miyoshi, A., Kitajima, Y., Sumi, K., Sato, K., Hagiwara, A., Koga, Y., & Miyazaki, K. (2004). Snail and SIP1 increase cancer invasion by upregulating MMP family in hepatocellular carcinoma cells. British Journal of Cancer, 90(6), 1265–1273. https://doi.org/10.1038/sj.bjc.6601685.
Jordà, M., Olmeda, D., Vinyals, A., Valero, E., Cubillo, E., Llorens, A., … Fabra, A. (2005). Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the snail transcription factor. Journal of Cell Science, 118(Pt 15), 3371–85. doi:https://doi.org/10.1242/jcs.02465.
Hu, F., Wang, C., Guo, S., Sun, W., Mi, D., Gao, Y., … Yang, S. (2011). δEF1 promotes osteolytic metastasis of MDA-MB-231 breast cancer cells by regulating MMP-1 expression. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, 1809(3), 200–210. doi:https://doi.org/10.1016/J.BBAGRM.2011.01.003.
Sanchez-Tillo, E., de Barrios, O., Siles, L., Cuatrecasas, M., Castells, A., & Postigo, A. (2011). Catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proceedings of the National Academy of Sciences, 108(48), 19204–19209. https://doi.org/10.1073/pnas.1108977108.
Wu, W.-S., You, R.-I., Cheng, C.-C., Lee, M.-C., Lin, T.-Y., & Hu, C.-T. (2017). Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Scientific Reports, 7(1), 17753. https://doi.org/10.1038/s41598-017-18101-7.
Drak Alsibai, K., & Meseure, D. (2018). Tumor microenvironment and noncoding RNAs as co-drivers of epithelial–mesenchymal transition and cancer metastasis. Developmental Dynamics. John Wiley & Sons, Ltd. doi:https://doi.org/10.1002/dvdy.24548.
Korpal, M., Ell, B. J., Buffa, F. M., Ibrahim, T., Blanco, M. A., Celià-Terrassa, T., … Kang, Y. (2011). Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nature Medicine, 17(9), 1101–1109. doi:https://doi.org/10.1038/nm.2401.
Valastyan, S., & Weinberg, R. A. (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell, 147(2), 275–292. https://doi.org/10.1016/j.cell.2011.09.024.
Brabletz, T. (2012). To differentiate or not—routes towards metastasis. Nature Reviews Cancer, 12(6), 425–436. https://doi.org/10.1038/nrc3265.
Polyak, K., & Weinberg, R. A. (2009, April). Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Reviews. Cancer. https://doi.org/10.1038/nrc2620.
Marusyk, A., & Polyak, K. (2010). Tumor heterogeneity: causes and consequences. Biochimica et Biophysica Acta, 1805(1), 105–117. https://doi.org/10.1016/j.bbcan.2009.11.002.
Meacham, C. E., & Morrison, S. J. (2013). Tumor heterogeneity and cancer cell plasticity. Nature, 501(7467), 328. https://doi.org/10.1038/NATURE12624.
Grosse-Wilde, A., Fouquier d’Hérouël, A., McIntosh, E., Ertaylan, G., Skupin, A., Kuestner, R. E., … Huang, S. (2015). Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS One, 10(5), e0126522. doi:https://doi.org/10.1371/journal.pone.0126522.
Jolly, M. K., Tripathi, S. C., Jia, D., Mooney, S. M., Celiktas, M., Hanash, S. M., … Levine, H. (2016). Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget, 7(19), 27067–27084. https://doi.org/10.18632/oncotarget.8166.
Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., … Blanpain, C. (2018). Identification of the tumour transition states occurring during EMT. Nature, 556(7702), 463–468. doi:https://doi.org/10.1038/s41586-018-0040-3.
Kröger, C., Afeyan, A., Mraz, J., Eaton, E. N., Reinhardt, F., Khodor, Y. L., … Weinberg, R. A. (2019). Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proceedings of the National Academy of Sciences, 201812876. doi:https://doi.org/10.1073/pnas.1812876116.
Wiedow, O., & Meyer-Hoffert, U. (2005). Neutrophil serine proteases: Potential key regulators of cell signalling during inflammation. Journal of Internal Medicine. John Wiley & Sons, ltd (10.1111). https://doi.org/10.1111/j.1365-2796.2005.01476.x.
Turk, B., Turk, D., & Turk, V. (2012). Protease signalling: the cutting edge. The EMBO Journal, 31(7), 1630–1643. https://doi.org/10.1038/emboj.2012.42.
Pišlar, A., Perišić Nanut, M., & Kos, J. (2015). Lysosomal cysteine peptidases – molecules signaling tumor cell death and survival. Seminars in Cancer Biology, 35, 168–179. https://doi.org/10.1016/J.SEMCANCER.2015.08.001.
Zhao, P., Metcalf, M., & Bunnett, N. W. (2014). Biased signaling of protease-activated receptors. Frontiers in Endocrinology, 5, 67. https://doi.org/10.3389/fendo.2014.00067.
Winer, A., Adams, S., & Mignatti, P. (2018). Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Molecular Cancer Therapeutics, 17(6), 1147–1155. https://doi.org/10.1158/1535-7163.MCT-17-0646.
Coussens, L. M., Fingleton, B., & Matrisian, L. M. (2002). Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science (New York, N.Y.), 295(5564), 2387–2392. https://doi.org/10.1126/science.1067100.
Löser, R., & Pietzsch, J. (2015). Cysteine cathepsins: their role in tumor progression and recent trends in the development of imaging probes. Frontiers in Chemistry, 3, 37. https://doi.org/10.3389/fchem.2015.00037.
Vandooren, J., Opdenakker, G., Loadman, P. M., & Edwards, D. R. (2016). Proteases in cancer drug delivery. Advanced Drug Delivery Reviews, 97, 144–155. https://doi.org/10.1016/j.addr.2015.12.020.
Vidak, E., Javoršek, U., Vizovišek, M., & Turk, B. (2019). Cysteine Cathepsins and their extracellular roles: shaping the microenvironment. Cells, 8(3), 264. https://doi.org/10.3390/cells8030264.
Weidle, U. H., Tiefenthaler, G., & Georges, G. (2014). Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genomics and Proteomics, 11(2), 67-79.
Sloane, B. F., List, K., Fingleton, B., & Matrisian, L. (2013). Proteases in cancer: significance for invasion and metastasis. In Proteases: structure and function (pp. 491–550). Vienna: Springer Vienna. https://doi.org/10.1007/978-3-7091-0885-7_15.
Sevenich, L., & Joyce, J. A. (2014). Pericellular proteolysis in cancer. Genes & Development, 28(21), 2331–2347. https://doi.org/10.1101/gad.250647.114.
Olson, O. C., & Joyce, J. A. (2015). Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nature Reviews Cancer, 15(12), 712–729. https://doi.org/10.1038/nrc4027.
Mohamed, M. M., & Sloane, B. F. (2006). Multifunctional enzymes in cancer. Nature Reviews Cancer, 6(10), 764–775. https://doi.org/10.1038/nrc1949.
Anja, P., Anahid, J., & Janko, K. (2018). Cysteine cathepsins: their biological and molecular significance in cancer stem cells. Seminars in Cancer Biology, 53, 168–177. https://doi.org/10.1016/J.SEMCANCER.2018.07.010.
Eatemadi, A., Aiyelabegan, H. T., Negahdari, B., Mazlomi, M. A., Daraee, H., Daraee, N., … Sadroddiny, E. (2017). Role of protease and protease inhibitors in cancer pathogenesis and treatment. doi:https://doi.org/10.1016/j.biopha.2016.12.021.
Friedl, P., & Wolf, K. (2010). Plasticity of cell migration: a multiscale tuning model. The Journal of Cell Biology, 188(1), 11–19. https://doi.org/10.1083/jcb.200909003.
Friedl, P., & Alexander, S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell Cell Press. https://doi.org/10.1016/j.cell.2011.11.016.
Kessenbrock, K., Plaks, V., & Werb, Z. (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell, 141(1), 52–67. https://doi.org/10.1016/j.cell.2010.03.015.
Duffy, M. J., McKiernan, E., O’Donovan, N., & McGowan, P. M. (2009). Role of ADAMs in cancer formation and progression. Clinical Cancer Research, 15(4), 1140–1144. https://doi.org/10.1158/1078-0432.CCR-08-1585.
Tanabe, L. M., & List, K. (2017). The role of type II transmembrane serine protease-mediated signaling in cancer. The FEBS Journal, 284(10), 1421–1436. https://doi.org/10.1111/febs.13971.
Noë, V., Fingleton, B., Jacobs, K., Crawford, H. C., Vermeulen, S., Steelant, W., … Mareel, M. (2001). Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. Journal of Cell Science, 114(Pt 1), 111–118.
Deryugina, E. I., Ratnikov, B., Monosov, E., Postnova, T. I., DiScipio, R., Smith, J. W., & Strongin, A. Y. (2001). MT1-MMP initiates activation of pro-MMP-2 and integrin αvβ3 promotes maturation of MMP-2 in breast carcinoma cells. Experimental Cell Research, 263(2), 209–223. https://doi.org/10.1006/EXCR.2000.5118.
Kajita, M., Itoh, Y., Chiba, T., Mori, H., Okada, A., Kinoh, H., & Seiki, M. (2001). Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. Journal of Cell Biology, 153(5), 893–904. https://doi.org/10.1083/jcb.153.5.893.
Symowicz, J., Adley, B. P., Gleason, K. J., Johnson, J. J., Ghosh, S., Fishman, D. A., … Stack, M. S. (2007). Engagement of collagen-binding integrins promotes matrix metalloproteinase-9–dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Research, 67(5), 2030–2039. doi:https://doi.org/10.1158/0008-5472.CAN-06-2808.
Lochter, A., Galosy, S., Muschler, J., Freedman, N., Werb, Z., & Bissell, M. J. (1997). Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. The Journal of Cell Biology, 139(7), 1861–1872.
Maretzky, T., Reiss, K., Ludwig, A., Buchholz, J., Scholz, F., Proksch, E., … Saftig, P. (2005). ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proceedings of the National Academy of Sciences of the United States of America, 102(26), 9182–7. doi:https://doi.org/10.1073/pnas.0500918102.
Covington, M. D., Burghardt, R. C., & Parrish, A. R. (2006). Ischemia-induced cleavage of cadherins in NRK cells requires MT1-MMP (MMP-14). American Journal of Physiology. Renal Physiology, 290(1), F43–F51. https://doi.org/10.1152/ajprenal.00179.2005.
Egeblad, M., & Werb, Z. (2002). New functions for the matrix metalloproteinases in cancer progression. Nature Reviews Cancer, 2(3), 161–174. https://doi.org/10.1038/nrc745.
Codony-Servat, J., Albanell, J., Lopez-Talavera, J. C., Arribas, J., & Baselga, J. (1999). Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Research, 59(6), 1196–1201.
Vecchi, M., Rudolph-Owen, L. A., Brown, C. L., Dempsey, P. J., & Carpenter, G. (1998). Tyrosine phosphorylation and proteolysis. Pervanadate-induced, metalloprotease-dependent cleavage of the ErbB-4 receptor and amphiregulin. The Journal of Biological Chemistry, 273(32), 20589–20595. https://doi.org/10.1074/JBC.273.32.20589.
Nath, D., Williamson, N. J., Jarvis, R., & Murphy, G. (2001). Shedding of c-met is regulated by crosstalk between a G-protein coupled receptor and the EGF receptor and is mediated by a TIMP-3 sensitive metalloproteinase. Journal of Cell Science, 114(Pt 6), 1213–1220. https://doi.org/10.1242/jcs.152595.
Illman, S. A., Lehti, K., Keski-Oja, J., & Lohi, J. (2006). Epilysin (MMP-28) induces TGF-beta mediated epithelial to mesenchymal transition in lung carcinoma cells. Journal of Cell Science, 119(Pt 18), 3856–3865. https://doi.org/10.1242/jcs.03157.
Zheng, G., Lyons, J. G., Thian, K. T., Wang, Y., Hsu, T. T., Min, D., … Harris, D. C. H. (2009). Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-β1 in renal tubular epithelial cells. American Journal of Pathology, 175(2), 580–591. doi:https://doi.org/10.2353/ajpath.2009.080983.
Cheng, S., & Lovett, D. H. (2003). Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. The American Journal of Pathology, 162(6), 1937–1949. https://doi.org/10.1016/S0002-9440(10)64327-1.
Tan, T. K., Zheng, G., Hsu, T. T., Wang, Y., Lee, V. W. S., Tian, X., … Harris, D. C. H. (2010). Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. American Journal of Pathology, 176(3), 1256–1270. doi:https://doi.org/10.2353/ajpath.2010.090188.
Cao, J., Chiarelli, C., Richman, O., Zarrabi, K., Kozarekar, P., & Zucker, S. (2008). Membrane type 1 matrix metalloproteinase induces epithelial-to-mesenchymal transition in prostate cancer. The Journal of Biological Chemistry, 283(10), 6232–6240. https://doi.org/10.1074/jbc.M705759200.
Sternlicht, M. D., Bissell, M. J., & Werb, Z. (2000). The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene, 19(8), 1102–1113. https://doi.org/10.1038/sj.onc.1203347.
Radisky, D. C., Levy, D. D., Littlepage, L. E., Liu, H., Nelson, C. M., Fata, J. E., … Bissell, M. J. (2005). Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature, 436(7047), 123–7. doi:https://doi.org/10.1038/nature03688.
Chen, Q. K., Lee, K., Radisky, D. C., & Nelson, C. M. (2013). Extracellular matrix proteins regulate epithelial-mesenchymal transition in mammary epithelial cells. Differentiation; Research in Biological Diversity, 86(3), 126–132. https://doi.org/10.1016/j.diff.2013.03.003.
Lee, K., Chen, Q. K., Lui, C., Cichon, M. A., Radisky, D. C., & Nelson, C. M. (2012). Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial-mesenchymal transition. Molecular Biology of the Cell, 23(20), 4097–4108. https://doi.org/10.1091/mbc.E12-02-0166.
Yu, G., Herazo-Maya, J. D., Nukui, T., Romkes, M., Parwani, A., Juan-Guardela, B. M., … Kass, D. J. (2014). Matrix metalloproteinase-19 promotes metastatic behavior in vitro and is associated with increased mortality in non-small cell lung cancer. American Journal of Respiratory and Critical Care Medicine, 190(7), 780–90. doi:https://doi.org/10.1164/rccm.201310-1903OC.
Pastushenko, I., & Blanpain, C. (2019). EMT transition states during tumor progression and metastasis. Trends in Cell Biology. Elsevier Current Trends. https://doi.org/10.1016/j.tcb.2018.12.001.
Hillebrand, L. E., Wickberg, S. M., Gomez-Auli, A., Follo, M., Maurer, J., Busch, H., … Reinheckel, T. (2018). MMP14 empowers tumor-initiating breast cancer cells under hypoxic nutrient-depleted conditions. The FASEB Journal, 33(3), 4124–4140. doi:https://doi.org/10.1096/fj.201801127r.
Pang, L., Li, Q., Li, S., He, J., Cao, W., Lan, J., … Li, F. (2016). Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: observations from clinical and in vitro analyses. Scientific Reports, 6(1), 22179. doi:https://doi.org/10.1038/srep22179.
Yang, C.-C., Zhu, L.-F., Xu, X.-H., Ning, T.-Y., Ye, J.-H., & Liu, L.-K. (2013). Membrane type 1 matrix metalloproteinase induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. BMC Cancer, 13(1), 171. https://doi.org/10.1186/1471-2407-13-171.
Nguyen, H.-L., Kadam, P., Helkin, A., Cao, K., Wu, S. J., Samara, G., …, Cao, J. (2016). MT1-MMP activation of TGF-? Signaling enables intercellular activation of an epithelial-mesenchymal transition program in cancer. Current Cancer Drug Targets, 16(7), 618–630. doi:https://doi.org/10.2174/1568009616666160216125634.
Qin, G., Luo, M., Chen, J., Dang, Y., Chen, G., Li, L., …, Yang, J. (2016). Reciprocal activation between MMP-8 and TGF-β1 stimulates EMT and malignant progression of hepatocellular carcinoma. Cancer Letters, 374(1), 85–95. doi:https://doi.org/10.1016/j.canlet.2016.02.001.
Bai, X., Li, Y.-Y., Zhang, H.-Y., Wang, F., He, H.-L., Yao, J.-C., …, Li, S.-S. (2017). Role of matrix metalloproteinase-9 in transforming growth factor-β1-induced epithelial-mesenchymal transition in esophageal squamous cell carcinoma. OncologyTargets and Therapy, 10, 2837–2847. doi:https://doi.org/10.2147/OTT.S134813.
Krstic, J., & Santibanez, J. F. (2014). Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. TheScientificWorldJournal, 2014, 521754. https://doi.org/10.1155/2014/521754.
Santibanez, J. F., Obradović, H., Kukolj, T., & Krstić, J. (2018). Transforming growth factor-β, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Developmental Dynamics, 247(3), 382–395. https://doi.org/10.1002/dvdy.24554.
Weber, S., & Saftig, P. (2012). Ectodomain shedding and ADAMs in development. Development, 139(20), 3693–3709. https://doi.org/10.1242/DEV.076398.
Murphy, G. (2008). The ADAMs: signalling scissors in the tumour microenvironment. Nature Reviews Cancer, 8(12), 932–941. https://doi.org/10.1038/nrc2459.
Becherer, J. D., & Blobel, C. P. (2003). Biochemical properties and functions of membrane-anchored metalloprotease-disintegrin proteins (ADAMs). Current Topics in Developmental Biology, 54, 101–123. https://doi.org/10.1016/S0070-2153(03)54006-6.
Dong, Y., Wu, Z., He, M., Chen, Y., Chen, Y., Shen, X., … Zeng, Z. (2018). ADAM9 mediates the interleukin-6-induced epithelial–mesenchymal transition and metastasis through ROS production in hepatoma cells. Cancer Letters, 421, 1–14. doi:https://doi.org/10.1016/J.CANLET.2018.02.010.
Pruessmeyer, J., & Ludwig, A. (2009). The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Seminars in Cell & Developmental Biology, 20(2), 164–174. https://doi.org/10.1016/J.SEMCDB.2008.09.005.
Atapattu, L., Saha, N., Chheang, C., Eissman, M. F., Xu, K., Vail, M. E., … Janes, P. W. (2016). An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth. Journal of Experimental Medicine, 213(9), 1741–1757. doi:https://doi.org/10.1084/JEM.20151095.
Cai, M., Wang, Z., Zhang, J., Zhou, H., Jin, L., Bai, R., & Weng, Y. (2015). Adam17, a target of Mir-326, promotes Emt-induced cells invasion in lung adenocarcinoma. Cellular Physiology and Biochemistry, 36(3), 1175–1185. https://doi.org/10.1159/000430288.
Ruff, M., Leyme, A., Le Cann, F., Bonnier, D., Le Seyec, J., Chesnel, F., … Théret, N. (2015). The disintegrin and metalloprotease ADAM12 is associated with TGF-β-induced epithelial to mesenchymal transition. PLoS One, 10(9), e0139179. doi:https://doi.org/10.1371/journal.pone.0139179.
Wang, J., Zhang, Z., Li, R., Mao, F., Sun, W., Chen, J., … Lei, T. (2018). ADAM12 induces EMT and promotes cell migration, invasion and proliferation in pituitary adenomas via EGFR/ERK signaling pathway. Biomedicine & Pharmacotherapy, 97, 1066–1077. doi:https://doi.org/10.1016/J.BIOPHA.2017.11.034.
Najy, A. J., Day, K. C., & Day, M. L. (2008). The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. Journal of Biological Chemistry, 283(26), 18393–18401. https://doi.org/10.1074/JBC.M801329200.
Bugge, T. H., Antalis, T. M., & Wu, Q. (2009). Type II transmembrane serine proteases. The Journal of Biological Chemistry, 284(35), 23177–23181. https://doi.org/10.1074/jbc.R109.021006.
Choi, S. Y., Bertram, S., Glowacka, I., Park, Y. W., & Pöhlmann, S. (2009, July 1). Type II transmembrane serine proteases in cancer and viral infections. Trends in Molecular Medicine. Elsevier Current Trends. https://doi.org/10.1016/j.molmed.2009.05.003.
Jung, H., Lee, K. P., Park, S. J., Park, J. H., Jang, Y., Choi, S.-Y., … Park, Y. W. (2008). TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial–mesenchymal transition. Oncogene, 27(18), 2635–2647. doi:https://doi.org/10.1038/sj.onc.1210914.
Wang, C.-H., Guo, Z.-Y., Chen, Z.-T., Zhi, X.-T., Li, D.-K., Dong, Z.-R., … Li, T. (2015). TMPRSS4 facilitates epithelial-mesenchymal transition of hepatocellular carcinoma and is a predictive marker for poor prognosis of patients after curative resection OPEN. doi:https://doi.org/10.1038/srep12366.
Li, S.-L., Chen, X., Wu, T., Zhang, X.-W., Li, H., Zhang, Y., & Ji, Z.-Z. (2018). Knockdown of TMPRSS3 inhibits gastric cancer cell proliferation, invasion and EMT via regulation of the ERK1/2 and PI3K/Akt pathways. Biomedicine & Pharmacotherapy, 107, 841–848. https://doi.org/10.1016/J.BIOPHA.2018.08.023.
Lucas, J. M., Heinlein, C., Kim, T., Hernandez, S. A., Malik, M. S., True, L. D., … Nelson, P. S. (2014). The androgen-regulated protease TMPRSS2 activates aProteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discovery, 4(11), 1310. doi:https://doi.org/10.1158/2159-8290.CD-13-1010.
Pawar, N. R., Buzza, M. S., & Antalis, T. M. (2019). Membrane-anchored serine proteases and protease-activated receptor-2–mediated signaling: co-conspirators in cancer progression. Cancer Research, 79(2), 301–310. https://doi.org/10.1158/0008-5472.CAN-18-1745.
Lehner, A., Magdolen, V., Schuster, T., Kotzsch, M., Kiechle, M., Meindl, A., … Gross, E. (2013). Downregulation of serine protease HTRA1 is associated with poor survival in breast cancer. PLoS One, 8(4), e60359. doi:https://doi.org/10.1371/journal.pone.0060359.
Zhu, F., Duan, Y.-F., Bao, W.-Y., Liu, W.-S., Yang, Y., & Cai, H.-H. (2015). HtrA1 regulates epithelial–mesenchymal transition in hepatocellular carcinoma. Biochemical and Biophysical Research Communications, 467(3), 589–594. https://doi.org/10.1016/j.bbrc.2015.09.105.
Wang, N., Eckert, K. A., Zomorrodi, A. R., Xin, P., Pan, W., Shearer, D. A., … Clawson, G. A. (2012). Down-regulation of HtrA1 activates the epithelial-mesenchymal transition and ATM DNA damage response pathways. PLoS One, 7(6), e39446. doi:https://doi.org/10.1371/journal.pone.0039446.
Glickman, M. H., & Ciechanover, A. (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews, 82(2), 373–428. https://doi.org/10.1152/physrev.00027.2001.
Pickart, C. M., & Eddins, M. J. (2004). Ubiquitin: structures, functions, mechanisms. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1695(1–3), 55–72. https://doi.org/10.1016/J.BBAMCR.2004.09.019.
Pfoh, R., Lacdao, I. K., & Saridakis, V. (2015). Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocrine-Related Cancer, 22(1), T35. https://doi.org/10.1530/ERC-14-0516.
Farshi, P., Deshmukh, R. R., Nwankwo, J. O., Arkwright, R. T., Cvek, B., Liu, J., & Dou, Q. P. (2015). Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opinion on Therapeutic Patents, 25(10), 1191–1208. https://doi.org/10.1517/13543776.2015.1056737.
Yuan, T., Yan, F., Ying, M., Cao, J., He, Q., Zhu, H., & Yang, B. (2018). Inhibition of ubiquitin-specific proteases as a novel anticancer therapeutic strategy. Frontiers in Pharmacology, 9, 1080. https://doi.org/10.3389/fphar.2018.01080.
Choi, B.-J., Park, S.-A., Lee, S.-Y., Cha, Y. N., & Surh, Y.-J. (2017). Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of snail: a potential role of Sox9. Scientific Reports, 7(1), 15918. https://doi.org/10.1038/s41598-017-15139-5.
Meng, J., Ai, X., Lei, Y., Zhong, W., Qian, B., Qiao, K., … Yang, C. (2019). USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics, 9(2), 573–587. doi:https://doi.org/10.7150/thno.27654.
Ouchida, A. T., Kacal, M., Zheng, A., Ambroise, G., Zhang, B., Norberg, E., & Vakifahmetoglu-Norberg, H. (2018). USP10 regulates the stability of the EMT-transcription factor slug/SNAI2. Biochemical and Biophysical Research Communications, 502(4), 429–434. https://doi.org/10.1016/J.BBRC.2018.05.156.
Zeng, Q., Li, Z., Zhao, X., Guo, L., Yu, C., Qin, J., … Yang, X. (2018). Ubiquitin-specific protease 7 promotes osteosarcoma cell metastasis by inducing epithelial-mesenchymal transition. Oncology Reports, 41(1), 543–551. doi:https://doi.org/10.3892/or.2018.6835.
Liu, S., de Boeck, M., van Dam, H., & ten Dijke, P. (2016). Regulation of the TGF-β pathway by deubiquitinases in cancer. The International Journal of Biochemistry & Cell Biology, 76, 135–145. https://doi.org/10.1016/J.BIOCEL.2016.05.001.
Wicks, S. J., Haros, K., Maillard, M., Song, L., Cohen, R. E., ten Dijke, P., & Chantry, A. (2005). The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene, 24(54), 8080–8084. https://doi.org/10.1038/sj.onc.1208944.
Eichhorn, P. J. A., Rodón, L., Gonzàlez-Juncà, A., Dirac, A., Gili, M., Martínez-Sáez, E., et al. (2012). USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nature Medicine, 18(3), 429–435. https://doi.org/10.1038/nm.2619.
Zhang, L., Zhou, F., Drabsch, Y., Gao, R., Snaar-Jagalska, B. E., Mickanin, C., … ten Dijke, P. (2012). USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nature Cell Biology, 14(7), 717–726. doi:https://doi.org/10.1038/ncb2522.
Wiener, R., Zhang, X., Wang, T., & Wolberger, C. (2012). The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature, 483(7391), 618–622. https://doi.org/10.1038/nature10911.
Dupont, S., Mamidi, A., Cordenonsi, M., Montagner, M., Zacchigna, L., Adorno, M., … Piccolo, S. (2009). FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell, 136(1), 123–135. doi:https://doi.org/10.1016/J.CELL.2008.10.051.
Zhao, Y., Thornton, A. M., Kinney, M. C., Ma, C. A., Spinner, J. J., Fuss, I. J., … Jain, A. (2011). The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor β (TGF-β) signaling and the development of regulatory T cells. The Journal of Biological Chemistry, 286(47), 40520. doi:https://doi.org/10.1074/JBC.M111.292961.
Chanda, A., Sarkar, A., & Bonni, S. (2018). The SUMO system and TGFβ signaling interplay in regulation of epithelial-mesenchymal transition: implications for cancer progression. Cancers, 10(8), E264. https://doi.org/10.3390/cancers10080264.
Chang, C.-C., Huang, Y.-S., Lin, Y.-M., Lin, C.-J., Jeng, J.-C., Liu, S.-M., … Shih, H.-M. (2018). The role of sentrin-specific protease 2 substrate recognition in TGF-β-induced tumorigenesis. Scientific Reports, 8(1), 9786. doi:https://doi.org/10.1038/s41598-018-28103-8.
Chandhoke, A. S., Karve, K., Dadakhujaev, S., Netherton, S., Deng, L., & Bonni, S. (2016). The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial–mesenchymal transition in a sumoylation-regulated manner. Cell Death and Differentiation, 23(5), 876. https://doi.org/10.1038/CDD.2015.152.
Stoka, V., Turk, V., & Turk, B. (2016). Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Research Reviews, 32, 22–37. https://doi.org/10.1016/J.ARR.2016.04.010.
Ketterer, S., Gomez-Auli, A., Hillebrand, L. E., Petrera, A., Ketscher, A., & Reinheckel, T. (2017). Inherited diseases caused by mutations in cathepsin protease genes. The FEBS Journal, 284(10), 1437–1454. https://doi.org/10.1111/febs.13980.
van Kasteren, S. I., & Overkleeft, H. S. (2014). Endo-lysosomal proteases in antigen presentation. Current Opinion in Chemical Biology, 23, 8–15. https://doi.org/10.1016/J.CBPA.2014.08.011.
Tan, G.-J., Peng, Z.-K., Lu, J.-P., & Tang, F.-Q. (2013). Cathepsins mediate tumor metastasis. World Journal of Biological Chemistry, 4(4), 91–101. https://doi.org/10.4331/wjbc.v4.i4.91.
Vizovišek, M., Fonović, M., & Turk, B. (2019). Cysteine cathepsins in extracellular matrix remodeling: extracellular matrix degradation and beyond. Matrix Biology, 75–76, 141–159. https://doi.org/10.1016/J.MATBIO.2018.01.024.
Aits, S., & Jä, M. (n.d.). Lysosomal cell death at a glance. Journal of Cell Science, 126, 1905–1912. https://doi.org/10.1242/jcs.091181.
Campden, R. I., & Zhang, Y. (2019). The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Archives of Biochemistry and Biophysics. https://doi.org/10.1016/J.ABB.2019.02.015.
Kern, U., Wischnewski, V., Biniossek, M. L., Schilling, O., & Reinheckel, T. (2015). Lysosomal protein turnover contributes to the acquisition of TGFβ-1 induced invasive properties of mammary cancer cells. Molecular Cancer, 14(1), 39. https://doi.org/10.1186/s12943-015-0313-5.
Kryczka, J., Papiewska-Pajak, I., Kowalska, M. A., Boncela, J., Kryczka, J., Papiewska-Pajak, I., … Boncela, J. (2019). Cathepsin B is upregulated and mediates ECM degradation in colon adenocarcinoma HT29 cells overexpressing Snail. Cells, 8(3), 203. doi:https://doi.org/10.3390/cells8030203.
Mitrović, A., Pečar Fonović, U., & Kos, J. (2017). Cysteine cathepsins B and X promote epithelial-mesenchymal transition of tumor cells. European Journal of Cell Biology, 96(6), 622–631. https://doi.org/10.1016/J.EJCB.2017.04.003.
Wang, J., Chen, L., Li, Y., & Guan, X.-Y. (2011). Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS One, 6(9), e24967. https://doi.org/10.1371/journal.pone.0024967.
Burton, L. J., Dougan, J., Jones, J., Smith, B. N., Randle, D., Henderson, V., & Odero-Marah, V. A. (2017). Targeting the nuclear Cathepsin L CCAAT displacement protein/cut Homeobox transcription factor-epithelial mesenchymal transition pathway in prostate and breast cancer cells with the Z-FY-CHO inhibitor. Molecular and Cellular Biology, 37(5), e00297–e00216. https://doi.org/10.1128/MCB.00297-16.
Kedinger, V., Sansregret, L., Harada, R., Vadnais, C., Cadieux, C., Fathers, K., … Nepveu, A. (2009). p110 CUX1 homeodomain protein stimulates cell migration and invasion in part through a regulatory cascade culminating in the repression of E-cadherin and occludin. The Journal of Biological Chemistry, 284(40), 27701–11. doi:https://doi.org/10.1074/jbc.M109.031849.
Han, M.-L., Zhao, Y.-F., Tan, C.-H., Xiong, Y.-J., Wang, W.-J., Wu, F., … Liang, Z.-Q. (2016). Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacologica Sinica, 37(12), 1606–1622. doi:https://doi.org/10.1038/aps.2016.93.
Wang, W., Xiong, Y., Ding, X., Wang, L., Zhao, Y., Fei, Y., … Liang, Z. (2019). Cathepsin L activated by mutant p53 and Egr-1 promotes ionizing radiation-induced EMT in human NSCLC. Journal of Experimental & Clinical Cancer Research, 38(1), 61. doi:https://doi.org/10.1186/s13046-019-1054-x.
Zhang, Q., Han, M., Wang, W., Song, Y., Chen, G., Wang, Z., & Liang, Z. (2015). Downregulation of cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factor-β-mediated epithelial-mesenchymal transition. Oncology Reports, 33(4), 1851–1859. https://doi.org/10.3892/or.2015.3754.
Bonnans, C., Chou, J., & Werb, Z. (2014). Remodelling the extracellular matrix in development and disease. Nature Reviews. Molecular Cell Biology, 15(12), 786–801. https://doi.org/10.1038/nrm3904.
Conlon, G. A., & Murray, G. I. (2019). Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. The Journal of Pathology, 247(5), 629–640. https://doi.org/10.1002/path.5225.
Overall, C. M., & Dean, R. A. (2006). Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer and Metastasis Reviews, 25(1), 69–75. https://doi.org/10.1007/s10555-006-7890-0.
Lawrence, R. E., & Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nature Cell Biology, 21(2), 133–142. https://doi.org/10.1038/s41556-018-0244-7.
López-Otín, C., & Matrisian, L. M. (2007). Emerging roles of proteases in tumour suppression. Nature Reviews Cancer, 7(10), 800–808. https://doi.org/10.1038/nrc2228.
Dennemärker, J., Lohmüller, T., Mayerle, J., Tacke, M., Lerch, M. M., Coussens, L. M., … Reinheckel, T. (2010). Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene, 29(11), 1611–21. doi:https://doi.org/10.1038/onc.2009.466.
Acknowledgements
The authors thank Eva Schill-Wendt for creating the artwork of the figure. The Deutsche Forschungsgemeinschaft (DFG) SFB 850 subproject B7, DFG-grant RE1584/6-2, and the German Cancer Consortium (DKTK) program Oncogenic Pathways project L627 support the work in the Reinheckel laboratory.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mitschke, J., Burk, U.C. & Reinheckel, T. The role of proteases in epithelial-to-mesenchymal cell transitions in cancer. Cancer Metastasis Rev 38, 431–444 (2019). https://doi.org/10.1007/s10555-019-09808-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-019-09808-2