
Abstract
While only a small part of the human genome encodes for proteins, biological functions for the so-called junk genome are increasingly being recognized through high-throughput technologies and mechanistic experimental studies. Indeed, novel mechanisms of gene regulation are being discovered that require coordinated interaction between DNA, RNA, and proteins. Therefore, interdisciplinary efforts are still needed to decipher these complex transcriptional networks. In this review, we discuss how non-coding RNAs (ncRNAs) are epigenetically regulated in cancer and metastases and consequently how ncRNAs participate in the sculpting of the epigenetic profile of a cancer cell, thus modulating the expression of other RNA molecules. In the latter case, ncRNAs not only affect the DNA methylation status of certain genomic loci but also interact with histone-modifying complexes, changing the structure of the chromatin itself. We present several examples of epigenetic changes causing aberrant expression of ncRNAs in the context of tumor progression. Interestingly, there are also important epigenetic changes and transcriptional regulatory effects derived from their aberrant expression. As ncRNAs can also be used as biomarkers for diagnosis and prognosis or explored as potential targets, we present insights into the use of ncRNAs for targeted cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The “central dogma of biology” proposed by Francis Crick considered RNA molecules to be mere intermediates between DNA and proteins [1]. Surprisingly, it was then later shown that less than 2% of the human genome is translated into proteins. Lacking a protein coding potential, the remaining genetic information was considered “junk” accumulated across evolution [2]. Curiously, recent studies highlight a stronger correlation between the size of these non-coding regions and biological/evolutionary complexity, than those established for protein-coding genes. Intriguingly, genes containing large introns have a significantly higher transcriptional activity in the nervous system and lower transcriptional activity in cancer. This suggests a regulatory potential of these non-coding regions coupled to tissue-specific gene expression patterns and corresponding mitosis rates [3]. Accordingly, the larger part of the human genome has been linked to active transcriptional loci under specific physiological contexts [4, 5]. Therefore, the genetic information enclosed in a particular sequence of DNA can be converted either into an RNA transcript, which encodes a protein, or into a transcript holding a different molecular function, which might be able to modulate the transcription (or translation) of other coding or non-coding genes [2, 6]. This complexity further increases when taking into account that a stretch of DNA can encode for a protein while simultaneously holding a coding-independent function, affecting other transcripts [7]. After transcription, the translation of mRNAs into proteins is assisted by ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs). Both housekeeping transcripts belong to the group of non-coding RNAs (ncRNAs) and have well-established roles in protein synthesis. Additionally, the non-coding transcriptome comprises other functional RNA molecules of a different nature. Despite their myriad of potential functions, they are roughly classified into two major classes according to their length: small non-coding RNAs (sncRNAs) and long non-coding RNAs (lncRNAs), having less or more than 200 nucleotides in length, respectively. However, this classification is unrelated to their biogenesis, function, and cellular location [8].
The conservation of the so-called junk genetic material across time suggests that it underwent an advantageous selection. Importantly, the introduction of parasitic sequences in the eukaryotic genome was evolutionarily simultaneous to the appearance of epigenetic suppressive mechanisms, such as DNA methylation. These mechanisms prevent the mobility of transposable elements and restrict their expression, thus avoiding chromosome instability, chromosomal translocations, and gene disruptions [9,10,11]. Since epigenetic marks are dynamic, some of these epigenetic silenced regions have gradually evolved to create tissue-specific regulatory networks, exhibiting enhancer-like activities [12]. Moreover, DNA methylation is thought to have evolved simultaneously to X-chromosome inactivation and genomic imprinting. In the latter case, gene silencing occurs at very specific genomic regions [13, 14]. In this mechanism, allele-specific maternal and paternal transcription is commonly achieved by allele-specific DNA methylation. One of such examples demonstrates that allele-specific methylation is coupled to an allele-specific transcription of the ncRNA KCNQ1OT1, which is then involved in the transcriptional silencing of other specific genes [15, 16] and indicates an effective cooperation between ncRNAs and epigenetics.
ncRNAs establish a complex layer of transcriptional and posttranscriptional regulation deeply shaped by an epigenetic landscape that is itself regulated by these molecules. This led to the revision of the concept of epigenetics, since it was historically based on two layers of gene regulation: DNA methylation and histone posttranslational modifications. The field is currently conscious of the functional association of ncRNAs with proteins, DNA, and other RNAs, sculpting the epigenetic landscape [17]. Accordingly, ncRNAs are able to recruit and interact with histone-modifying complexes and modulate the activity of DNA methyltransferases, regulating the transcriptional activity across the genome. In summary, ncRNAs affect directly or indirectly through epigenetic changes the gene expression of both coding and non-coding transcripts, and are themselves epigenetically regulated [17, 18].
Utilizing various epigenetic mechanisms, ncRNAs mediate development and cellular differentiation and guarantee cell-specific transcriptional requirements. Recent experimental approaches have demonstrated that the complex network of gene regulation, comprising these non-coding molecules and epigenetics, is often disrupted by genetic and epigenetic events in cancer [19,20,21]. Simultaneously, ncRNAs have been annotated as cancer-related biomarkers for diagnosis, prognosis, and personalized medicine [22,23,24,25,26].
2 Non-coding RNAs as players in gene expression regulation
MicroRNAs (miRNAs) are single-stranded ncRNA species of ~ 22 nucleotides in length derived from hairpin structures. They mediate posttranscriptional gene silencing by partial complementarity with the 3′UTR of a target mRNA. As more than 60% of the protein-coding genes are regulated by miRNAs, they represent one of the most extensively studied subclass of ncRNAs [27, 28]. Gene silencing by miRNA is achieved by triggering an endonucleolytic cleavage [29] or repressing translation [30]. Endogenous small interfering RNAs (siRNAs) have a similar length and biogenesis pathway as miRNAs, but while the latter are excised from stem-loop structures, siRNAs derive from long fully complementary double-stranded RNA precursors primarily of exogenous origin, such as retrotransposons and viral sequences. Both siRNAs and miRNAs are associated with translational repression and mRNA cleavage, depending on the homology to their mRNA target site. Perfect complementarity is commonly associated to mRNA degradation, typical of siRNAs, whereas bulge pairing sites lead generally to a translational silencing, typical of miRNAs [30, 31]. By contrast, PIWI-interacting RNAs (piRNAs) direct transposon cleavage, protecting the genome against transposon-induced insertional mutagenesis, guaranteeing genome integrity [32]. piRNAs are also implicated in epigenetic programming mechanisms [33, 34] and involved in a miRNA-like posttranscriptional silencing of mRNAs through transposon sequences that overlap with the 3′ or 5′UTRs of mRNAs [35, 36]. Curiously, the 3′UTR of more than one quarter of RefSeq transcripts overlap with retrotransposon sequences and those transcripts have lower expression compared to retrotransposon-free transcripts [35]. piRNAs are mainly transcribed in germline cells, but they also show expression in somatic tissues where the large majority is encoded in known transcripts instead of piRNA clusters, as described in germline cells [37]. Small nucleolar RNAs (snoRNAs) are generally encoded in introns of host genes, guiding posttranscriptional modifications of spliceosomal and ribosomal RNAs [38,39,40]. Inherently, they guarantee the accurate assembly and function of ribosomes [41]. Recently, it was described that snoRNAs can modulate 3′ end processing of mRNAs, thus controlling the expression of a subset of mRNAs [42]. They are located in the nucleolus [43] and play an important role in direct and indirect cellular functions such as splicing and translation [44].
lncRNAs are a heterogeneous class of ncRNAs that control transcription, translation, and mRNA stability, functions which are being highlighted in cell differentiation and development [18, 45, 46]. Lacking an open reading frame (ORF) and typically located in the chromatin and nucleus, the transcription of lncRNAs is generally associated (to a lower extent) with the expression of their antisense protein-coding genes, presenting a similar tissue-specific expression pattern [47]. Enhancer RNAs (eRNAs) are typically unspliced ncRNAs that are transcribed from enhancer loci. Their median size places them in the group of lncRNAs, and their expression correlates positively with the transcriptional activation of nearby genes [48, 49]. Circular RNAs (circRNAs) are covalently closed single-stranded ncRNAs (without 5′ cap and 3′ tail), mainly derived from back-splicing circularization of protein-coding exons, with a length corresponding to the incorporated exon(s). Their co-transcriptional biogenesis mechanism competes with pre-mRNA splicing [50], occurring preferentially in genes containing long flanking intronic sequences, containing complementary ALU repeats [51, 52]. The majority of these transcripts have a scarce, although cell-type-specific, expression pattern which supports the notion they are derived as by-products of pre-mRNA splicing [53]. circRNAs are stable and frequently show a developmental-stage and tissue-specific expression, suggesting that the cellular content of these molecules should also vary according to the mitotic index of a particular cell [54]. Overall, they represent 5–10% of the linear expression; however, some have estimated that particular circular isoforms have higher expression than the linear counterparts [55]. Important transcriptional regulatory functions were already described for this class of ncRNA. For instance, the circRNA derived from the CDR1 antisense transcript (CDR1as also called ciRS-7) holds 63 conserved binding sites for miR-7, acting as miRNA sponge [54, 56]. In turn, it is also targeted by miR-671 with higher complementarity that promotes its cleavage, adding complexity to this regulatory scenario [57]. The unusual stability of circRNAs allows their exploration as potential biomarkers in cancer. Accordingly, a recent study analyzing serum exosomes was able to discriminate patients with colon cancer from healthy controls based on the expression of circRNAs. Moreover, they verified that circRNAs were proportionally more represented in exosomes than their linear counterparts [58].
Taking into account the transcriptional and translational regulatory functions of ncRNAs by targeting other RNA transcripts, it is crucial to depict not only the epigenetic layer beyond their regulation but also their effects in the epigenetic landscape, establishing new bridges in this complex archipelago. Several studies highlighted the role of nuclear lncRNAs in guiding chromatin regulatory complexes to specific genomic loci, through multiple interactions between proteins, DNA, and RNAs [59]. In this context, transcriptional activation is achieved through the recruitment of chromatin modifiers such as the histone H3K4 methyltransferase complex [60], while transcriptional silencing is associated with H3K9me2/3, H3K27me3, and polycomb repressive complex 2 (PRC2) [61]. Some nuclear lncRNAs with these characteristics have already been described. For instance, HOTAIR is a lncRNA transcribed from the HOXC locus that interacts with PRC2 through its 5′ domain, mediating its occupancy at the HOXD locus. In turn, PRC2 promotes H3K27me3 deposition, repressing transcription in trans across 40 kb. Similarly, HOTAIR binds the LSD1/CoREST/REST complex, through the 3′ domain, guiding the enzymatic demethylation of H3K4me2 [62, 63]. Nevertheless, there are lncRNAs holding larger transcriptional silencing capacities. The ncRNA Xist is stably repressed in the active X chromosome in females (and in the unpaired X chromosome in males), being exclusively transcribed from the inactive one, coating and silencing the X-chromosome in cis [64]. X-inactivation maintenance is guaranteed not only by Xist expression but also by the support of other epigenetic mechanisms, namely the hypoacetylation of histone H4 and the hypermethylation of CpG islands (CGIs) [65]. Remarkably, the transcription of Xist is controlled by the methylation status of its own CGI, dependent on the activity of two other ncRNAs with antagonistic effects, Tsix promoting DNA methylation and Ftx associating with the lack of DNA methylation [66, 67].
3 Genetic variation of non-coding RNAs in cancer
The canonical studies in cancer biology highlighted genetic alterations in protein-coding genes such as mutations in TP53 [68], deletions of the RB1 locus [69], amplifications of MYC [70], and chromosomal rearrangements in the MLL locus [71]. Similarly, ncRNAs are also targeted by mutations and contain genomic variations associated with aberrant expression and activity. In chronic lymphocytic leukemia (CLL), patients undergo a frequent deletion at the 13q14 region that encodes for miR-15 and miR-16, which are implicated in apoptosis by targeting BCL2 [72, 73]. In a lower extent, these patients experience a 11q deletion that comprises the miR-34b/34c cluster locus (target of p53), promoting the downregulation of these miRNAs that are known to cooperate in the repression of malignant growth [72, 74]. In contrast, the chromosomal region containing the miR-17~92 cluster (13q31-q32) is amplified in diffuse large B cell lymphoma patients [75]. In a synergistic scenario, the expression of this locus is upregulated by Myc, promoting enhanced tumor growth [76]. On the other hand, these ncRNAs are also involved in chemoresistance [77] and radioresistance [78]. Likewise, miR-30d, miR-21, miR-17, and miR-155 undergo a high copy number variation (CNV) in non-small cell lung cancer (NSCLC), similarly to DICER1 and DROSHA that encode for proteins involved in their biogenesis [79]. ncRNAs are also affected by single-nucleotide polymorphisms (SNPs). A common G/C polymorphism within the pre-miR-146a sequence decreases the expression of the mature miRNA, being associated with higher predisposition to papillary thyroid carcinoma [80]. Similarly, the SNP rs61764370 is linked to cancer by disrupting the binding site for let-7 in the 3′UTR of the KRAS oncogene, increasing its expression [81, 82]. Despite several studies that have interrogated the expression of piRNAs in cancer, little is known about the possible genetic variations affecting piRNAs or the genes encoding for the proteins involved in their biogenesis/function. By analyzing the piRNA transcriptome of samples from The Cancer Genome Atlas (TCGA) consortium, several genetic variants were unveiled. Authors detected that 17 piRNA sequences overlap the position of already described mutations. Moreover, suggesting a possible oncogenic role of some piRNAs, a high expression of mitochondrial piRNAs was observed in tumor tissues, likely derived from an increased mitochondrial DNA (mtDNA) content in cancer [37]. snoRNAs are also affected by genetic alterations. SNORD50A and SNORD50B are recurrently deleted in different cancer types and are correlated with reduced survival [83]. It was observed that the snoRNA U50 is targeted by a homozygous 2-bp (TT) deletion in prostate cancer and by a recurrent heterozygous deletion in breast cancer. Accordingly, its overexpression led to reduced colony-formation capacity in vitro, suggesting a tumor suppressor nature for this ncRNA [84, 85]. Curiously, U50 is located at the chromosome breakpoint t(3;6)(q27;q15) described in human B cell lymphoma [86]. Another study showed that the genomic region encoding SNORA42 is frequently amplified in NSCLC, which is correlated with its higher expression, while the expression of the host gene remains unchanged. Functional studies indicated that SNORA42 confers an advantage in cell proliferation, contrary to its host gene [87]. On the other hand, there are also genomic variations affecting proteins that interact with ncRNAs, further suggesting their possible role in cancer. For instance, mutations in the dyskerin (DKC1) gene, are linked to cancer susceptibility [88]. The encoded enzyme associates with H/ACA box snoRNAs to catalyze the pseudouridylation of rRNAs. In turn, alterations in ribosome biogenesis are linked to tumor progression [89, 90].
It has been described that long intergenic non-coding RNA (lincRNA) loci hold cancer-associated SNPs and are affected by CNVs in cancer [91, 92]. One study showed that FAL1 is a lncRNA with frequent genomic amplification in epithelial tumors. The high genomic copy number was correlated with a higher RNA expression and associated with cancer progression in ovarian tumors [93]. Moreover, in prostate cancer, two lncRNAs, PCAN-R1 and PCAN-R2, were described to have higher expression in tumors correlated with a higher copy number. The oncogenic function of these lncRNAs was functionally revealed using knockdown experiments that exhibited a reduced cell proliferation [94]. Other genomic variations are associated with lncRNA transcriptional changes in cancer. For instance, the presence of the high-risk neuroblastoma-associated SNP rs6939340 is linked to a lower expression of the lncRNA NBAT-1 that is suggested to be implicated in metastatic progression and poor prognosis [95].
4 Epigenetic regulation of non-coding RNAs in cancer
Genetic and epigenetic changes cooperate to promote oncogenesis. While the first ones are implicated in the activation of oncogenes and inactivation of tumor suppressor genes, the second ones guide their transcriptional regulation. In this regard, the mechanisms of tumor formation cannot be fully elucidated without mentioning the occurrence of “epimutations,” such as aberrant histone modifications and DNA hyper- and hypomethylation events across the entire genome [96]. CpG hypomethylation is associated with a specific chromatin conformation that allows the accessibility of the genetic information to transcription factors. They promote the transcription of oncogenes such as BCL2 in leukemias [97]. Alternatively, CpG hypermethylation leads to the downregulation of important tumor suppressor genes, such as BRCA1 in breast cancer [98].
While cancer has been historically interrogated based on genetics and protein-coding genes, this view is now challenged by the realization that ncRNAs and epigenetics are indispensable to explain the entire tumorigenesis process. In this context, ncRNAs can be transcriptionally regulated by epigenetics and are themselves able to sculpt the epigenetic landscape of a normal or a malignant cell.
Several studies uncovered tumor suppressor and oncogenic ncRNAs epigenetically deregulated in cancer (Table 1) [128,129,130]. Genome-wide DNA hypomethylation is a common hallmark of cancer cells [131] and extends to the genomic loci of ncRNAs. Most commonly, studies have interrogated the existence of a transcriptional repression mediated by local CGI hypermethylation. Moreover, the discovery and scrutiny of epigenetic pathways altered during tumor formation and metastases are uncovering not only the complexity of a cancer cell but also possible biomarkers for diagnosis, prognosis, and targets for better therapies.
4.1 miRNAs
As a widely studied class of ncRNAs, there are several miRNAs described that undergo transcriptional inactivation by CGI hypermethylation. miRNAs with CGIs overlapping their promoter region are silenced through the transcriptional repression mediated by methyl-CpG-binding domain (MBD) proteins in a chromatin context characterized by the absence of histone modifications linked to active transcription (e.g., H3K4me3) [105, 119, 132]. For instance, miR-124a is epigenetically silenced in colon cancer, which is associated with the posttranslational de-repression of the oncogene CDK6, promoting the phosphorylation and inactivation of the tumor suppressor gene RB1 (Fig. 1a). Despite this miRNA being encoded in three different genomic loci, all the corresponding CGIs are hypermethylated in the colon cancer cell line HCT-116 in comparison with normal colon [105]. The epigenetic silencing of this miRNA was also noticed in several other cancer types [106,107,108,109,110,111]. Another example is given by the miR-132 promoter-associated CGI that was observed to be hypermethylated in ~ 40% of prostate cancer patients with corresponding transcriptional silencing. Functionally, this miRNA decreases cell adhesion, followed by death induction, and reduces cell migration and invasion. The pro-survival genes HB-EGF and TALIN2 were identified as direct targets of this miRNA, but their silencing did not entirely recapitulate the effects of miR-132 overexpression [112]. This miRNA is also epigenetically downregulated by CGI hypermethylation in colorectal [113] and pancreatic [114] cancers, and by SOX4/EZH2-mediated H3K27me3 in ovarian cancer [115]. In a later study, it was demonstrated that miR-132 silencing leads to a metabolic switch, increasing GLUT-1 protein expression which is implicated in lactate production and glucose uptake [133]. Curiously, CGIs are regulatory elements that commonly direct the expression of more than one transcript. For instance, miR-34b/c and BTG4 transcription is regulated by a CGI overlapping a bidirectional promoter. Its hypermethylation and decreased H3K4me3 are associated with the repression of both non-coding and coding transcripts, both suggested to be tumor suppressor genes in colorectal cancer [119]. miR-34b is also silenced by CGI hypermethylation in HCC [120] and lung adenocarcinomas (mir-34b/c), in the latter case associated with metastasis [121]. Despite the fact that hypomethylation events directing gene transcription are less studied, they have a tremendous importance in cancer and metastases. For instance, miR-191 is highly expressed in hepatocellular carcinoma (HCC), which correlates with the hypomethylation of the associated locus. It was suggested that this miRNA represses TIMP3 protein expression, contributing to the epithelial-to-mesenchymal transition (EMT) associated with a poor prognosis [117]. Importantly, another study suggested the existence of a dynamic epigenetic regulation associated with EMT and mesenchymal-epithelial transitions (MET). A CGI hypermethylation-associated repression of miR-200 loci was observed in transformed cells with mesenchymal phenotype. The repression of the miR-200 family allowed the expression of ZEB1 and ZEB2 which are transcriptional repressors of cell adhesion and polarity genes. In vitro experiments showed that miR-200ba/429 and miR-200c/141 repress migration, reducing tumoral growth and metastasis in vivo. Importantly, authors showed that the transitory CGI hypermethylation-associated repression of miR-200 loci induced by TGFβ treatment (EMT) was reverted by its withdrawal [118]. There are other ncRNAs that play a key role in metastasis and are deregulated in cancer. For instance, miR-1 undergoes a CGI hypermethylation-associated silencing in HCC [100] and colorectal cancer [101, 102]. This repression is also associated with reduced H3K4me3 levels. In vitro experiments demonstrated that the overexpression of this miRNA prevents cell proliferation, colony formation, cell motility, and cell invasion [101]. Similarly, another study showed that miR-145 is also silenced by CGI hypermethylation in glioma cell lines. This miRNA inhibits cell proliferation and cell invasion in vitro, suppresses xenograft growth in vivo, and directly targets SOX9 and ADD3. The ectopic expression of miR-145 reduces the expression of c-myc, N-myc, cyclin D1, E-cadherin, and N-cadherin, implicating an important role in cell adhesion and invasion [116].

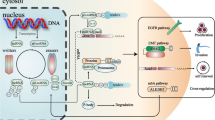
Epigenetic regulation of ncRNAs in cancer. There are ncRNAs epigenetically deregulated in cancer that contribute directly or indirectly to cancer progression and metastasis. a In normal cells, miR-124 can target the 3′UTR of CDK6 leading to its posttranscriptional silencing. In cancer, its CGI hypermethylation-associated repression is responsible for CDK6 de-repression which phosphorylates and inactivates the tumor suppressor RB1 [105]. b NBAT-1 is expressed in normal brain but downregulated in high-risk neuroblastoma patients, associated with the hypermethylation of its promoter region and the presence of the SNP rs6939340. Through the interaction of NBAT-1 with EZH2, this lncRNA is linked to the epigenetic silencing of target genes, decreasing cell proliferation and invasion [95]. c The lncRNA TP53TG1 was demonstrated to be repressed by CGI promoter hypermethylation in primary gastric tumors. Functionally, TP53TG1 interacts with YBX1, preventing its nuclear localization and the activation of growth-promoting genes [124]
In cancer, while both hypo- and hypermethylation events can coexist in parallel at different genomic loci, few studies addressed these changes simultaneously. In this context, a high-throughput study depicted and compared the transcriptome and methylome of miRNAs in HCC. Hypermethylation of miR-148a, miR-375, miR-195, miR-497, and miR-378 correlated with their silencing, while hypomethylation of miR-106b, miR-25, miR-93, miR-23a, and miR-27a was associated with their expression. Curiously, in silico analysis suggested that miR-148a targets both DNA methyltransferases DNMT1 and DNMT3B, potentially establishing a negative feedback loop. It was also hypothesized that the repression of miR-195/497 resulted from the promoter hypermethylation of the miRNAs themselves and of the transcription factors NEUROG2 and DDIT3 needed for their expression [103].
Super-enhancers are transcriptional regulatory loci that drive the robust expression of certain genes, guarantying cell and tissue identity [134, 135]. Similar to what happens in proximal regulatory regions such as CGIs, super-enhancers are also targeted by cancer-related hypermethylation events that are correlated with the transcriptional silencing of their related genes. In lung and breast cancers, bioinformatic analysis showed that the super-enhancer controlling the lncRNA MIRLET7BHG is coupled with the transcriptional repression of the corresponding encoded tumor suppressors let-7a-3 and let-7b [99]. Curiously, there are super-enhancers with a de novo regulatory role in malignant cells [135,136,137], having a potential to drive the expression of other coding and non-coding genes.
Finally, it is essential to mention the importance of the methylation at CGI shores and the existence of mirtrons which are intronic miRNA precursors processed in a Drosha-independent manner [138]. A screening performed in urothelial cell carcinoma (UCC) showed that both miRNAs and mirtrons are susceptible to hypermethylation-mediated silencing which was more common and dense in CpG shores than CGIs. Authors also explored the lower urinary expression of the epigenetically silenced miRNAs (miRs-152/328/1224) as potential biomarkers for the diagnosis of UCC [104].
4.1.1 piRNAs
Several studies were conducted to analyze the expression and potential role of both piRNAs and associated proteins in cancer. Functionally, the knockdown of PiwiL2 in murine bone marrow mesenchymal stem cells was found to reduce the expression of tumor suppressor genes, increasing cell proliferation [139]. Concordantly, lower expression of PIWIL1, PIWIL2, and PIWIL4 was associated with poor prognosis in renal cell carcinoma [140]. In soft tissue sarcoma patients, lower expression of PIWIL2 and PIWIL4 correlates with a worse prognosis [141]. These genes were also found to be downregulated in NSCLC, where patients with lower levels of PIWIL4 had shorter overall survival. In the same study, PIWIL1 was found to be expressed in a set of NSCLC cases with worse prognosis. Upon 5-Aza-dC treatment, authors showed a dose-dependent expression of PIWIL1, in two NSCLC cell lines. Moreover, they correlated the expression of PIWIL1 with a higher percentage of unmethylated CpGs within its CGI, suggesting that PIWIL1 expression could be partially regulated by DNA methylation [142]. Despite the fact that the expression of PIWI-proteins has been documented in somatic tissues, very few studies reported the existence of piRNAs in normal or cancer somatic tissues [143]. In primary testicular tumors, PIWIL1, PIWIL2, PIWIL4, and TDRD1 were found to be epigenetically silenced by CGI hypermethylation [144], which is associated with a loss of piRNAs [144, 145] and hypomethylation events at the LINE-1 loci [144, 146]. Curiously, the epigenetic silencing of the genes encoding for the PIWI proteins was also described in non-genetic male infertility syndromes [147] which are epidemiologically linked to testicular cancer [148, 149].
4.2 lncRNAs
There are also alterations in the methylation profile of the promoter region of lnRNAs, in cancer [92]. The comparison between the transcriptomes of low- and high-risk neuroblastomas has established a correlation between lower NBAT-1 expression and poor clinical outcomes. Authors discovered that the hypermethylation of the promoter region of NBAT-1 was linked to its lower expression in high-risk neuroblastoma patients. Later, NBAT-1 was functionally associated to the differentiation of neuronal tumor cells and to the decrease in cell proliferation and invasion, through the epigenetic silencing of target genes mediated by its interaction with EZH2 (Fig. 1b) [95]. In colon cancer, the promoter CGI hypermethylation of Vimentin (VIM) and its head-to-head antisense transcript was shown to lead to their transcriptional silencing. A detailed study suggested that the antisense transcription allows the formation of an R-loop structure, enhancing transcription by maintaining an open local chromatin conformation. In this context, both R-loop destabilization and antisense knockdown promote chromatin compaction and prevent the binding of transcriptional regulators of the NF-κB pathway [125]. Another example of epigenetic deregulation of lncRNAs is given by a recent study showing that p53-induced lncRNA, TP53TG1, is silenced by promoter hypermethylation in gastric and colon tumors with an associated poor prognosis. Authors showed that TP53TG1 interacts with the DNA/RNA binding protein YBX1, impeding its nuclear localization and preventing the activation of oncogenes. It was suggested that upon cancer-specific silencing of TP53TG1, YBX1 is able to activate growth-promoting genes and create chemoresistance (Fig. 1c) [124].
4.3 T-UCRs, snoRNAs, and snoRNA-host genes
Transcribed-ultraconserved regions (T-UCRs) are lncRNAs encoded from DNA sequences absolutely conserved between orthologous regions of the human, rat, and mouse genomes. It was described that T-UCRs Uc.160+, Uc283+A, and Uc.346+ undergo cancer-specific CGI hypermethylation-associated silencing which is a common event in several tumor types [127]. Interestingly, Uc.283+A binds to pri-miR-195, impairing miR-195 maturation and function [150]. Additionally, snoRNAs are also affected by epigenetic mechanisms in cancer. For instance, three snoRNA host genes were found to undergo a cancer-specific promoter hypermethylation-associated silencing with an associated low expression of the mature snoRNAs: SNORD123, U70C, and ACA59B. Interestingly, the methylation of a unique CGI was inversely correlated with the transcription of three different transcripts, namely SNORD123, its host gene LOC100505806, and SEMA5A which is transcribed in the opposite direction. Curiously, SNORD123 and ACA59B are snoRNAs conserved across vertebrates without a known target (orphan snoRNAs), suggesting that these snoRNAs may have a role in cancer by an unknown mechanism not related with ribosomal and spliceosomal RNA-guided modifications [126]. By contrast, U70C was found to be repressed in CLL patients [151] and deregulated in X-linked dyskeratosis congenita, a congenital disorder associated with cancer susceptibility [152]. In this case, this snoRNA directs a modification of 18S rRNA which is suggested to be associated with cancer [153,154,155]. The downregulation of snoRNAs was also described in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) [156]. Importantly, apart from their classical functions, they may have a role in gene silencing acting through an antisense-like mechanism in the nucleus, promoting pre-mRNA degradation, preventing splicing, or inhibiting the carriage of the transcript [157, 158].
There are lncRNAs that can act as endogenous competitors for miRNAs, being able to de-repress their targets. Growth arrest specific 5 gene (GAS5) is a lncRNA and snoRNA host gene with tumor suppressor functions that is downregulated in several solid tumors [159,160,161,162,163,164,165,166,167]. In the liver, this lncRNA was described as a competing endogenous RNA (ceRNA) for miR-222, increasing the protein levels of the tumor suppressor p27, a target of this miRNA [168]. Moreover, GAS5 exerts its tumor suppressor functions by interacting with E2F1, promoting its binding and activating the P27Kip1 promoter [169]. This lcnRNA may also act as endogenous sponge for the miR-21 oncogene [170]. Similarly, a recent study has described an antisense transcript encoded in the GAS5 genomic locus, GAS5-AS1, with a reduced expression in NSCLC tumors. Functional assays were unable to show an effect of this lncRNA in terms of proliferation, cell cycle progression, or apoptosis. However, it was demonstrated that this antisense transcript directs a pronounced decrease in cell migration and invasion, reducing ZEB1, N-Cadherin, VIM, and/or SNAIL1, crucial for EMT. Authors have suggested that the epigenetic silencing of GAS5-AS1 in NSCLC is, at least partially, due to histone deacetylation, while GAS5 is silenced through DNA methylation [123].
4.4 Genomic-imprinted ncRNAs
Genomic imprinting is an epigenetic mechanism associated with DNA methylation and histone posttranslational modifications, through which genes are transcriptionally regulated in a parental allele-specific manner. Loss of imprinting with aberrant DNA methylation profiles at genomic-imprinted loci is associated with transcriptional changes of their encoded genes in cancer and other diseases [171]. DLK1-DIO3-imprinted locus (14q32) is controlled by two intergenic, differentially methylated regions (DMRs). It encodes three protein-coding genes, DLK1, RTL1, and DIO3, from the paternal allele and several long and short ncRNAs from the maternally inherited allele [172]. The methylation signature of this locus was investigated by comparing lung cancer versus non-tumoral tissue, being observed an inverse correlation between DNA methylation and expression of the corresponding genes. Accordingly, while the hypermethylation of DIO3 was associated with its transcriptional silencing, the hypomethylation of SNORD113-5, SNORD113-7, SNORD114-9, and miR-889 correlated with their higher expression in smoking-induced lung cancer. Concordantly, these methylome differences were extended to other protein-coding genes, snoRNAs, miRNAs, and lncRNAs [173]. miR-411, miR-370, and miR-376a, encoded in this locus, are upregulated in lung cancer and linked to a more aggressive phenotype, as well as poor survival. miR-411 downregulation decreased cell migration [174]. Independent of the methylation profile, other cancer types have shown an increase in the expression of the miRNA members of this imprinted locus, namely in HCC [175] and uterine carcinoma [176]. Contradictorily, a downregulation of these miRNAs was observed in colorectal cancer [177], gastric adenocarcinoma [178], medulloblastoma [179], and papillary thyroid cancer [180]. Concordantly, a genome-wide analysis unveiled the silencing of the miR-379/miR-656 cluster across different human cancers. This was observed in a high percentage of samples from glioblastoma multiforme, kidney renal clear cell carcinoma, breast invasive carcinoma, and ovarian serous cystadenocarcinoma [181]. Intriguingly, miRNAs expressed from this imprinted locus are preferentially exported in exosomes, being almost absent in the cells where they are produced, which suggest that the levels of the abovementioned miRNAs may not reflect their transcriptional activity [182]. Another study showed that this locus also encodes 138 piRNAs. Seven of these piRNAs are exclusively encoded within this imprinted locus and are somatically expressed in lung (non-malignant and tumor samples), suggesting their potential role in the anomalous methylation profile of the imprinted locus during lung cancer progression. By comparing paired tumor and non-malignant lung tissue, the authors noticed that four of these piRNAs were upregulated in lung adenocarcinoma and one in lung squamous cell carcinoma [183]. Concerning the lncRNAs of this locus, they are also deregulated in cancer. MEG3 encodes for a lncRNA that is highly expressed in brain. In vitro assays revealed that MEG3 has an anti-proliferative activity, inhibiting DNA synthesis, suppressing colony formation, and activating p53-mediated transactivation. Curiously, its transcriptional silencing is a common event in meningiomas through its allelic loss (in higher-grade tumors) or through an increase in CpG methylation within its promoter or the imprinting control region [122].
5 Non-coding RNAs as epigenomic regulators
HOTAIR is a lncRNA upregulated in different types of cancer [184] and has important functions through its interactions with other RNA molecules or recruiting PRC2 and LSD1/CoREST/REST chromatin-modifying complexes. The transient LSD1-mediated demethylation of H3K4 promotes the assembly of the Myc-induced transcription initiation complex [185]. HBXIP, an oncoprotein that directly interacts with c-Myc, was suggested to recruit HOTAIR together with LSD1, mechanistically mediating the c-Myc transcriptional activation through the c-Myc/HBXIP/Hotair/LSD1 complex, where HOTAIR serves as a scaffold, in breast cancer cells (Fig. 2a) [186]. In esophageal squamous cell carcinoma (ESCC), HOTAIR promotes H3K27me3 deposition at the promoter region of WIF-1, an inhibitor of the Wnt/β-catenin signaling pathway, leading to the epigenetic silencing of WIF-1 and consequent de-repression of Wnt target genes. This observation supports the employment of HOTAIR expression as a prognostic factor for metastatic progression in ESCC (Fig. 2a) [187]. Moreover, HOTAIR upregulation was associated with a genome-wide retargeting of PRC2 linked to both breast and colorectal cancer metastases [195,196,197]. Besides the capacity to recruit two distinct chromatin-modifying complexes, HOTAIR can also function as a ceRNA. For instance, higher expression of HOTAIR was demonstrated to be associated with a malignant phenotype and poor prognosis in gastric cancer patients. HOTAIR promotes migration and invasion, and its knockdown restrains cell proliferation and induces apoptosis. In this context, HOTAIR upregulation is correlated with higher expression of HER2, due to its function as a sponge for miR-331-3p, de-repressing its target gene HER2 (Fig. 2b) [188]. Despite mir-141 undergoing a CGI hypermethylation-associated silencing in gliomas, it was also verified that HOTAIR acts as a sponge for this miRNA, positively regulating SKA2, which is implicated in cancer progression (Fig. 2b) [189]. Similarly, by sponging miR-152, HOTAIR induces HLA-G upregulation, pointing to a potential role of this lncRNA to escape cancer immune surveillance (Fig. 2b) [190, 198]. In esophageal cancer, HOTAIR sequestrates miR-148a, which de-represses Snail2, and promotes EMT (Fig. 2b) [191]. In summary, HOTAIR acts not only by regulating gene expression through epigenetic changes but also as a sponge for miRNAs, allowing the expression of their targets. The complexity of this epigenetic scenario increases, taking into account that each chromatin-modifying complex may interact with various ncRNA molecules. HOTAIR binds EZH2, the enzymatic subunit of the repressive polycomb complex PRC2. However, since EZH2 is upregulated in several human cancer types and associated with aggressiveness and poor survival, it was essential to depict how polycomb complexes are guided to their specific target genomic locations. By cross-linking methods, several intronic RNA sequences were identified as being able to bind EZH2, regulating the transcriptional activity of their host gene. In this regard, the ectopic expression of the EZH2-bound intronic RNA sequence associated with SMYD3 resulted in a higher genomic occupancy of EZH2 in the corresponding genomic locus, reducing the transcription of the corresponding host gene [199].

ncRNAs as posttranscriptional and epigenetic modulators of gene expression in cancer. HOTAIR and other ncRNAs act as regulatory molecules in a wide variety of biological processes. a, Left: In cancer, HBXIP interacts with c-Myc and recruits HOTAIR together with LSD1 (mediates the transient demethylation of H3K4me2). The complex c-Myc/HBXIP/Hotair/LSD1 is responsible for c-Myc transcriptional activation [186]. a, Right: HOTAIR can also serve as a scaffold for PRC2 complex promoting H3K27me3 in the promoter region of WIF-1, which is an inhibitor of the Wnt/β-catenin signaling pathway. The epigenetic silencing of WIF-1 associates with the activation of the WNT-β-catenin pathway [187]. b Additionally, HOTAIR not only regulates chromatin dynamics but also influences gene expression posttranscriptionally. This lncRNA acts as a ceRNA, sponging miR-331-3p, mir-141, miR-152, and miR-148a, de-repressing the cancer- and metastases-related proteins HER2 [188], SKA2 [189], HLA-G [190], and Snail2 [191], respectively. c DNMT enzymes catalyze the conversion of cytosine to 5-mC whereas TET enzymes catalyze the conversion of 5-mC to 5-hmC. DNA demethylation (loss of 5mC) can be achieved either as a passive process through DNA replication in the absence of functional DNA methylation maintenance or actively through TET-mediated 5mC oxidation. miR-29b targets DNMT3A, DNMT3B, and indirectly DNMT1, by targeting its transactivator SP1, promoting a global DNA hypomethylation [192]. Simultaneously, this miRNA is able to target the 3′UTR of TET1, TET2, and TET3 decreasing, as a consequence, the cellular 5-hmC content [193]. miR-29b was found to be downregulated in AML patients with balanced 11q23 translocation [194] but upregulated in TET2-wild-type AML patients [193], suggesting that this miRNA should have an important role in the epigenetic profile of AML cells. Importantly, the oncogenic or tumor suppressor role of this miRNA in AML is controversial
There are several other examples of ncRNAs that are able to modulate the epigenome in cancer and metastases. For instance, FAL1 is a lncRNA frequently amplified in epithelial tumors. Its association with the polycomb complex protein BMI1 promotes its binding to the CDKN1A promoter, repressing p21 expression in cancer [93]. By contrast, NBAT-1 can be epigenetically silenced in cancer and, per se, the expression of this lncRNA is biologically responsible for the epigenetic silencing of target genes such as SOX9, VCAN, and OSMR, through the interaction with EZH2, suggesting an important role in tumor progression [95]. ZFAS1 is a snoRNA host lncRNA described as a tumor suppressor gene in breast cancer [23, 200] and as an oncogene in colorectal cancer [201] and HCC [202]. The latter study showed an upregulation of ZFAS1 in primary tumors compared to adjacent non-malignant tissue. The oncogenic function was attributed to miR-150 sequestration, de-repressing ZEB1, MMP14, and MMP16, which may act to promote metastasis [202]. Mechanistically, it was also shown that ZFAS1 is associated with the inhibition of the CpG methylation of the miR-9 promoter-related CGI, through a DNMT1-dependent mechanism [203]. Accordingly, the upregulation of miR-9 in HCC was associated with an aggressive phenotype and poor prognosis [204]. In addition, it was shown that silencing of E-cadherin mediated by miR-9 leads to the activation of β-catenin signaling, activating pro-metastatic genes in breast cancer [205]. Other still uncharacterized snoRNA-host genes and “orphan” snoRNAs may hold unexpected functions in the cellular context with potential roles in cancer. As many snoRNAs are intronically encoded in genes coding for ribosomal proteins, guiding themselves posttranscriptional modifications in rRNA, it is worth thinking that some “orphan” snoRNAs might have a function somehow associated with their host gene. Curiously, a recent study identified novel “orphan” snoRNAs, encoded within host genes with epigenetic functions [206], whose expression is deregulated in cancer, e.g., DNMT3A [207] and KAT6B [208], anticipating that they might have an epigenetic-associated role in cancer.
ncRNAs have different epigenetic roles in cancer, promoting and preventing specific epigenetic changes. For instance, a ncRNA encompassing the full mRNA sequence of CEBPA interacts with DNMT1 blocking the DNA methylation of the CEBPA locus [209]. In contrast, it was suggested that the lncRNA HNF1A-AS1, upregulated in lung adenocarcinomas, binds to DNMT1, mediating its binding to the E-cadherin promoter, sustaining its repression. Reduced levels of HNF1A-AS1 increased E-cadherin and decreased N-cadherin and β-catenin, showing the involvement of this lncRNA in EMT. The ectopic expression of HNF1A-AS1 promoted cell proliferation, and its downregulation inhibited cell migration and invasion in vitro and decreased tumor growth and metastasis in vivo [210]. Similarly, HNF1A-AS1 is upregulated in bladder cancer and HCC, where this lncRNA promotes proliferation by acting as a ceRNA, sponging miR-30b-5p and de-repressing its target gene Bcl-2 [211, 212]. HNF1A-AS1 also exerts its oncogenic functions, acting as a sponge for miRNA-34a, positively regulating SIRT1 in colon cancer [213]. Importantly, there are ncRNAs deregulated in cancer that directly target epigenetic genes, leading to global genomic effects. This is the case for miR-29b, which is downregulated in AML patients with balanced 11q23 translocation [194]. Taking into account that DNMTs catalyze the conversion of cytosine to 5-methylcytosine (5-mC), it was functionally shown that miR-29b targets DNMT3A, DNMT3B, and SP1, a transactivator of DNMT1, promoting a genome-wide DNA hypomethylation, and induces the transcription of tumor suppressor genes in AML (Fig. 2c) [192]. By contrast, miR-29b, among other miRNAs, was shown to be upregulated in TET2-wild-type AML patients. Considering that TET2 catalyzes the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC), being involved in active and passive DNA demethylation, functional assays have demonstrated that miR-29b targets and decreases TET2 expression (in addition to TET1 and TET3), and reduces the cellular levels of 5-hmC in hematopoietic cells (Fig. 2c) [193]. Thus, the expression levels of miR-29b should have an important role in the epigenetic profile of AML cells.
Recently, piRNAs were suggested to guide gene-specific CpG methylation at non-transposable element genetic loci, which may be partially mediated by their direct binding to genomic DNA or nascent mRNA transcripts, close to target CpG sites [214]. For instance, the piwi/piRNA complex was linked to the methylation of a CGI overlapping the promoter region of CREB2, in neurons [215]. On the other hand, the SNP rs1326306 G>T at the piR-021285 locus increased the risk of breast cancer, since the polymorphism within the mature sequence of a piRNA might alter its potential to methylate its target loci. Transfection of breast cancer cell lines with piRNA mimics showed the existence of significant methylation differences between wild-type and variant piR-021285 mimics. Among other genes, the variant piR-021285 mimic induced lower ARHGAP11A 5′UTR/first exon methylation, which was associated with a higher mRNA expression. This variant also increased cell invasiveness that might be associated with higher levels of ARHGAP11A, since the expression of this gene has been found to be upregulated in migratory breast cancer cells [216].
6 Future perspectives and conclusions
This review provides examples and new insights into the epigenetic regulation of ncRNAs and how ncRNAs by their own can produce important changes in the epigenetic landscape of a cancer cell, modulating the expression of other cancer-related coding and non-coding genes. The meticulous scrutiny of the transcriptional networks established in cancer by ncRNAs and epigenetics opens the opportunity to establish novel biomarkers and new candidate targets, resulting in a better and more personalized cancer treatment through new pharmacological approaches. Translationally, the use of the current epigenetic drugs is still very limited due to genome-wide effects [217]. In this context, ncRNAs could be exploited not only as specific biomarkers for early diagnosis and personalized treatment but also as targets with specific downstream transcriptional and translational effects. RNA interference (RNAi) is a promising approach to functionally and specifically downregulate almost any RNA molecule without genome-wide side effects [218,219,220]. For instance, Miravirsen is a miRNA-targeting drug, which employs locked nucleic acid-modified oligonucleotides to repress miR-122, required for hepatitis C virus infection [221, 222]. In addition, miRNA replacement was also approached using RNA mimics, namely in cancer [223]. Considering the epigenetic roles of some RNA molecules and their aptitudes to control gene expression, the establishment of mechanistic gene regulatory networks supported by the development of novel approaches to modulate the expression of some ncRNAs could lead to the development of novel epigenetic and non-epigenetic therapeutic strategies in cancer.
References
Crick, F. H. (1958). On protein synthesis. Symposia of the Society for Experimental Biology, 12, 138–163.
Alexander, R. P., Fang, G., Rozowsky, J., Snyder, M., & Gerstein, M. B. (2010). Annotating non-coding regions of the genome. Nature Reviews. Genetics, 11(8), 559–571.
Taft, R. J., Pheasant, M., & Mattick, J. S. (2007). The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays, 29(3), 288–299.
ENCODE. (2012). Project Consortium, an integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74.
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., Tanzer, A., Lagarde, J., Lin, W., Schlesinger, F., Xue, C., Marinov, G. K., Khatun, J., Williams, B. A., Zaleski, C., Rozowsky, J., Roder, M., Kokocinski, F., Abdelhamid, R. F., Alioto, T., Antoshechkin, I., Baer, M. T., Bar, N. S., Batut, P., Bell, K., Bell, I., Chakrabortty, S., Chen, X., Chrast, J., Curado, J., Derrien, T., Drenkow, J., Dumais, E., Dumais, J., Duttagupta, R., Falconnet, E., Fastuca, M., Fejes-Toth, K., Ferreira, P., Foissac, S., Fullwood, M. J., Gao, H., Gonzalez, D., Gordon, A., Gunawardena, H., Howald, C., Jha, S., Johnson, R., Kapranov, P., King, B., Kingswood, C., Luo, O. J., Park, E., Persaud, K., Preall, J. B., Ribeca, P., Risk, B., Robyr, D., Sammeth, M., Schaffer, L., See, L. H., Shahab, A., Skancke, J., Suzuki, A. M., Takahashi, H., Tilgner, H., Trout, D., Walters, N., Wang, H., Wrobel, J., Yu, Y., Ruan, X., Hayashizaki, Y., Harrow, J., Gerstein, M., Hubbard, T., Reymond, A., Antonarakis, S. E., Hannon, G., Giddings, M. C., Ruan, Y., Wold, B., Carninci, P., Guigo, R., & Gingeras, T. R. (2012). Landscape of transcription in human cells. Nature, 489(7414), 101–108.
Dhanasekaran, K., Kumari, S., & Kanduri, C. (2013). Noncoding RNAs in chromatin organization and transcription regulation: an epigenetic view. Sub-Cellular Biochemistry, 61, 343–372.
Tay, Y., Kats, L., Salmena, L., Weiss, D., Tan, S. M., Ala, U., Karreth, F., Poliseno, L., Provero, P., Di Cunto, F., Lieberman, J., Rigoutsos, I., & Pandolfi, P. P. (2011). Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell, 147(2), 344–357.
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., Stadler, P. F., Hertel, J., Hackermuller, J., Hofacker, I. L., Bell, I., Cheung, E., Drenkow, J., Dumais, E., Patel, S., Helt, G., Ganesh, M., Ghosh, S., Piccolboni, A., Sementchenko, V., Tammana, H., & Gingeras, T. R. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science, 316(5830), 1484–1488.
Walsh, C. P., Chaillet, J. R., & Bestor, T. H. (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nature Genetics, 20(2), 116–117.
Liang, G., Chan, M. F., Tomigahara, Y., Tsai, Y. C., Gonzales, F. A., Li, E., Laird, P. W., & Jones, P. A. (2002). Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Molecular and Cellular Biology, 22(2), 480–491.
Slotkin, R. K., & Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nature Reviews. Genetics, 8(4), 272–285.
Xie, M., Hong, C., Zhang, B., Lowdon, R. F., Xing, X., Li, D., Zhou, X., Lee, H. J., Maire, C. L., Ligon, K. L., Gascard, P., Sigaroudinia, M., Tlsty, T. D., Kadlecek, T., Weiss, A., O'Geen, H., Farnham, P. J., Madden, P. A., Mungall, A. J., Tam, A., Kamoh, B., Cho, S., Moore, R., Hirst, M., Marra, M. A., Costello, J. F., & Wang, T. (2013). DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nature Genetics, 45(7), 836–841.
Reik, W., & Lewis, A. (2005). Co-evolution of X-chromosome inactivation and imprinting in mammals. Nature Reviews. Genetics, 6(5), 403–410.
Paulsen, M., & Ferguson-Smith, A. C. (2001). DNA methylation in genomic imprinting, development, and disease. The Journal of Pathology, 195(1), 97–110.
Li, E., Beard, C., & Jaenisch, R. (1993). Role for DNA methylation in genomic imprinting. Nature, 366(6453), 362–365.
Du, M., Zhou, W., Beatty, L. G., Weksberg, R., & Sadowski, P. D. (2004). The KCNQ1OT1 promoter, a key regulator of genomic imprinting in human chromosome 11p15.5. Genomics, 84(2), 288–300.
Peschansky, V. J., & Wahlestedt, C. (2014). Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics, 9(1), 3–12.
Morris, K. V., & Mattick, J. S. (2014). The rise of regulatory RNA. Nature Reviews. Genetics, 15(6), 423–437.
Morris, K. V., Chan, S. W., Jacobsen, S. E., & Looney, D. J. (2004). Small interfering RNA-induced transcriptional gene silencing in human cells. Science, 305(5688), 1289–1292.
Mendell, J. T. (2005). MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle, 4(9), 1179–1184.
Esteller, M. (2011). Non-coding RNAs in human disease. Nature Reviews. Genetics, 12(12), 861–874.
Liz, J., & Esteller, M. (2016). lncRNAs and microRNAs with a role in cancer development. Biochimica et Biophysica Acta, 1859(1), 169–176.
Askarian-Amiri, M. E., Crawford, J., French, J. D., Smart, C. E., Smith, M. A., Clark, M. B., Ru, K., Mercer, T. R., Thompson, E. R., Lakhani, S. R., Vargas, A. C., Campbell, I. G., Brown, M. A., Dinger, M. E., & Mattick, J. S. (2011). SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA, 17(5), 878–891.
Ronnau, C. G., Verhaegh, G. W., Luna-Velez, M. V., & Schalken, J. A. (2014). Noncoding RNAs as novel biomarkers in prostate cancer. BioMed Research International, 2014, 591703.
Fatima, R., Akhade, V. S., Pal, D., & Rao, S. M. (2015). Long noncoding RNAs in development and cancer: potential biomarkers and therapeutic targets. Mol Cell Ther, 3, 5.
Hayes, J., Peruzzi, P. P., & Lawler, S. (2014). MicroRNAs in cancer: biomarkers, functions and therapy. Trends in Molecular Medicine, 20(8), 460–469.
Lee, R. C., Feinbaum, R. L., & Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75(5), 843–854.
Friedman, R. C., Farh, K. K., Burge, C. B., & Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Research, 19(1), 92–105.
Llave, C., Xie, Z., Kasschau, K. D., & Carrington, J. C. (2002). Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science, 297(5589), 2053–2056.
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., & Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes & Development, 20(5), 515–524.
Zeng, Y., Yi, R., & Cullen, B. R. (2003). MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proceedings of the National Academy of Sciences of the United States of America, 100(17), 9779–9784.
Vagin, V. V., Sigova, A., Li, C., Seitz, H., Gvozdev, V., & Zamore, P. D. (2006). A distinct small RNA pathway silences selfish genetic elements in the germline. Science, 313(5785), 320–324.
Huang, X. A., Yin, H., Sweeney, S., Raha, D., Snyder, M., & Lin, H. (2013). A major epigenetic programming mechanism guided by piRNAs. Developmental Cell, 24(5), 502–516.
Yin, H., & Lin, H. (2007). An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature, 450(7167), 304–308.
Faulkner, G. J., Kimura, Y., Daub, C. O., Wani, S., Plessy, C., Irvine, K. M., Schroder, K., Cloonan, N., Steptoe, A. L., Lassmann, T., Waki, K., Hornig, N., Arakawa, T., Takahashi, H., Kawai, J., Forrest, A. R., Suzuki, H., Hayashizaki, Y., Hume, D. A., Orlando, V., Grimmond, S. M., & Carninci, P. (2009). The regulated retrotransposon transcriptome of mammalian cells. Nature Genetics, 41(5), 563–571.
Lim, A. K., Lorthongpanich, C., Chew, T. G., Tan, C. W., Shue, Y. T., Balu, S., Gounko, N., Kuramochi-Miyagawa, S., Matzuk, M. M., Chuma, S., Messerschmidt, D. M., Solter, D., & Knowles, B. B. (2013). The nuage mediates retrotransposon silencing in mouse primordial ovarian follicles. Development, 140(18), 3819–3825.
Martinez, V. D., Vucic, E. A., Thu, K. L., Hubaux, R., Enfield, K. S., Pikor, L. A., Becker-Santos, D. D., Brown, C. J., Lam, S., & Lam, W. L. (2015). Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Scientific Reports, 5, 10423.
Kiss-Laszlo, Z., Henry, Y., Bachellerie, J. P., Caizergues-Ferrer, M., & Kiss, T. (1996). Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell, 85(7), 1077–1088.
Cavaille, J., Nicoloso, M., & Bachellerie, J. P. (1996). Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature, 383(6602), 732–735.
Ganot, P., Bortolin, M. L., & Kiss, T. (1997). Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell, 89(5), 799–809.
Decatur, W. A., & Fournier, M. J. (2002). rRNA modifications and ribosome function. Trends in Biochemical Sciences, 27(7), 344–351.
Huang, C., Shi, J., Guo, Y., Huang, W., Huang, S., Ming, S., Wu, X., Zhang, R., Ding, J., Zhao, W., Jia, J., Huang, X., Xiang, A. P., Shi, Y., & Yao, C. (2017). A snoRNA modulates mRNA 3′ end processing and regulates the expression of a subset of mRNAs. Nucleic Acids Research, 45(15), 8647–8660.
Gerbi, S. A. (1995). Small nucleolar RNA. Biochemistry and Cell Biology, 73(11–12), 845–858.
Kiss, T. (2001). Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. The EMBO Journal, 20(14), 3617–3622.
Fatica, A., & Bozzoni, I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nature Reviews. Genetics, 15(1), 7–21.
Hu, W., Alvarez-Dominguez, J. R., & Lodish, H. F. (2012). Regulation of mammalian cell differentiation by long non-coding RNAs. EMBO Reports, 13(11), 971–983.
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., Guernec, G., Martin, D., Merkel, A., Knowles, D. G., Lagarde, J., Veeravalli, L., Ruan, X., Ruan, Y., Lassmann, T., Carninci, P., Brown, J. B., Lipovich, L., Gonzalez, J. M., Thomas, M., Davis, C. A., Shiekhattar, R., Gingeras, T. R., Hubbard, T. J., Notredame, C., Harrow, J., & Guigo, R. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Research, 22(9), 1775–1789.
Kim, T. K., Hemberg, M., Gray, J. M., Costa, A. M., Bear, D. M., Wu, J., Harmin, D. A., Laptewicz, M., Barbara-Haley, K., Kuersten, S., Markenscoff-Papadimitriou, E., Kuhl, D., Bito, H., Worley, P. F., Kreiman, G., & Greenberg, M. E. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature, 465(7295), 182–187.
Andersson, R., Gebhard, C., Miguel-Escalada, I., Hoof, I., Bornholdt, J., Boyd, M., Chen, Y., Zhao, X., Schmidl, C., Suzuki, T., Ntini, E., Arner, E., Valen, E., Li, K., Schwarzfischer, L., Glatz, D., Raithel, J., Lilje, B., Rapin, N., Bagger, F. O., Jorgensen, M., Andersen, P. R., Bertin, N., Rackham, O., Burroughs, A. M., Baillie, J. K., Ishizu, Y., Shimizu, Y., Furuhata, E., Maeda, S., Negishi, Y., Mungall, C. J., Meehan, T. F., Lassmann, T., Itoh, M., Kawaji, H., Kondo, N., Kawai, J., Lennartsson, A., Daub, C. O., Heutink, P., Hume, D. A., Jensen, T. H., Suzuki, H., Hayashizaki, Y., Muller, F., Forrest, A. R. R., Carninci, P., Rehli, M., & Sandelin, A. (2014). An atlas of active enhancers across human cell types and tissues. Nature, 507(7493), 455–461.
Ashwal-Fluss, R., Meyer, M., Pamudurti, N. R., Ivanov, A., Bartok, O., Hanan, M., Evantal, N., Memczak, S., Rajewsky, N., & Kadener, S. (2014). circRNA biogenesis competes with pre-mRNA splicing. Molecular Cell, 56(1), 55–66.
Cocquerelle, C., Daubersies, P., Majerus, M. A., Kerckaert, J. P., & Bailleul, B. (1992). Splicing with inverted order of exons occurs proximal to large introns. The EMBO Journal, 11(3), 1095–1098.
Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., Marzluff, W. F., & Sharpless, N. E. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA, 19(2), 141–157.
Guo, J. U., Agarwal, V., Guo, H., & Bartel, D. P. (2014). Expanded identification and characterization of mammalian circular RNAs. Genome Biology, 15(7), 409.
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., Maier, L., Mackowiak, S. D., Gregersen, L. H., Munschauer, M., Loewer, A., Ziebold, U., Landthaler, M., Kocks, C., le Noble, F., & Rajewsky, N. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 495(7441), 333–338.
Salzman, J., Chen, R. E., Olsen, M. N., Wang, P. L., & Brown, P. O. (2013). Cell-type specific features of circular RNA expression. PLoS Genetics, 9(9), e1003777.
Hansen, T. B., Jensen, T. I., Clausen, B. H., Bramsen, J. B., Finsen, B., Damgaard, C. K., & Kjems, J. (2013). Natural RNA circles function as efficient microRNA sponges. Nature, 495(7441), 384–388.
Hansen, T. B., Wiklund, E. D., Bramsen, J. B., Villadsen, S. B., Statham, A. L., Clark, S. J., & Kjems, J. (2011). miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. The EMBO Journal, 30(21), 4414–4422.
Li, Y., Zheng, Q., Bao, C., Li, S., Guo, W., Zhao, J., Chen, D., Gu, J., He, X., & Huang, S. (2015). Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Research, 25(8), 981–984.
Guttman, M., & Rinn, J. L. (2012). Modular regulatory principles of large non-coding RNAs. Nature, 482(7385), 339–346.
Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y., Lajoie, B. R., Protacio, A., Flynn, R. A., Gupta, R. A., Wysocka, J., Lei, M., Dekker, J., Helms, J. A., & Chang, H. Y. (2011). A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature, 472(7341), 120–124.
Boros, J., Arnoult, N., Stroobant, V., Collet, J. F., & Decottignies, A. (2014). Polycomb repressive complex 2 and H3K27me3 cooperate with H3K9 methylation to maintain heterochromatin protein 1alpha at chromatin. Molecular and Cellular Biology, 34(19), 3662–3674.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E., & Chang, H. Y. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell, 129(7), 1311–1323.
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y., Segal, E., & Chang, H. Y. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science, 329(5992), 689–693.
Brown, C. J., Ballabio, A., Rupert, J. L., Lafreniere, R. G., Grompe, M., Tonlorenzi, R., & Willard, H. F. (1991). A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature, 349(6304), 38–44.
Csankovszki, G., Nagy, A., & Jaenisch, R. (2001). Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. The Journal of Cell Biology, 153(4), 773–784.
Navarro, P., Page, D. R., Avner, P., & Rougeulle, C. (2006). Tsix-mediated epigenetic switch of a CTCF-flanked region of the Xist promoter determines the Xist transcription program. Genes & Development, 20(20), 2787–2792.
Chureau, C., Chantalat, S., Romito, A., Galvani, A., Duret, L., Avner, P., & Rougeulle, C. (2011). Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Human Molecular Genetics, 20(4), 705–718.
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., Xie, M., Zhang, Q., McMichael, J. F., Wyczalkowski, M. A., Leiserson, M. D. M., Miller, C. A., Welch, J. S., Walter, M. J., Wendl, M. C., Ley, T. J., Wilson, R. K., Raphael, B. J., & Ding, L. (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502(7471), 333–339.
Friend, S. H., Bernards, R., Rogelj, S., Weinberg, R. A., Rapaport, J. M., Albert, D. M., & Dryja, T. P. (1986). A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature, 323(6089), 643–646.
Collins, S., & Groudine, M. (1982). Amplification of endogenous myc-related DNA sequences in a human myeloid leukaemia cell line. Nature, 298(5875), 679–681.
Meyer, C., Schneider, B., Jakob, S., Strehl, S., Attarbaschi, A., Schnittger, S., Schoch, C., Jansen, M. W., van Dongen, J. J., den Boer, M. L., Pieters, R., Ennas, M. G., Angelucci, E., Koehl, U., Greil, J., Griesinger, F., Zur Stadt, U., Eckert, C., Szczepanski, T., Niggli, F. K., Schafer, B. W., Kempski, H., Brady, H. J., Zuna, J., Trka, J., Nigro, L. L., Biondi, A., Delabesse, E., Macintyre, E., Stanulla, M., Schrappe, M., Haas, O. A., Burmeister, T., Dingermann, T., Klingebiel, T., & Marschalek, R. (2006). The MLL recombinome of acute leukemias. Leukemia, 20(5), 777–784.
Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K., Rassenti, L., Kipps, T., Negrini, M., Bullrich, F., & Croce, C. M. (2002). Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America, 99(24), 15524–15529.
Cimmino, A., Calin, G. A., Fabbri, M., Iorio, M. V., Ferracin, M., Shimizu, M., Wojcik, S. E., Aqeilan, R. I., Zupo, S., Dono, M., Rassenti, L., Alder, H., Volinia, S., Liu, C. G., Kipps, T. J., Negrini, M., & Croce, C. M. (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America, 102(39), 13944–13949.
Corney, D. C., Flesken-Nikitin, A., Godwin, A. K., Wang, W., & Nikitin, A. Y. (2007). MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Research, 67(18), 8433–8438.
Ota, A., Tagawa, H., Karnan, S., Tsuzuki, S., Karpas, A., Kira, S., Yoshida, Y., & Seto, M. (2004). Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Research, 64(9), 3087–3095.
Tagawa, H., Karube, K., Tsuzuki, S., Ohshima, K., & Seto, M. (2007). Synergistic action of the microRNA-17 polycistron and Myc in aggressive cancer development. Cancer Science, 98(9), 1482–1490.
Rao, E., Jiang, C., Ji, M., Huang, X., Iqbal, J., Lenz, G., Wright, G., Staudt, L. M., Zhao, Y., McKeithan, T. W., Chan, W. C., & Fu, K. (2012). The miRNA-17 approximately 92 cluster mediates chemoresistance and enhances tumor growth in mantle cell lymphoma via PI3K/AKT pathway activation. Leukemia, 26(5), 1064–1072.
Jiang, P., Rao, E. Y., Meng, N., Zhao, Y., & Wang, J. J. (2010). MicroRNA-17-92 significantly enhances radioresistance in human mantle cell lymphoma cells. Radiation Oncology, 5, 100.
Czubak, K., Lewandowska, M. A., Klonowska, K., Roszkowski, K., Kowalewski, J., Figlerowicz, M., & Kozlowski, P. (2015). High copy number variation of cancer-related microRNA genes and frequent amplification of DICER1 and DROSHA in lung cancer. Oncotarget, 6(27), 23399–23416.
Jazdzewski, K., Murray, E. L., Franssila, K., Jarzab, B., Schoenberg, D. R., & de la Chapelle, A. (2008). Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proceedings of the National Academy of Sciences of the United States of America, 105(20), 7269–7274.
Paranjape, T., Heneghan, H., Lindner, R., Keane, F. K., Hoffman, A., Hollestelle, A., Dorairaj, J., Geyda, K., Pelletier, C., Nallur, S., Martens, J. W., Hooning, M. J., Kerin, M., Zelterman, D., Zhu, Y., Tuck, D., Harris, L., Miller, N., Slack, F., & Weidhaas, J. (2011). A 3′-untranslated region KRAS variant and triple-negative breast cancer: a case-control and genetic analysis. The Lancet Oncology, 12(4), 377–386.
Kim, M., Chen, X., Chin, L. J., Paranjape, T., Speed, W. C., Kidd, K. K., Zhao, H., Weidhaas, J. B., & Slack, F. J. (2014). Extensive sequence variation in the 3′ untranslated region of the KRAS gene in lung and ovarian cancer cases. Cell Cycle, 13(6), 1030–1040.
Siprashvili, Z., Webster, D. E., Johnston, D., Shenoy, R. M., Ungewickell, A. J., Bhaduri, A., Flockhart, R., Zarnegar, B. J., Che, Y., Meschi, F., Puglisi, J. D., & Khavari, P. A. (2016). The noncoding RNAs SNORD50A and SNORD50B bind K-Ras and are recurrently deleted in human cancer. Nature Genetics, 48(1), 53–58.
Dong, X. Y., Guo, P., Boyd, J., Sun, X., Li, Q., Zhou, W., & Dong, J. T. (2009). Implication of snoRNA U50 in human breast cancer. Journal of Genetics and Genomics, 36(8), 447–454.
Dong, X. Y., Rodriguez, C., Guo, P., Sun, X., Talbot, J. T., Zhou, W., Petros, J., Li, Q., Vessella, R. L., Kibel, A. S., Stevens, V. L., Calle, E. E., & Dong, J. T. (2008). SnoRNA U50 is a candidate tumor-suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Human Molecular Genetics, 17(7), 1031–1042.
Tanaka, R., Satoh, H., Moriyama, M., Satoh, K., Morishita, Y., Yoshida, S., Watanabe, T., Nakamura, Y., & Mori, S. (2000). Intronic U50 small-nucleolar-RNA (snoRNA) host gene of no protein-coding potential is mapped at the chromosome breakpoint t(3;6)(q27;q15) of human B-cell lymphoma. Genes to Cells, 5(4), 277–287.
Mei, Y. P., Liao, J. P., Shen, J., Yu, L., Liu, B. L., Liu, L., Li, R. Y., Ji, L., Dorsey, S. G., Jiang, Z. R., Katz, R. L., Wang, J. Y., & Jiang, F. (2012). Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene, 31(22), 2794–2804.
Alter, B. P., Giri, N., Savage, S. A., & Rosenberg, P. S. (2009). Cancer in dyskeratosis congenita. Blood, 113(26), 6549–6557.
Montanaro, L., Trere, D., & Derenzini, M. (2008). Nucleolus, ribosomes, and cancer. The American Journal of Pathology, 173(2), 301–310.
Shiue, C. N., Berkson, R. G., & Wright, A. P. (2009). c-Myc induces changes in higher order rDNA structure on stimulation of quiescent cells. Oncogene, 28(16), 1833–1842.
Jiang, Z., Zhou, Y., Devarajan, K., Slater, C. M., Daly, M. B., & Chen, X. (2012). Identifying putative breast cancer-associated long intergenic non-coding RNA loci by high density SNP array analysis. Frontiers in Genetics, 3, 299.
Yan, X., Hu, Z., Feng, Y., Hu, X., Yuan, J., Zhao, S. D., Zhang, Y., Yang, L., Shan, W., He, Q., Fan, L., Kandalaft, L. E., Tanyi, J. L., Li, C., Yuan, C. X., Zhang, D., Yuan, H., Hua, K., Lu, Y., Katsaros, D., Huang, Q., Montone, K., Fan, Y., Coukos, G., Boyd, J., Sood, A. K., Rebbeck, T., Mills, G. B., Dang, C. V., & Zhang, L. (2015). Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell, 28(4), 529–540.
Hu, X., Feng, Y., Zhang, D., Zhao, S. D., Hu, Z., Greshock, J., Zhang, Y., Yang, L., Zhong, X., Wang, L. P., Jean, S., Li, C., Huang, Q., Katsaros, D., Montone, K. T., Tanyi, J. L., Lu, Y., Boyd, J., Nathanson, K. L., Li, H., Mills, G. B., & Zhang, L. (2014). A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell, 26(3), 344–357.
Du, Z., Fei, T., Verhaak, R. G., Su, Z., Zhang, Y., Brown, M., Chen, Y., & Liu, X. S. (2013). Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nature Structural & Molecular Biology, 20(7), 908–913.
Pandey, G. K., Mitra, S., Subhash, S., Hertwig, F., Kanduri, M., Mishra, K., Fransson, S., Ganeshram, A., Mondal, T., Bandaru, S., Ostensson, M., Akyurek, L. M., Abrahamsson, J., Pfeifer, S., Larsson, E., Shi, L., Peng, Z., Fischer, M., Martinsson, T., Hedborg, F., Kogner, P., & Kanduri, C. (2014). The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell, 26(5), 722–737.
Oey, H., & Whitelaw, E. (2014). On the meaning of the word ‘epimutation’. Trends in Genetics, 30(12), 519–520.
Hanada, M., Delia, D., Aiello, A., Stadtmauer, E., & Reed, J. C. (1993). bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood, 82(6), 1820–1828.
Esteller, M., Silva, J. M., Dominguez, G., Bonilla, F., Matias-Guiu, X., Lerma, E., Bussaglia, E., Prat, J., Harkes, I. C., Repasky, E. A., Gabrielson, E., Schutte, M., Baylin, S. B., & Herman, J. G. (2000). Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. Journal of the National Cancer Institute, 92(7), 564–569.
Heyn, H., Vidal, E., Ferreira, H. J., Vizoso, M., Sayols, S., Gomez, A., Moran, S., Boque-Sastre, R., Guil, S., Martinez-Cardus, A., Lin, C. Y., Royo, R., Sanchez-Mut, J. V., Martinez, R., Gut, M., Torrents, D., Orozco, M., Gut, I., Young, R. A., & Esteller, M. (2016). Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biology, 17, 11.
Datta, J., Kutay, H., Nasser, M. W., Nuovo, G. J., Wang, B., Majumder, S., Liu, C. G., Volinia, S., Croce, C. M., Schmittgen, T. D., Ghoshal, K., & Jacob, S. T. (2008). Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Research, 68(13), 5049–5058.
Suzuki, H., Takatsuka, S., Akashi, H., Yamamoto, E., Nojima, M., Maruyama, R., Kai, M., Yamano, H. O., Sasaki, Y., Tokino, T., Shinomura, Y., Imai, K., & Toyota, M. (2011). Genome-wide profiling of chromatin signatures reveals epigenetic regulation of microRNA genes in colorectal cancer. Cancer Research, 71(17), 5646–5658.
Chen, W. S., Leung, C. M., Pan, H. W., Hu, L. Y., Li, S. C., Ho, M. R., & Tsai, K. W. (2012). Silencing of miR-1-1 and miR-133a-2 cluster expression by DNA hypermethylation in colorectal cancer. Oncology Reports, 28(3), 1069–1076.
He, X. X., Kuang, S. Z., Liao, J. Z., Xu, C. R., Chang, Y., Wu, Y. L., Gong, J., Tian, D. A., Guo, A. Y., & Lin, J. S. (2015). The regulation of microRNA expression by DNA methylation in hepatocellular carcinoma. Molecular BioSystems, 11(2), 532–539.
Dudziec, E., Miah, S., Choudhry, H. M., Owen, H. C., Blizard, S., Glover, M., Hamdy, F. C., & Catto, J. W. (2011). Hypermethylation of CpG islands and shores around specific microRNAs and mirtrons is associated with the phenotype and presence of bladder cancer. Clinical Cancer Research, 17(6), 1287–1296.
Lujambio, A., Ropero, S., Ballestar, E., Fraga, M. F., Cerrato, C., Setien, F., Casado, S., Suarez-Gauthier, A., Sanchez-Cespedes, M., Git, A., Spiteri, I., Das, P. P., Caldas, C., Miska, E., & Esteller, M. (2007). Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Research, 67(4), 1424–1429.
Furuta, M., Kozaki, K. I., Tanaka, S., Arii, S., Imoto, I., & Inazawa, J. (2010). miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis, 31(5), 766–776.
Wilting, S. M., van Boerdonk, R. A., Henken, F. E., Meijer, C. J., Diosdado, B., Meijer, G. A., le Sage, C., Agami, R., Snijders, P. J., & Steenbergen, R. D. (2010). Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Molecular Cancer, 9, 167.
Lv, X. B., Jiao, Y., Qing, Y., Hu, H., Cui, X., Lin, T., Song, E., & Yu, F. (2011). miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chinese Journal of Cancer, 30(12), 821–830.
Wang, P., Chen, L., Zhang, J., Chen, H., Fan, J., Wang, K., Luo, J., Chen, Z., Meng, Z., & Liu, L. (2014). Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene, 33(4), 514–524.
Chen, X., He, D., Dong, X. D., Dong, F., Wang, J., Wang, L., Tang, J., Hu, D. N., Yan, D., & Tu, L. (2013). MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Investigative Ophthalmology & Visual Science, 54(3), 2248–2256.
Tivnan, A., Zhao, J., Johns, T. G., Day, B. W., Stringer, B. W., Boyd, A. W., Tiwari, S., Giles, K. M., Teo, C., & McDonald, K. L. (2014). The tumor suppressor microRNA, miR-124a, is regulated by epigenetic silencing and by the transcriptional factor, REST in glioblastoma. Tumour Biology, 35(2), 1459–1465.
Formosa, A., Lena, A. M., Markert, E. K., Cortelli, S., Miano, R., Mauriello, A., Croce, N., Vandesompele, J., Mestdagh, P., Finazzi-Agro, E., Levine, A. J., Melino, G., Bernardini, S., & Candi, E. (2013). DNA methylation silences miR-132 in prostate cancer. Oncogene, 32(1), 127–134.
Qin, J., Ke, J., Xu, J., Wang, F., Zhou, Y., Jiang, Y., & Wang, Z. (2015). Downregulation of microRNA-132 by DNA hypermethylation is associated with cell invasion in colorectal cancer. Onco Targets Ther, 8, 3639–3648.
Zhang, S., Hao, J., Xie, F., Hu, X., Liu, C., Tong, J., Zhou, J., Wu, J., & Shao, C. (2011). Downregulation of miR-132 by promoter methylation contributes to pancreatic cancer development. Carcinogenesis, 32(8), 1183–1189.
Lin L, Wang Z, Jin H, Shi H, Lu Z, Qi Z (2016) MiR-212/132 is epigenetically downregulated by SOX4/EZH2-H3K27me3 feedback loop in ovarian cancer cells. Tumour Biol. 37(12), 15719–15727.
Rani, S. B., Rathod, S. S., Karthik, S., Kaur, N., Muzumdar, D., & Shiras, A. S. (2013). MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro-Oncology, 15(10), 1302–1316.
He, Y., Cui, Y., Wang, W., Gu, J., Guo, S., Ma, K., & Luo, X. (2011). Hypomethylation of the hsa-miR-191 locus causes high expression of hsa-mir-191 and promotes the epithelial-to-mesenchymal transition in hepatocellular carcinoma. Neoplasia, 13(9), 841–853.
Davalos, V., Moutinho, C., Villanueva, A., Boque, R., Silva, P., Carneiro, F., & Esteller, M. (2012). Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene, 31(16), 2062–2074.
Toyota, M., Suzuki, H., Sasaki, Y., Maruyama, R., Imai, K., Shinomura, Y., & Tokino, T. (2008). Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Research, 68(11), 4123–4132.
Xie, K., Liu, J., Chen, J., Dong, J., Ma, H., Liu, Y., & Hu, Z. (2014). Methylation-associated silencing of microRNA-34b in hepatocellular carcinoma cancer. Gene, 543(1), 101–107.
Daugaard, I., Knudsen, A., Kjeldsen, T. E., Hager, H., & Hansen, L. L. (2017). The association between miR-34 dysregulation and distant metastases formation in lung adenocarcinoma. Experimental and Molecular Pathology, 102(3), 484–491.
Zhang, X., Gejman, R., Mahta, A., Zhong, Y., Rice, K. A., Zhou, Y., Cheunsuchon, P., Louis, D. N., & Klibanski, A. (2010). Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Research, 70(6), 2350–2358.
Wu, Y., Lyu, H., Liu, H., Shi, X., Song, Y., & Liu, B. (2016). Downregulation of the long noncoding RNA GAS5-AS1 contributes to tumor metastasis in non-small cell lung cancer. Scientific Reports, 6, 31093.
Diaz-Lagares, A., Crujeiras, A. B., Lopez-Serra, P., Soler, M., Setien, F., Goyal, A., Sandoval, J., Hashimoto, Y., Martinez-Cardus, A., Gomez, A., Heyn, H., Moutinho, C., Espada, J., Vidal, A., Paules, M., Galan, M., Sala, N., Akiyama, Y., Martinez-Iniesta, M., Farre, L., Villanueva, A., Gross, M., Diederichs, S., Guil, S., & Esteller, M. (2016). Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(47), E7535–E7544.
Boque-Sastre, R., Soler, M., Oliveira-Mateos, C., Portela, A., Moutinho, C., Sayols, S., Villanueva, A., Esteller, M., & Guil, S. (2015). Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proceedings of the National Academy of Sciences of the United States of America, 112(18), 5785–5790.
Ferreira, H. J., Heyn, H., Moutinho, C., & Esteller, M. (2012). CpG island hypermethylation-associated silencing of small nucleolar RNAs in human cancer. RNA Biology, 9(6), 881–890.
Lujambio, A., Portela, A., Liz, J., Melo, S. A., Rossi, S., Spizzo, R., Croce, C. M., Calin, G. A., & Esteller, M. (2010). CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene, 29(48), 6390–6401.
Cao, J. (2014). The functional role of long non-coding RNAs and epigenetics. Biol Proced Online, 16, 11.
Suzuki, H., Maruyama, R., Yamamoto, E., & Kai, M. (2012). DNA methylation and microRNA dysregulation in cancer. Molecular Oncology, 6(6), 567–578.
Lopez-Serra, P., & Esteller, M. (2012). DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene, 31(13), 1609–1622.
Ehrlich, M. (2009). DNA hypomethylation in cancer cells. Epigenomics, 1(2), 239–259.
Urdinguio, R. G., Fernandez, A. F., Lopez-Nieva, P., Rossi, S., Huertas, D., Kulis, M., Liu, C. G., Croce, C. M., Calin, G. A., & Esteller, M. (2010). Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics, 5(7), 656–663.
Qu, W., Ding, S. M., Cao, G., Wang, S. J., Zheng, X. H., & Li, G. H. (2016). miR-132 mediates a metabolic shift in prostate cancer cells by targeting Glut1. FEBS Open Bio, 6(7), 735–741.
Whyte, W. A., Orlando, D. A., Hnisz, D., Abraham, B. J., Lin, C. Y., Kagey, M. H., Rahl, P. B., Lee, T. I., & Young, R. A. (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 153(2), 307–319.
Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-Andre, V., Sigova, A. A., Hoke, H. A., & Young, R. A. (2013). Super-enhancers in the control of cell identity and disease. Cell, 155(4), 934–947.
Loven, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., Bradner, J. E., Lee, T. I., & Young, R. A. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell, 153(2), 320–334.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646–674.
Ruby, J. G., Jan, C. H., & Bartel, D. P. (2007). Intronic microRNA precursors that bypass Drosha processing. Nature, 448(7149), 83–86.
Wu, Q., Ma, Q., Shehadeh, L. A., Wilson, A., Xia, L., Yu, H., & Webster, K. A. (2010). Expression of the Argonaute protein PiwiL2 and piRNAs in adult mouse mesenchymal stem cells. Biochemical and Biophysical Research Communications, 396(4), 915–920.
Iliev, R., Stanik, M., Fedorko, M., Poprach, A., Vychytilova-Faltejskova, P., Slaba, K., Svoboda, M., Fabian, P., Pacik, D., Dolezel, J., & Slaby, O. (2016). Decreased expression levels of PIWIL1, PIWIL2, and PIWIL4 are associated with worse survival in renal cell carcinoma patients. Onco Targets Ther, 9, 217–222.
Greither, T., Koser, F., Kappler, M., Bache, M., Lautenschlager, C., Gobel, S., Holzhausen, H. J., Wach, S., Wurl, P., & Taubert, H. (2012). Expression of human Piwi-like genes is associated with prognosis for soft tissue sarcoma patients. BMC Cancer, 12, 272.
Navarro, A., Tejero, R., Vinolas, N., Cordeiro, A., Marrades, R. M., Fuster, D., Caritg, O., Moises, J., Munoz, C., Molins, L., Ramirez, J., & Monzo, M. (2015). The significance of PIWI family expression in human lung embryogenesis and non-small cell lung cancer. Oncotarget, 6(31), 31544–31556.
Bamezai, S., Rawat, V. P., & Buske, C. (2012). Concise review: The Piwi-piRNA axis: pivotal beyond transposon silencing. Stem Cells, 30(12), 2603–2611.
Ferreira, H. J., Heyn, H., Garcia del Muro, X., Vidal, A., Larriba, S., Munoz, C., Villanueva, A., & Esteller, M. (2014). Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics, 9(1), 113–118.
Rounge, T. B., Furu, K., Skotheim, R. I., Haugen, T. B., Grotmol, T., & Enerly, E. (2015). Profiling of the small RNA populations in human testicular germ cell tumors shows global loss of piRNAs. Molecular Cancer, 14, 153.
Ushida, H., Kawakami, T., Minami, K., Chano, T., Okabe, H., Okada, Y., & Okamoto, K. (2012). Methylation profile of DNA repetitive elements in human testicular germ cell tumor. Molecular Carcinogenesis, 51(9), 711–722.
Heyn, H., Ferreira, H. J., Bassas, L., Bonache, S., Sayols, S., Sandoval, J., Esteller, M., & Larriba, S. (2012). Epigenetic disruption of the PIWI pathway in human spermatogenic disorders. PLoS One, 7(10), e47892.
Hotaling, J. M., & Walsh, T. J. (2009). Male infertility: a risk factor for testicular cancer. Nature Reviews. Urology, 6(10), 550–556.
Peng, X., Zeng, X., Peng, S., Deng, D., & Zhang, J. (2009). The association risk of male subfertility and testicular cancer: a systematic review. PLoS One, 4(5), e5591.
Liz, J., Portela, A., Soler, M., Gomez, A., Ling, H., Michlewski, G., Calin, G. A., Guil, S., & Esteller, M. (2014). Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Molecular Cell, 55(1), 138–147.
Ronchetti, D., Mosca, L., Cutrona, G., Tuana, G., Gentile, M., Fabris, S., Agnelli, L., Ciceri, G., Matis, S., Massucco, C., Colombo, M., Reverberi, D., Recchia, A. G., Bossio, S., Negrini, M., Tassone, P., Morabito, F., Ferrarini, M., & Neri, A. (2013). Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Medical Genomics, 6, 27.
Bellodi, C., McMahon, M., Contreras, A., Juliano, D., Kopmar, N., Nakamura, T., Maltby, D., Burlingame, A., Savage, S. A., Shimamura, A., & Ruggero, D. (2013). H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Reports, 3(5), 1493–1502.
McMahon, M., Ayllon, V., Panov, K. I., & O'Connor, R. (2010). Ribosomal 18 S RNA processing by the IGF-I-responsive WDR3 protein is integrated with p53 function in cancer cell proliferation. The Journal of Biological Chemistry, 285(24), 18309–18318.
Peng, Q., Wu, J., Zhang, Y., Liu, Y., Kong, R., Hu, L., Du, X., & Ke, Y. (2010). 1A6/DRIM, a novel t-UTP, activates RNA polymerase I transcription and promotes cell proliferation. PLoS One, 5(12), e14244.
Uemura, M., Zheng, Q., Koh, C. M., Nelson, W. G., Yegnasubramanian, S., & De Marzo, A. M. (2012). Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene, 31(10), 1254–1263.
Valleron, W., Laprevotte, E., Gautier, E. F., Quelen, C., Demur, C., Delabesse, E., Agirre, X., Prosper, F., Kiss, T., & Brousset, P. (2012). Specific small nucleolar RNA expression profiles in acute leukemia. Leukemia, 26(9), 2052–2060.
Ono, M., Yamada, K., Avolio, F., Scott, M. S., van Koningsbruggen, S., Barton, G. J., & Lamond, A. I. (2010). Analysis of human small nucleolar RNAs (snoRNA) and the development of snoRNA modulator of gene expression vectors. Molecular Biology of the Cell, 21(9), 1569–1584.
Ono, M., Scott, M. S., Yamada, K., Avolio, F., Barton, G. J., & Lamond, A. I. (2011). Identification of human miRNA precursors that resemble box C/D snoRNAs. Nucleic Acids Research, 39(9), 3879–3891.
Mourtada-Maarabouni, M., Pickard, M. R., Hedge, V. L., Farzaneh, F., & Williams, G. T. (2009). GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene, 28(2), 195–208.
Pickard, M. R., Mourtada-Maarabouni, M., & Williams, G. T. (2013). Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochimica et Biophysica Acta, 1832(10), 1613–1623.
Qiao, H. P., Gao, W. S., Huo, J. X., & Yang, Z. S. (2013). Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pacific Journal of Cancer Prevention, 14(2), 1077–1082.
Lu, X., Fang, Y., Wang, Z., Xie, J., Zhan, Q., Deng, X., Chen, H., Jin, J., Peng, C., Li, H., & Shen, B. (2013). Downregulation of gas5 increases pancreatic cancer cell proliferation by regulating CDK6. Cell and Tissue Research, 354(3), 891–896.
Liu, Z., Wang, W., Jiang, J., Bao, E., Xu, D., Zeng, Y., Tao, L., & Qiu, J. (2013). Downregulation of GAS5 promotes bladder cancer cell proliferation, partly by regulating CDK6. PLoS One, 8(9), e73991.
Shi, X., Sun, M., Liu, H., Yao, Y., Kong, R., Chen, F., & Song, Y. (2015). A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Molecular Carcinogenesis, 54(Suppl 1), E1–E12.
Sun, M., Jin, F. Y., Xia, R., Kong, R., Li, J. H., Xu, T. P., Liu, Y. W., Zhang, E. B., Liu, X. H., & De, W. (2014). Decreased expression of long noncoding RNA GAS5 indicates a poor prognosis and promotes cell proliferation in gastric cancer. BMC Cancer, 14, 319.
Yin, D., He, X., Zhang, E., Kong, R., De, W., & Zhang, Z. (2014). Long noncoding RNA GAS5 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer. Medical Oncology, 31(11), 253.
Cao, S., Liu, W., Li, F., Zhao, W., & Qin, C. (2014). Decreased expression of lncRNA GAS5 predicts a poor prognosis in cervical cancer. International Journal of Clinical and Experimental Pathology, 7(10), 6776–6783.
Yu, F., Zheng, J., Mao, Y., Dong, P., Lu, Z., Li, G., Guo, C., Liu, Z., & Fan, X. (2015). Long non-coding RNA growth arrest-specific transcript 5 (GAS5) inhibits liver fibrogenesis through a mechanism of competing endogenous RNA. The Journal of Biological Chemistry, 290(47), 28286–28298.
Luo, G., Liu, D., Huang, C., Wang, M., Xiao, X., Zeng, F., Wang, L., & Jiang, G. (2017). LncRNA GAS5 inhibits cellular proliferation by targeting P27Kip1. Molecular Cancer Research, 15(7), 789–799.
Zhang, Z., Zhu, Z., Watabe, K., Zhang, X., Bai, C., Xu, M., Wu, F., & Mo, Y. Y. (2013). Negative regulation of lncRNA GAS5 by miR-21. Cell Death and Differentiation, 20(11), 1558–1568.
Jelinic, P., & Shaw, P. (2007). Loss of imprinting and cancer. The Journal of Pathology, 211(3), 261–268.
da Rocha, S. T., Edwards, C. A., Ito, M., Ogata, T., & Ferguson-Smith, A. C. (2008). Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends in Genetics, 24(6), 306–316.
Molina-Pinelo S SA, Moreno-Mata N, Ferrer I, Suarez R, Andres-Leon E, Rodriguez-Paredes M, Gutekunst J, Jantus-Lewintre E, Camps C, Carnero A, Paz-Ares L (2016) Impact of DLK1–DIO3 imprinted cluster hypomethylation in smoker patients with lung cancer. Oncotarget https://doi.org/10.18632/oncotarget.10611.
Nadal, E., Zhong, J., Lin, J., Reddy, R. M., Ramnath, N., Orringer, M. B., Chang, A. C., Beer, D. G., & Chen, G. (2014). A microRNA cluster at 14q32 drives aggressive lung adenocarcinoma. Clinical Cancer Research, 20(12), 3107–3117.
Luk, J. M., Burchard, J., Zhang, C., Liu, A. M., Wong, K. F., Shek, F. H., Lee, N. P., Fan, S. T., Poon, R. T., Ivanovska, I., Philippar, U., Cleary, M. A., Buser, C. A., Shaw, P. M., Lee, C. N., Tenen, D. G., Dai, H., & Mao, M. (2011). DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. The Journal of Biological Chemistry, 286(35), 30706–30713.
Devor, E. J., JN, D. E. M., Ramachandran, S., Goodheart, M. J., & Leslie, K. K. (2012). Global dysregulation of the chromosome 14q32 imprinted region in uterine carcinosarcoma. Experimental and Therapeutic Medicine, 3(4), 677–682.
El-Daly, S. M., Abba, M. L., Patil, N., & Allgayer, H. (2016). miRs-134 and -370 function as tumor suppressors in colorectal cancer by independently suppressing EGFR and PI3K signalling. Scientific Reports, 6, 24720.
Azarbarzin, S., Hosseinpour Feizi, M. A., Safaralizadeh, R., Ravanbakhsh, R., Kazemzadeh, M., Fateh, A., Karimi, N., & Moaddab, Y. (2016). The value of miR-299-5p in diagnosis and prognosis of intestinal-type gastric adenocarcinoma. Biochemical Genetics, 54(4), 413–420.
Lucon, D. R., Rocha Cde, S., Craveiro, R. B., Dilloo, D., Cardinalli, I. A., Cavalcanti, D. P., Aguiar Sdos, S., Maurer-Morelli, C., & Yunes, J. A. (2013). Downregulation of 14q32 microRNAs in primary human desmoplastic medulloblastoma. Frontiers in Oncology, 3, 254.
Geraldo, M. V., Nakaya, H. I., & Kimura, E. T. (2017). Down-regulation of 14q32-encoded miRNAs and tumor suppressor role for miR-654-3p in papillary thyroid cancer. Oncotarget, 8(6), 9597–9607.
Laddha, S. V., Nayak, S., Paul, D., Reddy, R., Sharma, C., Jha, P., Hariharan, M., Agrawal, A., Chowdhury, S., Sarkar, C., & Mukhopadhyay, A. (2013). Genome-wide analysis reveals downregulation of miR-379/miR-656 cluster in human cancers. Biology Direct, 8, 10.
Tsang, E. K., Abell, N. S., Li, X., Anaya, V., Karczewski, K. J., Knowles, D. A., Sierra, R. G., Smith, K. S., & Montgomery, S. B. (2017). Small RNA sequencing in cells and exosomes identifies eQTLs and 14q32 as a region of active export. G3 (Bethesda), 7(1), 31–39.
Enfield, K. S., Martinez, V. D., Marshall, E. A., Stewart, G. L., Kung, S. H., Enterina, J. R., & Lam, W. L. (2016). Deregulation of small non-coding RNAs at the DLK1-DIO3 imprinted locus predicts lung cancer patient outcome. Oncotarget, 7(49), 80957–80966.
Hajjari, M., & Salavaty, A. (2015). HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol Med, 12(1), 1–9.
Amente, S., Bertoni, A., Morano, A., Lania, L., Avvedimento, E. V., & Majello, B. (2010). LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene, 29(25), 3691–3702.
Li, Y., Wang, Z., Shi, H., Li, H., Li, L., Fang, R., Cai, X., Liu, B., Zhang, X., & Ye, L. (2016). HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Research, 76(2), 293–304.
Ge, X. S., Ma, H. J., Zheng, X. H., Ruan, H. L., Liao, X. Y., Xue, W. Q., Chen, Y. B., Zhang, Y., & Jia, W. H. (2013). HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Science, 104(12), 1675–1682.
Liu, X. H., Sun, M., Nie, F. Q., Ge, Y. B., Zhang, E. B., Yin, D. D., Kong, R., Xia, R., Lu, K. H., Li, J. H., De, W., Wang, K. M., & Wang, Z. X. (2014). Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Molecular Cancer, 13, 92.
Bian, E. B., Ma, C. C., He, X. J., Wang, C., Zong, G., Wang, H. L., & Zhao, B. (2016). Epigenetic modification of miR-141 regulates SKA2 by an endogenous ‘sponge’ HOTAIR in glioma. Oncotarget, 7(21), 30610–30625.
Song, B., Guan, Z., Liu, F., Sun, D., Wang, K., & Qu, H. (2015). Long non-coding RNA HOTAIR promotes HLA-G expression via inhibiting miR-152 in gastric cancer cells. Biochemical and Biophysical Research Communications, 464(3), 807–813.
Xu, F., & Zhang, J. (2017). Long non-coding RNA HOTAIR functions as miRNA sponge to promote the epithelial to mesenchymal transition in esophageal cancer. Biomedicine & Pharmacotherapy, 90, 888–896.
Garzon, R., Liu, S., Fabbri, M., Liu, Z., Heaphy, C. E., Callegari, E., Schwind, S., Pang, J., Yu, J., Muthusamy, N., Havelange, V., Volinia, S., Blum, W., Rush, L. J., Perrotti, D., Andreeff, M., Bloomfield, C. D., Byrd, J. C., Chan, K., Wu, L. C., Croce, C. M., & Marcucci, G. (2009). MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood, 113(25), 6411–6418.
Cheng, J., Guo, S., Chen, S., Mastriano, S. J., Liu, C., D'Alessio, A. C., Hysolli, E., Guo, Y., Yao, H., Megyola, C. M., Li, D., Liu, J., Pan, W., Roden, C. A., Zhou, X. L., Heydari, K., Chen, J., Park, I. H., Ding, Y., Zhang, Y., & Lu, J. (2013). An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Reports, 5(2), 471–481.
Garzon, R., Volinia, S., Liu, C. G., Fernandez-Cymering, C., Palumbo, T., Pichiorri, F., Fabbri, M., Coombes, K., Alder, H., Nakamura, T., Flomenberg, N., Marcucci, G., Calin, G. A., Kornblau, S. M., Kantarjian, H., Bloomfield, C. D., Andreeff, M., & Croce, C. M. (2008). MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood, 111(6), 3183–3189.
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., Tsai, M. C., Hung, T., Argani, P., Rinn, J. L., Wang, Y., Brzoska, P., Kong, B., Li, R., West, R. B., van de Vijver, M. J., Sukumar, S., & Chang, H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature, 464(7291), 1071–1076.
Kogo, R., Shimamura, T., Mimori, K., Kawahara, K., Imoto, S., Sudo, T., Tanaka, F., Shibata, K., Suzuki, A., Komune, S., Miyano, S., & Mori, M. (2011). Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Research, 71(20), 6320–6326.
Wu, Z. H., Wang, X. L., Tang, H. M., Jiang, T., Chen, J., Lu, S., Qiu, G. Q., Peng, Z. H., & Yan, D. W. (2014). Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncology Reports, 32(1), 395–402.
Pistoia, V., Morandi, F., Wang, X., & Ferrone, S. (2007). Soluble HLA-G: are they clinically relevant? Seminars in Cancer Biology, 17(6), 469–479.
Guil, S., Soler, M., Portela, A., Carrere, J., Fonalleras, E., Gomez, A., Villanueva, A., & Esteller, M. (2012). Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nature Structural & Molecular Biology, 19(7), 664–670.
Zhang, Z., Weaver, D. L., Olsen, D., deKay, J., Peng, Z., Ashikaga, T., & Evans, M. F. (2016). Long non-coding RNA chromogenic in situ hybridisation signal pattern correlation with breast tumour pathology. Journal of Clinical Pathology, 69(1), 76–81.
Thorenoor, N., Faltejskova-Vychytilova, P., Hombach, S., Mlcochova, J., Kretz, M., Svoboda, M., & Slaby, O. (2016). Long non-coding RNA ZFAS1 interacts with CDK1 and is involved in p53-dependent cell cycle control and apoptosis in colorectal cancer. Oncotarget, 7(1), 622–637.
Li, T., Xie, J., Shen, C., Cheng, D., Shi, Y., Wu, Z., Deng, X., Chen, H., Shen, B., Peng, C., Li, H., Zhan, Q., & Zhu, Z. (2015). Amplification of long noncoding RNA ZFAS1 promotes metastasis in hepatocellular carcinoma. Cancer Research, 75(15), 3181–3191.
Wang, T., Ma, S., Qi, X., Tang, X., Cui, D., Wang, Z., Chi, J., Li, P., & Zhai, B. (2016). Long noncoding RNA ZNFX1-AS1 suppresses growth of hepatocellular carcinoma cells by regulating the methylation of miR-9. Onco Targets Ther, 9, 5005–5014.
Cai, L., & Cai, X. (2014). Up-regulation of miR-9 expression predicate advanced clinicopathological features and poor prognosis in patients with hepatocellular carcinoma. Diagnostic Pathology, 9, 1000.
Ma, L., Young, J., Prabhala, H., Pan, E., Mestdagh, P., Muth, D., Teruya-Feldstein, J., Reinhardt, F., Onder, T. T., Valastyan, S., Westermann, F., Speleman, F., Vandesompele, J., & Weinberg, R. A. (2010). miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nature Cell Biology, 12(3), 247–256.
Kishore, S., Gruber, A. R., Jedlinski, D. J., Syed, A. P., Jorjani, H., & Zavolan, M. (2013). Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biology, 14(5), R45.
Choi, M. S., Shim, Y. H., Hwa, J. Y., Lee, S. K., Ro, J. Y., Kim, J. S., & Yu, E. (2003). Expression of DNA methyltransferases in multistep hepatocarcinogenesis. Human Pathology, 34(1), 11–17.
Simo-Riudalbas, L., Perez-Salvia, M., Setien, F., Villanueva, A., Moutinho, C., Martinez-Cardus, A., Moran, S., Berdasco, M., Gomez, A., Vidal, E., Soler, M., Heyn, H., Vaquero, A., de la Torre, C., Barcelo-Batllori, S., Vidal, A., Roz, L., Pastorino, U., Szakszon, K., Borck, G., Moura, C. S., Carneiro, F., Zondervan, I., Savola, S., Iwakawa, R., Kohno, T., Yokota, J., & Esteller, M. (2015). KAT6B is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Research, 75(18), 3936–3945.
Di Ruscio, A., Ebralidze, A. K., Benoukraf, T., Amabile, G., Goff, L. A., Terragni, J., Figueroa, M. E., De Figueiredo Pontes, L. L., Alberich-Jorda, M., Zhang, P., Wu, M., D'Alo, F., Melnick, A., Leone, G., Ebralidze, K. K., Pradhan, S., Rinn, J. L., & Tenen, D. G. (2013). DNMT1-interacting RNAs block gene-specific DNA methylation. Nature, 503(7476), 371–376.
Wu, Y., Liu, H., Shi, X., Yao, Y., Yang, W., & Song, Y. (2015). The long non-coding RNA HNF1A-AS1 regulates proliferation and metastasis in lung adenocarcinoma. Oncotarget, 6(11), 9160–9172.
Zhan Y, Li Y, Guan B, Wang Z, Peng D, Chen Z, He A, He S, Gong Y, Li X, Zhou L (2017) Long non-coding RNA HNF1A-AS1 promotes proliferation and suppresses apoptosis of bladder cancer cells through upregulating Bcl-2. Oncotarget. 8(44), 76656–76665.
Liu, Z., Wei, X., Zhang, A., Li, C., Bai, J., & Dong, J. (2016). Long non-coding RNA HNF1A-AS1 functioned as an oncogene and autophagy promoter in hepatocellular carcinoma through sponging hsa-miR-30b-5p. Biochemical and Biophysical Research Communications, 473(4), 1268–1275.
Fang C, Qiu S, Sun F, Li W, Wang Z, Yue B, Wu X, Yan D (2017) Long non-coding RNA HNF1A-AS1 mediated repression of miR-34a/SIRT1/p53 feedback loop promotes the metastatic progression of colon cancer by functioning as a competing endogenous RNA. Cancer Lett, 410, 50–62.
Fu, A., Jacobs, D. I., & Zhu, Y. (2014). Epigenome-wide analysis of piRNAs in gene-specific DNA methylation. RNA Biology, 11(10), 1301–1312.
Rajasethupathy, P., Antonov, I., Sheridan, R., Frey, S., Sander, C., Tuschl, T., & Kandel, E. R. (2012). A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell, 149(3), 693–707.
Fu, A., Jacobs, D. I., Hoffman, A. E., Zheng, T., & Zhu, Y. (2015). PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis, 36(10), 1094–1102.
Ghoshal, K., & Bai, S. (2007). DNA methyltransferases as targets for cancer therapy. Drugs Today (Barc), 43(6), 395–422.
Liu, X. Q., Song, W. J., Sun, T. M., Zhang, P. Z., & Wang, J. (2011). Targeted delivery of antisense inhibitor of miRNA for antiangiogenesis therapy using cRGD-functionalized nanoparticles. Molecular Pharmaceutics, 8(1), 250–259.
Love, T. M., Moffett, H. F., & Novina, C. D. (2008). Not miR-ly small RNAs: big potential for microRNAs in therapy. The Journal of Allergy and Clinical Immunology, 121(2), 309–319.
Novina, C. D., & Chabner, B. A. (2008). RNA-directed therapy: the next step in the miRNA revolution. The Oncologist, 13(1), 1–3.
Lanford, R. E., Hildebrandt-Eriksen, E. S., Petri, A., Persson, R., Lindow, M., Munk, M. E., Kauppinen, S., & Orum, H. (2010). Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science, 327(5962), 198–201.
Burnett, J. C., & Rossi, J. J. (2012). RNA-based therapeutics: current progress and future prospects. Chemistry & Biology, 19(1), 60–71.
Adams, B. D., Parsons, C., & Slack, F. J. (2016). The tumor-suppressive and potential therapeutic functions of miR-34a in epithelial carcinomas. Expert Opinion on Therapeutic Targets, 20(6), 737–753.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ferreira, H.J., Esteller, M. Non-coding RNAs, epigenetics, and cancer: tying it all together. Cancer Metastasis Rev 37, 55–73 (2018). https://doi.org/10.1007/s10555-017-9715-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9715-8