
Abstract
Lung cancer is the leading cause of cancer-related deaths worldwide. The initiation and progression of lung cancer is the result of the interaction between permanent genetic and dynamic epigenetic alterations. DNA methylation is the best studied epigenetic mark in human cancers. Altered DNA methylation in cancer was identified in 1983. Within 30 years of this discovery, DNA methylation inhibitors are used clinically to treat a variety of cancers, highlighting the importance of the epigenetic basis of cancer. In addition, histone modifications, nucleosome remodeling, and micro RNA (miRNA)-mediated gene regulation are also fundamental to tumor genesis. Distinct chromatin alterations occur in all stages of lung cancer, including initiation, growth, and metastasis. Therefore, stage-specific epigenetic changes can be used as powerful and reliable tools for early diagnosis of lung cancer and to monitor patient prognosis. Moreover, since epigenetic changes are dynamic and reversible, chromatin modifiers are promising targets for the development of more effective therapeutic strategies against cancer. This review summarizes the chromatin alterations in lung cancer, focusing on the diagnostic and therapeutic approaches targeting epigenetic modifications that could help to reduce the high case-fatality rate of this dreadful disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Lung cancer accounts for approximately 25 % of cancer-related deaths worldwide and is responsible for more deaths than the other most prevalent cancers together, breast, prostate, and colorectal (Fig. 1a). Unfortunately, the mortality rate closely parallels the incidence rate for lung cancer because of persistently low patient survival (Fig. 1b) [1]. There are two major forms of lung cancer: small cell lung cancer (SCLC), representing approximately 20 %, and non-small cell lung cancer (NSCLC) which accounts for the remaining 80 % of lung cancers (Fig. 1c) [1–3]. Histologically, NSCLC can further be classified into adenocarcinoma, which is the most prevalent form (40 % prevalence), followed by squamous cell carcinoma (25 % prevalence), and large cell carcinoma which represents only 10 % of the cases [1, 3, 4].

Lung cancer statistics. a Lung cancer is responsible for more cancer-related deaths than the next three most prevalent cancers combined. Cancer deaths by site for the year 2012–13 were plotted for lung cancer (black bar) against that of prostate (blue), breast (pink), and colorectal cancer (green bar) combined. b Gender-specific incidence (solid lines) and mortality (dashed lines) of lung cancer for each year from 1971 till 2012 indicate that lung cancer mortality parallels the incidence rate for both males and females. c The major histological subtypes of lung cancer. Data are obtained from Globocan, SEER Cancer statistics, National Cancer Institute, and Center for Disease Control and Prevention. d Cancer is a result of the interplay between permanent genetic mutations and dynamic epigenetic alterations
The initiation and subsequent progression of lung cancer is a result of the accumulation of a combination of permanent genetic alterations, including point mutations, deletions, translocation, and/or amplifications as well as dynamic epigenetic alterations, which are influenced by environmental factors [1]. Epigenetic changes refer to the sum of heritable alterations of the chromatin, which is a complex of proteins and DNA in the nucleus of the cell and influences all DNA-dependent processes such as replication, repair, recombination, and transcription [5, 6]. Chromatin-mediated regulation of transcription involves DNA methylation, histone modifications, nucleosome remodeling, interaction with the nuclear matrix, and regulation via small noncoding RNAs [7, 8]. Chromatin-mediated transcription regulation establishes heritable patterns of differential gene expression and silencing from the same genome [9]. Proper DNA methylation is essential for cell differentiation and embryonic development. DNA methylation plays a critical role in repressing gene activity and maintaining genome stability by preventing recombination events between repetitive sequences. In eukaryotes, DNA methylation occurs at C5 in CpG dinucleotides [10]. While DNA methylation is a relatively stable change in somatic cells, histone modifications are more diverse and complex and can change rapidly during the course of the cell cycle.
Nucleosomes are the basic repeating structural and functional units of chromatin. Each nucleosome consist of 1.7 turns of DNA (146 bp) surrounding a histone octamer, which consists of two H2A-H2B dimers and one (H3-H4)2 tetramer. The additional histone H1 wraps another 20 bp, making up two complete turns of DNA per nucleosome [8]. Histone proteins can undergo post-translational modifications at their N terminal tails, which comprises acetylation, methylation, phosphorylation, ubiquitination, and sumoylation [9, 11]. A recent model suggests that a specific combination of histone modifications called the “histone code” imparts the expression status of a region of the chromatin [12]. Knudson’s two-hit hypothesis of 1971 states that in order of a cell to become oncogenic, both copies of a tumor suppressor gene should be mutated in the same cell [13]. Although somatic genetic mutations are known to play an important role in oncogenesis, epigenetic alterations are more frequent and they can either repress the expression of tumor suppressor genes or activate expression of oncogenes. Epigenetic modifications observed in lung carcinogenesis include aberrant DNA methylation patterns, histone modifications, and regulation by noncoding RNAs (ncRNA) [7] (Fig. 1d). Furthermore, these alterations can occur in defined nuclear positions and chromosome domains after exposure to environmental risk factors, as smoking, drugs, and chronic inflammation. Cigarette smoking is strongly associated with lung cancer, especially squamous and small cell lung cancer. More than 80 % of all lung cancers can be attributed to cigarette smoking [1]. An increase in the expression of DNA methyltransferases DNMT1, DNMT3A, and DNMT3B is known in NSCLC, especially among smokers and correlates with gene silencing of tumor suppressor genes, for instance CDKN2A, FHIT, and RARβ [14, 15]. In this review, we highlight the importance of chromatin alterations in lung cancer, focusing on the diagnostic and therapeutic approaches targeting epigenetic modifications that could help to reduce the high case-fatality rate of this dreadful disease.
2 Chromatin structure and lung cancer
Profound changes in DNA methylation patterns are an important characteristic of cancer cells. DNA hypomethylation at CpG dinucleotides was the first epigenetic abnormality to be identified in cancer cells, over three decades ago [16]. Furthermore, it was observed that the degree of hypomethylation of genomic DNA correlated with the severity of the cancer, such that genome-wide DNA methylation decreased as the tumor progressed from a benign proliferating mass to metastatic invasive cancer [17]. High-resolution CpG methylation mapping revealed that DNA hypomethylation in lung cancers occurred specifically at repetitive sequences [18], including heterochromatin repeats (e.g., satellite DNA), SINEs (short interspersed nuclear elements), LINEs (long interspersed nuclear elements), LTR (long terminal repeat) elements, and segmental duplications in subtelomeric regions (Fig. 2, left). In contrast, single-copy sequences were rarely demethylated. SINEs and LINEs together make up approximately 45 % of the human genome [19] and are usually methylated in normal tissues. However, the cancer-specific hypomethylation at repeat regions was not conserved between the individual tumors indicating randomness for targeting repeat sequences for demethylation in cancer. To evaluate whether genome-wide DNA hypomethylation was a cause or consequence of tumorigenesis, transgenic mice carrying a hypomorphic Dnmt1 allele were generated [20]. These mice displayed significant genome-wide DNA hypomethylation. The causal role of DNA methylation to tumor formation was demonstrated as these mice developed aggressive T cell lymphoma, consisting of a high frequency of aneuploidy, i.e., the loss or gain of one or more chromosomes [20]. One explanation for the mechanistic contribution of reduced DNA methylation to carcinogenesis is that hypomethylation of genomic DNA favors mitotic recombination between repetitive sequences resulting in chromosomal instability. Mitotic recombination normally occurs at a high frequency in human cells [21, 22]. Since recombination depends on the homology between nucleotide sequences, repetitive sequences are specially permissive to recombination events, resulting in gross chromosomal anomalies, including chromosomal rearrangements, deletions, and/or translocations [17]. Further evidence confirming this hypothesis was obtained using a diploid human cancer cell line depleted of DNMT1 and/or DNMT3b. These DNMT deficient cell lines displayed aneuploidy. Detailed analysis of the mitotic spreads revealed multiple translocations and chromosomal rearrangements in these DNMT knockout cell lines. This hypothesis is also supported by previous work wherein DNA methylation suppressed crossing over in Ascobolus [23]. Moreover, mouse ES cells deficient in DNMT1 displayed a higher efficiency of gene targeting, supporting the role of DNA methylation in preventing recombination [24].

Chromatin structure in normal and lung cancer cells. Upper panel, normal cells have methylated DNA (red circles) at repetitive genomic regions such as satellite DNA, LINE, and SINE repeats as well as transposon elements, thereby maintaining genomic stability. In addition, CpG islands usually remain unmethylated, allowing the transcription of tumor suppressor genes. The histone tails from promoter regions are marked by “active” histone marks, such as histone 3 lysine 9 acetylation (H3K9Ac, green circle) and H3 lysine 4 trimethylation (H3K4me3, yellow star). Gene body and intronic regions are also methylated preventing spurious transcription initiations. Lower panel, in cancer cells, there is dramatic reduction in DNA methylation at the repeat regions which confers genomic instability. LINE, SINE, and LTRs can undergo mitotic recombination resulting in chromosomal anomalies, including rearrangements, deletions, translocations, and aneuploidy. Further, DNA demethylation of transposons results in active retrotransposition of the transposable elements to sites in the genome wherein they can deregulate gene expression or result in chromosomal rearrangements. In addition, CpG islands at promoters are highly methylated resulting in transcriptional repression of several genes, including tumor suppressor genes. Further, there is also an accumulation of “repressive” histone marks, H3 lysine 27 trimethylation (H3K27me3, orange circles) which results in heterochromatin formation. Lastly, there is a loss of DNA methylation at coding regions and introns allowing transcription to initiate from incorrect start sites as well as interfere with splicing resulting in the formation of alternative transcripts
In addition to genomic instability due to increased recombination at repetitive sequences, DNA hypomethylation contributes to carcinogenesis by reactivation of transposable elements (Fig. 2, middle). LINEs belong to the class of transposable elements that lack LTRs at their ends. LINEs, which are part of the LINE-1 (or L1) family, constitute approximately 17 % of the human genome and are the only transposable elements capable of autonomous transposition [19, 25]. While the majority of the L1 elements have been rendered inactive in the human genome, there are still 80–100 functional active retrotransposition-competent L1s [26]. High-throughput transposon sequencing analyses have been used to determine extent and abundance of young retrotransposon insertions in human populations. On comparing lung tumor and adjacent normal lung DNA from patients, it was observed that somatic L1 insertions occur at high frequencies in human lung cancer genome [27]. Moreover, L1-permissive tumors displayed a specific hypomethylation signature indicating that altered DNA methylation may be responsible for the extraordinary levels of L1 transpositions observed in lung tumors. Although it has been known that DNA methylation plays an important role in suppressing retrotranspositions in the human genome [28], the direct contribution of DNA hypomethylation to somatic L1 retrotranspositions has not been evaluated in humans. Once reactivated, L1 elements can mutate and activate oncogenes or suppress tumor suppressor genes [28, 29]. In addition, transposition can also facilitate gross chromosomal rearrangements commonly observed in human tumors, further destabilizing the genome in cancer cells [30].
While global DNA hypomethylation is a common early event in lung cancer, there is also an equally high incidence of gene-specific promoter CpG island hypermethylation (Fig. 2, right). CpG island hypermethylation results in silencing of the target genes, which include tumor suppressor genes and DNA repair genes as well as genes involved in cell cycle control. Recently, it was shown that abnormal promoter methylation not only affects protein coding genes but also affects various noncoding RNAs that may play a role in malignant growth [31]. In addition, it was shown that a subset of genes related to epithelial-mesenchymal-transition (EMT) are significantly repressed in NSCLC after DNA methylation and that increased-gene specific DNA methylation correlates with EMT [32]. EMT is a fundamental and conserved process characterized by loss of cell adhesion and increased cell motility (Fig. 3a). EMT is essential for numerous developmental processes including mesoderm formation and neural tube formation and wound healing [33]. However, initiation of metastasis involves invasion, which has many phenotypic similarities to EMT, including a loss of cell-cell adhesion and an increase in cell mobility [33]. EMT is regulated by a variety of growth factors including epidermal growth factor (EGF), platelet-derived growth factors (PDGFs), fibroblast growth factor-2 (FGF-2), and transforming growth factor-beta (TGF-β) [34]. EMT is characterized by the loss of CDH1 (E-cadherin) a trans-membrane protein that is required for adherent junctions [35]. Following the loss of epithelial markers, there is a corresponding increase in mesenchymal markers, for instance VIM (vimentin), CDH12 (N-cadherin) FN1 (fibronectin), ACTA2 (alpha-smooth muscle actin), and increased activity of MMP (matrix metalloproteinases) [36, 37]. In the last few years, extensive studies have shown that a multilayer regulatory network of transcription factors controls EMT. The most studied network is the regulation through SNAIL (SNAI1 and SNAI2), ZEB (ZEB1 and ZEB2), and TWIST (TWIST1) family members, which are together referred to as EMT-transcription factors (EMT-TF) [38]. In a recent publication, it was shown that one of the master regulators of EMT, TWIST, binds to the CDH1 promoter and recruits the CHD4/nucleosome remodeling and deacetylase complex (CHD4/NuRD complex, also known as Mi2/NuRD complex) by direct interaction to several of its components as MTA2, CHD4, and RBBP7 (Fig. 3b) [39]. In addition, MTA2 directly interacts to and recruits the histone deacetylase HDAC2. Finally, the TWIST/CHD4/NuRD complex represses CHD1 expression by nucleosome remodeling as well as deacetylation of histones. The biological relevance of this mechanism of transcription regulation was demonstrated within the context of metastasis of two types of cancers, lung and breast cancer, since depletion of the components of the TWIST/CHD4/NuRD complex suppressed cell migration and invasion in cell culture and murine models of cancer metastasis. This work shows that not only DNA methylation but also other chromatin modifications, as nucleosome remodeling and histone modifications, plays a role during cancer metastasis. Further confirmation of this line of ideas was obtained by forced expression of TWIST in the mammary gland of Balb/c mice, which leads to metastasis in the lung [40]. Interestingly, activated RAS signaling is required for the in vivo function of TWIST during cancer, for instance via the KRASG12D mutant [41]. Metastasis is a multistep process which involves the dissociation of tumor cells from the epithelium, invasion to the connective tissues, through the adjacent basement membrane, intravasation, and subsequent extravasation and growth at a distant site [42]. EMT has been shown to increase the motility and invasiveness of cancer cells, the earliest events involved in the metastatic spread. Therefore, a better understanding of the molecular program leading to EMT will be critical for disease management, especially for late-stage diseases.

EMT occurs during lung cancer metastases and is controlled by multilayer regulatory networks. a EMT is the initiating step for lung metastasis, where epithelial cells are transformed to mesenchymal cells and migrate in the surrounding tissue. Multiple extracellular signaling pathways can regulate EMT through modulation of TWIST, SNAIL, and ZEB family members. b Loss of CDH1 (also known as E-cadherin) is necessary to induce EMT. The transcription factor TWIST targets the CHD4/NuRD complex containing MTA2, RBBP7 and HDAC2 to the proximal promoter of CDH1. The TWIST/CHD4/NuRD complex represses CDH1 expression by chromatin remodeling and histone deacetylation. TWIST requires active RAS signaling to promote EMT. Modified from [39]
3 Chromatin modifications as biomarkers for early lung cancer diagnostics
Clinical manifestations of lung cancer are diverse and patients are mostly asymptomatic at early stages. Further, symptoms are subtle and non-specific, resembling more common benign etiologies. Accordingly, lung cancer is more frequently diagnosed at advanced stages when patient prognosis is poor [3, 43]. Current diagnostic strategies involve imaging tests, including chest X-ray, sputum cytology, and tissue biopsies. However, most of them are invasive and are usually performed after development of symptoms which is most often at late stages. In 1990, spiral computed tomography (also called helical CT) was introduced as a promising technique for early lung cancer detection [44]. It is more sensitive than chest X-ray and allows imaging of tumors that are less than several centimeters in diameter. However, despite its success and sensitivity, it suffers from serious limitations. A large-scale clinical trial demonstrated a very high rate of detection of benign calcified nodules and thus high rate of false-positive detection [45]. Therapeutic options against lung cancer are more successful at early stages of the disease, as it is evident by the high 5-year patient survival rate of 52 %. Subsequent invasion of cancer cells in the surrounding tissue and to the lymph nodes decreases the 5-year survival rate to 25 %. Metastatic spread to different organs leads to a dramatic drop to 4 % patient survival [46]. The marginal increase in the 5-year patient survival from 12 % (1975–77) to 17 % (2002–08) for all ethnic groups [46] demonstrates the urgent need of new strategies to improve/enhance specificity and sensitivity of early lung cancer detection in order to improve patient prognosis. Biomarkers such as DNA methylation and micro RNA patterns to detect cancer have several advantages over genetic mutation screening. The incidence of anomalous DNA methylation is undoubtedly higher than that of somatic mutations, which can take place in various gene regions and are difficult to detect. While locus-specific DNA demethylation may not be ideal, global DNA hypomethylation can be used as a diagnostic marker for lung cancer. Further, gene-specific promoter DNA methylation can be used as a reliable and powerful marker for early lung cancer diagnosis, and as well to monitor patient prognosis.
3.1 Evaluating the status of DNA methylation in tissue and body fluids
In 24 % of bronchoalveolar lavage (BAL) samples obtained from NSCLC patients, methylation of the promoter of the cell cycle regulator CDKN2A (also known as p16INK4a) was observed [47]. Promoter methylation of CDKN2A and PTPRN2 has been shown to be one of the earliest events in cellular hyperplasia [48]. Subsequently, studies have shown aberrant promoter hypermethylation of RASSF1A, CDH13, MGMT, and APC in lung cancer [49–52]. Methylation of homeobox gene family member, SHOX2, in bronchial aspirates was recently identified in a 250-patient case-control study with 78 % sensitivity and 96 % specificity [53].
Hypermethylation of each CDKN2A, CDX2, HOXA1, and OPCML individually distinguished lung adenocarcinoma from healthy donors with a sensitivity of 67–86 % and a specificity of 74–82 % and showed significant DNA methylation even in stage I tumor samples [54]. Hypermethylation of the DAPK promoter was found in 34 % of cancer samples. Taking into consideration the different histological subtypes of NSCLC, DAPK promoter methylation was observed more frequently in squamous cell carcinoma than in adenocarcinoma and large cell carcinoma; however, these differences were not statistically significant [55].
An alternative to BAL is the collection of sputum, where tumor cells can be identified by atypical cell morphology. In general, sputum cytology using the Saccomanno method can be used to accurately diagnose primary lung cancer patients. Cytological typing accuracy among NSCLC was highest for squamous cell carcinoma (98 %) and reduced to 73 and 67 % in adenocarcinoma and large cell carcinoma, respectively. Cytologic typing accuracy was 91 % for SCLC [56]. Analysis of promoter methylation of RASSF1A combined with 3OST2 in sputum specimen demonstrated a sensitivity of 85 % with a specificity of 74 % [57]. The promoter methylation of 31 genes was analyzed in sputum of lung cancer patients in two independent cohorts to define a gene promoter methylation signature for lung cancer risk assessment [58]. Accurate diagnosis was made for 71–77 % of the patients using the promoter methylation signature of seven of these genes (PAX5β, PAX5α, Dal-1, GATA5, SULF2, and CXCL14). Whang et al. observed promoter hypermethylation of MLH1 in 55 % of the tumor samples obtained from stage I and II patients. Further evaluation demonstrated a similar promoter hypermethylation in 38 % of the sputum samples. Finally, they reported a 72 % concordance of sputum samples with matched tissue biopsies [59]. A different study investigated CDKN2A methylation in 80.2 % of tumor tissues and showed a frequency of 74.7 % in sputum specimens [60]. Several studies have evaluated the correlation between tissue and sputum samples. Hypermethylation of the best studied gene, CDKN2A, was found to be higher in tumor samples than in sputum with an interquartile range of 84–37 to 74–32 %, respectively [60–63].
The advantage of sputum collection is that the procedure can be done by the patient itself at home and samples can be sent for analysis. However, unless patients are trained for proper sputum collection, sampling may be inadequate because of overrepresentation of epithelial cells resulting in underestimation of the methylation level in cancer cells. Nevertheless, this method needs at least five sputum specimens to achieve the highest possible proportion of correct positive diagnoses [56]. Sputum cytology is still implemented as standard diagnostic tool for lung cancer diagnosis [64]. Although in developed countries, sputum cytology was replaced by tumor biopsies/tumor cytology. Over the last decade, research on sputum cytology for risk assessment and recurrence of early lung cancer brought new insights and implemented stringent and advanced molecular techniques that are highly sensitive.
DNA from necrotic and apoptotic cancer cells has been found in serum and plasma. Several genes have been evaluated in lung cancer patients to identify specific and sensitive targets for early lung cancer detection in clinical trials. In NSCLC, 75–87 % of serum samples correspond to their matched tissue samples for promoter hypermethylation of RASSF1A, CDKN2A, RARb, CDH13, FHIT, and BLU [65]. These authors evaluated cancer risk using this panel of six genes yielding a sensitivity of 73 % and specificity of 82 %. Remarkably, on comparing tumor tissues with the corresponding matched plasma samples, 75 % concordance was obtained. Promoter methylation of CDKN2A, DAPK, PAX5b, and GATA5 was analyzed in blood but it was 0.2 to 0.6-fold lower than in tissue samples [62]. Subsequent studies have observed methylation rates of CDKN2A from 22.2 to 75.7 % [60, 66, 67] in blood samples. Furthermore, hypermethylation for DAPK was found in 35 % of the bronchial epithelium and 41 % of the blood samples of smokers, whereas the remaining samples from nonsmokers were unaffected, showing smoking-/lung cancer-associated methylation changes [68]. The clinical significance of detecting methylation in blood could facilitate the evaluation of tumor progression next to routine screening. Nevertheless, finding such biomarkers in blood samples would indicate that the lung tumor has already become invasive [69], reflecting an advanced cancer stage. In addition to the studies mentioned above, there have been some reports that have found no correlation between promoter methylation and tumor stages in blood samples.
3.2 Breath prints as promising source for early lung cancer diagnostics
Current cytological examinations only detect around 50 % of lung cancer cases which results in immense work for both the clinic and patient. Largely, lung cancer is detected 2–12 month after the first presentation. In addition, current diagnostic tools are invasive and a further source of distress to patients. Recently, considerable effort has been put on identifying noninvasive approaches for early lung cancer diagnosis. An interesting approach to identify lung cancer was using specially trained “sniffer dogs,” which are able to detect volatile organic compounds (VOC) in exhaled breath [70]. These dogs successfully identified lung cancer patients within the group of controls and chronic obstructive pulmonary disease (COPD) patients. Further advancement in the technique is capturing and measuring the compound of interest in the exhaled breath. Breath capture methods range from directly breathing into an analysis platform (electronic Nose, eNose) or the relatively simple collection of exhaled breath through cooling devices (exhaled breath condensates, EBCs). Breath-based proteomics using the “electronic nose” are constructed from highly sensitive biological olfactory components that are coupled to electronic transducers [71]. Moreover, EBC-based lung cancer diagnosis has recently become more relevant, especially since studies have reported that EBCs can also be used to detect DNA mutations and DNA methylation patterns in lung cancer patients [72]. Recently, Xiao et al. [73] demonstrated promoter hypermethylation of CDKN2A in EBC of 40 % of the NSCLC patients that were analyzed using fluorescent quantitative methylation-specific PCR (F-MSP). However, DNA methylation of DAPK, PAX5beta, and RASSF1A has been also assayed in EBCs of lung cancer patients showing high variability between each individual [74]. The discrepancies between different reports might be explained through the fact that EBC is a highly diluted mixtures of compounds. Thus, EBC-based diagnosis of lung cancer requires appropriate stringent standardization protocols in order to reduce variability and increase sensitivity of the technique. Nevertheless, collecting EBCs is a promising new strategy of diagnosis of lung diseases, including lung cancer.
4 MicroRNAs as biomarker in lung cancer diagnostics
Micro RNAs (miRNAs) are short, noncoding RNAs that control gene expression by affecting the stability of their target messenger RNA (mRNA) [75]. In normal cells, they are responsible for the fine-tuning of homeostatic gene expression and help to confer robustness to cellular processes, which is required for inducing and keeping cell fate decisions [76]. Since 2002, miRNAs have been implicated in oncogenic transformation and their expression is altered at early stages of lung cancer. Further, approximately half of the miRNAs are located in fragile genomic regions which are known to be amplified or deleted in human cancer [77]. In cancer cells, a global reduction of miRNAs and alterations in the miRNA processing machinery have been found [78]. It is not surprising that miRNAs initially investigated as key players for cellular differentiation are inactivated in cancer cells. For instance, let-7, which is the best studied miRNA, was identified to be essential for proper development in C. elegans. Besides, the hsamiR-let-7 homologue in human has been shown to play major roles in lung cancer [79]. Northern blot analysis on a broad range of primary lung cancer patient samples showed a significant reduction of hsa-miR-let-7 expression. The human RAS gene contains multiple hsa-miR-let-7 complementary sites that allow hsa-miR-let-7 to negatively regulate RAS levels [80]. Thus, reduced hsa-miR-let-7 expression might increase RAS levels in human lung cancer suggesting a possible mechanism for how hsa-miR-let-7 promotes tumorigenesis. Interestingly, regression analysis showed that loss of hsa-miR-let-7 is independent from the pathological stage of lung cancer. Nonetheless, loss of hsa-miR-let-7 is associated with reduced survival after curative resection [81]. Paradoxically, some miRNAs have been found to be overexpressed in lung cancer. Hayashita et al. reported that hsa-miR-17HG cluster is especially overexpressed in SCLC [82]. Southern blot analysis revealed that the malignancy of the hsa-miR-17HG cluster is achieved by gene amplification. Differential expression of this miRNA cluster aberrantly regulated its target genes which are important for tumor pathogenesis.
Large-scale analysis for miRNA expression in tumor samples is useful to find phenotypic signatures of particular cancer types. Extensive work is being carried out to identify miRNA signatures for different lung cancer subtypes and different cancer stages. Donnem et al. published miRNA signatures specific for NSCLC [83]. They found significant alterations for 128 miRNAs, where the most highly misregulated miRNAs (hsa-miR-21, hsa-miR-106a, hsa-miR-126, hsa-miR-185, miR-210, and miR-15) were involved in angiogenesis. In addition, Boeri et al. detected miRNAs (hsa-miR-28-3p, hsa-miR-30c, hsa-miR-92a, hsa-miR-140-5p, hsa-miR-451, and hsa-miR-660) in blood of asymptomatic lung cancer patients up to 2 years before diagnosis [84], thereby demonstrating the use of miRNA profiles for early lung cancer diagnosis. However, the use of miRNA signatures for the diagnosis of specific cancers and stages still has limitations which are similar to transcriptome profiling. It is challenging to find the right platform with adequate bioinformatic tools for comprehensive studies in order to obtain reproducible results and to reduce costs of clinical investigations [85]. Furthermore, miRNA classifiers tend to have modest discrimination ability by comparing different studies [86]. The large variability between the different studies suggests the need of further improvement to develop stringent protocols in order to gain clinically relevant outcomes.
5 Conventional and alternative lung cancer therapy
Standard treatments for lung cancer include surgery, platinum-based chemotherapy, radiotherapy, combined chemo radiotherapy, and targeted therapy, either alone or in combination (Fig. 4). At early stages of the disease, surgery to remove the tumor and the nearby lymph nodes is the most consistent and successful treatment. Tumors can be removed by: anatomic segmentectomy, lobectomy, or pneumonectomy [2, 87–89]. When surgery is no longer an option, radiotherapy and/or chemotherapy may be suggested [90, 91]. Although external beam radiotherapy is normally used to treat all types of lung cancer, poor prognosis is still a major problem in NSCLC [2, 88]. Stereotactic body radiation is a noninvasive method targeting small tumors (T1-2, N0, M0). High amounts of small, highly focused, and accurate radiation beams are used to deliver potent doses of radiotherapy in just few fractions [2, 90, 92, 93]. For patients with advanced and metastatic NSCLC, chemotherapy is the main therapeutic strategy [91].

Molecular diagnosis and treatment of lung cancer at different stages of lung cancer. Lung cancer is a highly aggressive disease. At stage I, the disease is “localized” characterized by a primary tumor (shown in brown) which has not invaded into deeper lung tissues or the neighboring lymph nodes (in blue). As the disease progresses, at stages II and III, the cancer cell mass is larger and it may have invaded into the surrounding tissue and/or the neighboring lymph node (shown in brown). The disease is locally advanced or “regional” at these stages. Stage IV represents distant metastatic spread to other organs. The lower panels indicate the treatment approaches and the potential use of molecular markers at each stage
Identification of new potential biomarkers has led to a novel strategy, targeted therapy. Well-known biomarkers are mutations in receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR) [94]. Effective inhibitors of the constitutively active EGFR mutants are gefitinib, erlotinib, lapatinib, and cetuximab. Survival benefits have also been shown using bevacizumab, which is a monoclonal antibody that targets the vascular endothelial growth factor (VEGF) [94]. Moreover, in patients suffering from advanced NSCLC, bevacizumab improves the overall survival when added to paclitaxel-carboplatin [95]. Contrary to the lung cancer treatments mentioned above, which act directly against cancer cells or tumors, immunotherapy is a more sophisticated method that stimulates the patient’s immune system to target cancer cells. Modalities of immunotherapy comprise vaccination which promote or increase the immune response in patients suffering from NSCLC [96]. Melanoma-associated antigen A3 (MAGEA3) and mucinous glycoprotein-1 (MUC1) are tumor-associated antigens expressed in NSCLC. Both, MAGEA3 and MUC1 vaccines have shown evidence of activity and are currently being evaluated in phase II and phase III trials, respectively [96].
5.1 Chromatin modifiers as targeted therapy in lung cancer
Due to the reversibility of epigenetic modifications, chromatin modifiers are potential targets for the development of more effective therapeutic strategies against cancer [97–99]. Current treatments targeting chromatin regulators approved by the Food and Drug Administration (FDA) and European Medicines Evaluation Agency (EMEA) include histone deacetylase (HDAC) inhibitors, DNA methyl transferase (DNMT) inhibitors, and Janus kinase 2 (JAK2) inhibitors [97, 100, 101]. DNA methylation is considered to be a powerful therapeutic target in lung cancer [102], and the most extensively studied of these agents are DNA methyltransferase inhibitors (DNMTi) [103].
Azacytidine (5-azacytidine, Vidaza) and decitabine (5-aza′-2-deoxycytidine, Dacogen) are the most extensively used DNMTi in experimental and clinical studies [97, 102, 104]. Azacytidine is activated through phosphorylation and is incorporated into DNA and RNA. Decitabine is only integrated into DNA, which makes it a more potent inhibitor [105]. Both azacytidine and decitabine are analogs to the base cytidine (Fig. 5, top panel) and induce passive DNA demethylation as they incorporate into the DNA during replication. Besides inhibiting DNMT activity, azacytidine treatment also has an effect on histone modification patterns [106]. This was demonstrated in a study where the use of azacytidine alone or in combination with etinostat reduced the expression of the histone methyltransferase EZH2 and consequently increases the expression of EZH2-target genes [106]. Unfortunately, although the results with both azacytidine and decitabine have been promising, the use of these chemical compounds to induce DNA hypomethylation alone appears to not be completely effective [102].

Chromatin modifiers as therapeutic targets in lung cancer. In a cancer cell (left), chromatin modifications can be targeted for therapy. Top panel, 5′azadeoxycytidine induces passive DNA demethylation as it is incorporated into the DNA during replication and inhibits the DNA methlytransferase (DNMT, red oval). Middle panel, histone deacetylase (HDAC, brown oval) inhibitors are chemical compounds that are used to increase the level of histone acetylation by the subsequent activity of the histone acetyltransferases (HAT, green oval). Lower panel, cell-type-specific siRNA delivery to cancer cells. The single chain variable fragment (scFv) targeting a specific receptor on the cancer cell can be conjugated to the cationic peptides (such as Poly Arginine) which would bind the siRNA. Once the complex is internalized into the cell, the siRNA will be released resulting in the repression of the target mRNA
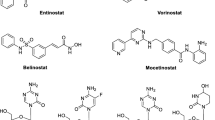
Trichostatin A (TSA), valproic acid, sodium phenyl butyrate, and romidepsin (Fig. 5, middle panel) belong to the first generation of histone deacetylase inhibitors (HDACis) which are usually administered intravenously [107]. Various HDACis, such as LBH589, scriptaid, valproic acid, apicidin, OSU-HDAC-44, and MAS-275, have successively induced cell death when tested in preclinical models of cancer [108, 109]. Valproic acid has been studied for the treatment of SCLC [110]; however, side effects are a limitation due to the high concentrations needed to induce antitumor activity. Two derivatives of this agent, ACS2 and ACS33, showed a stronger HDAC inhibitory activity and a higher cytotoxic activity than valporic acid itself [110, 111].
Romidepsin increased the efficacy of erlotinib in NSCLC cell lines, and this combination also induced apoptosis suggesting a possible benefit on patients suffering from NSCLC not predicted to respond to tyrosine kinase inhibitor (TKi) therapy [112]. It was recently shown that romidepsin induced cell cycle arrest and apoptosis and reduced expression of MMP2 and MMP9 in NSCLS cells [113].
TSA has been shown to be effective against prostate and breast cancer cells both in vitro and in vivo [114]. It does not alter embryonic or postnatal mouse development and has nontoxic effects in adult mice. TSA induced apoptosis in NSCLC cell lines as compared to control cells. Further, it was shown that while the tumor cells were apoptotic, characterized by increased expression of GSN, while control cells were predominantly in cell cycle arrest [115]. In SCLC cells, TSA has been showed to induce morphological differentiation, as well as inhibition in a dose-dependent manner of cell growth via cell cycle arrest resulting in apoptosis [116]. Further, pretreatment of SCLC patients with TSA enhanced the efficacy of chemotherapy [116].
As compared to the first generation of HDACs, second-generation agents can be used as oral formulations. Benzamides and hydroxamates are the two categories included in the second generation of HDACis [107]. Entinostat is the furthest developed benzamide, while vorinostat is the most studied hydroxamate agent [117]. Etinostat specifically inhibits HDAC1 and HDAC3 which are predominantly localized in the nucleus [117]. Vorinostat, also known as suberanilohydroxamic acid (SAHA), on the other hand targets the activity of all 11 known human HDACs. It has been shown to cause cell growth inhibition, differentiation, and apoptosis of different tumor types both in vitro and in vivo [118]. Vorinostat also reduced the expression of human telomerase reverse transcriptase (hTERT) in A549 cells by inducing DNA demethylation at the first exon of hTERT [119].
Although HDACis show promising results for cancer therapy, combinatorial therapeutic options are more beneficial and preferred [120]. For instance, the use of both azacytidine and entinostat in combination inhibits mutant K-ras/Tp53 lung adenocarcinoma in in vivo cancer models [121]. Likewise, combination of TSA and decitabine restores the expression of MLH1, TIMP3, and CDKN2A in colorectal cancer cells [102]. A forthcoming therapeutic approach to treat lung cancers might be the combination of three different agents such as gefitinib (an EGFR TKi), azacytidine, and TSA [122]. Therefore, combinatorial therapies using epigenetic modulators are a promising alternative when applied with other modalities of cancer treatment including immunotherapy [117].
6 Conclusions and future perspectives
Early lung cancer diagnosis is crucial to improve patient prognosis and reduce the extremely high case-fatality rate (95 %). Efforts to develop noninvasive, accurate, fast, and straight forward methods to screen individuals of the high-risk groups, which include current and former smokers, individuals exposed to environmental smoke, cooking fumes, indoor smoky coal emissions, asbestos, some metals (e.g., nickel, arsenic, and cadmium), radon, and ionizing radiation are essential. Environmental hazards may induce subtle genetic and epigenetic changes which could be detected by routine screening based on molecular markers. Molecular diagnoses based on exhaled breath, sputum, or blood are promising techniques, which when included into routine clinical practice can improve and complement the success of CT and CXR for early lung cancer diagnosis and especially help to distinguish between false and true positives. EBC-based expression analysis can also be enhanced to discriminate between different NSCLC subtypes by incorporating expression analysis of known markers of the different NSCLC subtypes. Furthermore, it might be combined with other known genetic and epigenetic markers for the detection of hyper-proliferative non-cancer-related diseases as idiopathic pulmonary fibrosis (IPF) or chronic obstructive pulmonary disease (COPD). Further, it could be used to monitor the response of a patient to specific treatments in order to fine-tune the therapy to improve the prognosis.
In clinical oncology, antibodies have been used as a prominent therapy against cancer [123]. Development of a large variety of recombinant antibodies is of great advantage since they are essential tools for research, diagnosis, and therapy with improved specificities than those provided by conventional antibody technology [124]. The smallest antigen-binding fragment of an antibody is the Fv fragment, which maintains its complete receptor binding site [125]. Single-chain variable fragments (scFv) against specific antigens in cancer have been selected for their particular powerful effect in immunotherapy, for instance against T cell neoplasias [123, 126, 127]. A similar approach can be used to develop therapies against lung adenocarcinoma, which has been shown to originate from alveolar type II (ATII) cells [102, 128]. Using recombinant antibodies against membrane proteins of ATII cells is an attractive approach for cell-targeted delivery of drugs. Small interfering RNAs (siRNAs) are considered as the new generation of biodrugs due to their specific and efficient response in RNA interference (RNAi) [129]. Using siRNAs coupled to ATII cell-specific antibodies will ensure cell-targeted delivery minimizing both the side effects and toxicity observed with chemotherapy (Fig. 5, lower panel). Once the target cell is localized, the siRNA complex is actively internalized inducing the desired biological effect. Using siRNAs to target specific epigenetic regulators, such as DNMTs or HDACs, could be superior to the use of chemical compounds to interfere with their function. Further, developments combining specific siRNA and cell-targeted delivery would improve patient care significantly, especially reducing side effects.
References
Balgkouranidou, I., Liloglou, T., & Lianidou, E. S. (2013). Lung cancer epigenetics: emerging biomarkers. Biomarkers in Medicine, 7(1), 49–58.
Molina, J. R., et al. (2008). Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic Proceedings, 83(5), 584–594.
Herbst, R. S., Heymach, J. V., & Lippman, S. M. (2008). Lung cancer. New England Journal of Medicine, 359(13), 1367–1380.
Hoffman, P. C., Mauer, A. M., & Vokes, E. E. (2000). Lung cancer. Lancet, 355(9202), 479–485.
Nemeth, A., & Langst, G. (2004). Chromatin higher order structure: opening up chromatin for transcription. Briefings in Functional Genomics & Proteomics, 2(4), 334–343.
Zhang, Y. (2011). Recent progress in the epigenetics and chromatin field. Cell Research, 21(3), 373–374.
Brzezianska, E., Dutkowska, A., & Antczak, A. (2013). The significance of epigenetic alterations in lung carcinogenesis. Molecular Biology Reports, 40(1), 309–325.
Ozturk, N., Singh, I., Mehta, A., Braun, T., & Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Frontiers in Cell and Developmental Biology.
Esteller, M. (2008). Epigenetics in cancer. New England Journal of Medicine, 358(11), 1148–1159.
Deaton, A. M., & Bird, A. (2011). CpG islands and the regulation of transcription. Genes and Development, 25(10), 1010–1022.
Cedar, H., & Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nature Reviews Genetics, 10(5), 295–304.
Wang, Y., et al. (2004). Beyond the double helix: writing and reading the histone code. Novartis Foundation Symposium, 259, 3–17. discussion 17–21, 163–9.
Knudson, A. G., Jr. (1971). Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America, 68(4), 820–823.
Kim, H., et al. (2006). Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer, 107(5), 1042–1049.
Lin, R. K., et al. (2007). Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer, 55(2), 205–213.
Feinberg, A. P., & Vogelstein, B. (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301(5895), 89–92.
Weber, M., et al. (2005). Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics, 37(8), 853–862.
Rauch, T. A., et al. (2008). High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proceedings of the National Academy of Sciences of the United States of America, 105(1), 252–257.
Lander, E. S., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921.
Gaudet, F., et al. (2003). Induction of tumors in mice by genomic hypomethylation. Science, 300(5618), 489–492.
Gupta, P. K., et al. (1997). High frequency in vivo loss of heterozygosity is primarily a consequence of mitotic recombination. Cancer Research, 57(6), 1188–1193.
Holt, D., et al. (1999). Interindividual variation in mitotic recombination. American Journal of Human Genetics, 65(5), 1423–1427.
Maloisel, L., & Rossignol, J. L. (1998). Suppression of crossing-over by DNA methylation in Ascobolus. Genes and Development, 12(9), 1381–1389.
Kim, M., et al. (2004). Dnmt1 deficiency leads to enhanced microsatellite instability in mouse embryonic stem cells. Nucleic Acids Research, 32(19), 5742–5749.
Beck, C. R., et al. (2011). LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics, 12, 187–215.
Sargurupremraj, M., & Wjst, M. (2013). Transposable elements and their potential role in complex lung disorder. Respiratory Research, 14, 99.
Iskow, R. C., et al. (2010). Natural mutagenesis of human genomes by endogenous retrotransposons. Cell, 141(7), 1253–1261.
Dupuy, A. J., et al. (2005). Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature, 436(7048), 221–226.
Collier, L. S., et al. (2005). Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature, 436(7048), 272–276.
Xing, J., et al. (2009). Mobile elements create structural variation: analysis of a complete human genome. Genome Research, 19(9), 1516–1526.
Lujambio, A., et al. (2010). CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene, 29(48), 6390–6401.
Lin, S. H., et al. (2014). Genes suppressed by DNA methylation in non-small cell lung cancer reveal the epigenetics of epithelial-mesenchymal transition. BMC Genomics, 15, 1079.
Thiery, J. P., et al. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890.
Kalluri, R., & Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation, 119(6), 1420–1428.
Le Bras, G. F., Taubenslag, K. J., & Andl, C. D. (2012). The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhesion & Migration, 6(4), 365–373.
Richardson, F., et al. (2012). The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Research, 32(2), 537–552.
Xiao, D., & He, J. (2010). Epithelial mesenchymal transition and lung cancer. Journal of Thoracic Disease, 2(3), 154–159.
Singh, A., & Settleman, J. (2010). EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene, 29(34), 4741–4751.
Fu, J., et al. (2011). The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Research, 21(2), 275–289.
Yang, J., et al. (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell, 117(7), 927–939.
Tran, P. T., et al. (2012). Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genetics, 8(5), e1002650.
De Craene, B., & Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer, 13(2), 97–110.
Van’t Westeinde, S. C., & van Klaveren, R. J. (2011). Screening and early detection of lung cancer. Cancer Journal, 17(1), 3–10.
Mulshine, J. L. (2003). Screening for lung cancer: in pursuit of pre-metastatic disease. Nature Reviews Cancer, 3(1), 65–73.
Marshall, H. M., et al. (2013). Screening for lung cancer with low-dose computed tomography: a review of current status. Journal of Thoracic Disease, 5(Suppl 5), S524–S539.
Siegel, R., Naishadham, D., & Jemal, A. (2013). Cancer statistics, 2013. CA: A Cancer Journal for Clinicians, 63(1), 11–30.
Ahrendt, S. A., et al. (1999). Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. Journal of the National Cancer Institute, 91(4), 332–339.
Selamat, S. A., et al. (2011). DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS One, 6(6), e21443.
Yanagawa, N., et al. (2003). Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Science, 94(7), 589–592.
Pulling, L. C., et al. (2003). Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: more common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression. Cancer Research, 63(16), 4842–4848.
Kim, J. S., et al. (2005). Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer, 104(9), 1825–1833.
Lin, Q., et al. (2009). RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non-small cell lung cancer in China. Journal of Cancer Research and Clinical Oncology, 135(12), 1675–1684.
Kneip, C., et al. (2011). SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. Journal of Thoracic Oncology, 6(10), 1632–1638.
Tsou, J. A., et al. (2007). Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Molecular Cancer, 6, 70.
Niklinska, W., et al. (2009). Prognostic significance of DAPK and RASSF1A promoter hypermethylation in non-small cell lung cancer (NSCLC). Folia Histochemica et Cytobiologica, 47(2), 275–280.
Risse, E. K., et al. (1985). Sputum cytology by the Saccomanno method in diagnosing lung malignancy. Diagnostic Cytopathology, 1(4), 286–291.
Hubers, A. J., et al. (2014). Combined sputum hypermethylation and eNose analysis for lung cancer diagnosis. Journal of Clinical Pathology, 67(8), 707–711.
Leng, S., et al. (2012). Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clinical Cancer Research, 18(12), 3387–3395.
Wang, Y. C., et al. (2003). Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. Journal of Clinical Investigation, 111(6), 887–895.
Liu, Y., et al. (2003). Hypermethylation of p16INK4a in Chinese lung cancer patients: biological and clinical implications. Carcinogenesis, 24(12), 1897–1901.
Destro, A., et al. (2004). K-ras and p16(INK4A)alterations in sputum of NSCLC patients and in heavy asymptomatic chronic smokers. Lung Cancer, 44(1), 23–32.
Belinsky, S. A., et al. (2007). Predicting gene promoter methylation in non-small-cell lung cancer by evaluating sputum and serum. British Journal of Cancer, 96(8), 1278–1283.
Cirincione, R., et al. (2006). Methylation profile in tumor and sputum samples of lung cancer patients detected by spiral computed tomography: a nested case–control study. International Journal of Cancer, 118(5), 1248–1253.
Ammanagi, A. S., et al. (2012). Sputum cytology in suspected cases of carcinoma of lung (Sputum cytology a poor man’s bronchoscopy!). Lung India, 29(1), 19–23.
Hsu, H. S., et al. (2007). Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer, 110(9), 2019–2026.
Suga, Y., et al. (2008). Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncology Reports, 20(5), 1137–1142.
Bearzatto, A., et al. (2002). p16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancer. Clinical Cancer Research, 8(12), 3782–3787.
Russo, A. L., et al. (2005). Differential DNA hypermethylation of critical genes mediates the stage-specific tobacco smoke-induced neoplastic progression of lung cancer. Clinical Cancer Research, 11(7), 2466–2470.
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646–674.
Boedeker, E., Friedel, G., & Walles, T. (2012). Sniffer dogs as part of a bimodal bionic research approach to develop a lung cancer screening. Interactive Cardiovascular and Thoracic Surgery, 14(5), 511–515.
Rattray, N. J., et al. (2014). Taking your breath away: metabolomics breathes life in to personalized medicine. Trends in Biotechnology, 32(10), 538–548.
Dent, A. G., Sutedja, T. G., & Zimmerman, P. V. (2013). Exhaled breath analysis for lung cancer. Journal of Thoracic Disease, 5(Suppl 5), S540–S550.
Xiao, P., et al. (2014). Methylation of P16 in exhaled breath condensate for diagnosis of non-small cell lung cancer. Lung Cancer, 83(1), 56–60.
Han, W., et al. (2009). Gene promoter methylation assayed in exhaled breath, with differences in smokers and lung cancer patients. Respiratory Research, 10, 86.
Filipowicz, W., Bhattacharyya, S. N., & Sonenberg, N. (2008). Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Reviews Genetics, 9(2), 102–114.
Ebert, M. S., & Sharp, P. A. (2012). Roles for microRNAs in conferring robustness to biological processes. Cell, 149(3), 515–524.
Calin, G. A., et al. (2004). MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proceedings of the National Academy of Sciences of the United States of America, 101(32), 11755–11760.
Lu, J., et al. (2005). MicroRNA expression profiles classify human cancers. Nature, 435(7043), 834–838.
Trang, P., et al. (2010). Regression of murine lung tumors by the let-7 microRNA. Oncogene, 29(11), 1580–1587.
Johnson, S. M., et al. (2005). RAS is regulated by the let-7 microRNA family. Cell, 120(5), 635–647.
Takamizawa, J., et al. (2004). Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Research, 64(11), 3753–3756.
Hayashita, Y., et al. (2005). A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Research, 65(21), 9628–9632.
Donnem, T., et al. (2012). MicroRNA signatures in tumor tissue related to angiogenesis in non-small cell lung cancer. PLoS One, 7(1), e29671.
Boeri, M., et al. (2011). MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proceedings of the National Academy of Sciences of the United States of America, 108(9), 3713–3718.
Pritchard, C. C., Cheng, H. H., & Tewari, M. (2012). MicroRNA profiling: approaches and considerations. Nature Reviews Genetics, 13(5), 358–369.
Nair, V. S., Maeda, L. S., & Ioannidis, J. P. (2012). Clinical outcome prediction by microRNAs in human cancer: a systematic review. Journal of the National Cancer Institute, 104(7), 528–540.
Cagle, P. T., & Chirieac, L. R. (2012). Advances in treatment of lung cancer with targeted therapy. Archives of Pathology and Laboratory Medicine, 136(5), 504–509.
(2011) In The diagnosis and treatment of lung cancer (Update). Cardiff (UK).
Schuchert, M. J., et al. (2010). Sublobar resection for early-stage lung cancer. Seminars in Thoracic and Cardiovascular Surgery, 22(1), 22–31.
Sonke, J. J., & Belderbos, J. (2010). Adaptive radiotherapy for lung cancer. Seminars in Radiation Oncology, 20(2), 94–106.
Pfister, D. G., et al. (2004). American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. Journal of Clinical Oncology, 22(2), 330–353.
Simone, C. B., 2nd, et al. (2013). Stereotactic body radiation therapy for lung cancer. Chest, 143(6), 1784–1790.
Timmerman, R., et al. (2010). Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA, 303(11), 1070–1076.
Ma, P.C. (2012). Personalized targeted therapy in advanced non-small cell lung cancer. Cleveland Clinic Journal of Medicine, (79 Electronic Suppl 1), eS56–60.
Sandler, A., et al. (2006). Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. New England Journal of Medicine, 355(24), 2542–2550.
Davies, M. (2014). New modalities of cancer treatment for NSCLC: focus on immunotherapy. Cancer Management and Research, 6, 63–75.
Hatzimichael, E., & Crook, T. (2013). Cancer epigenetics: new therapies and new challenges. Journal of Drug Delivery, 2013, 529312.
Jones, P. A., & Baylin, S. B. (2007). The epigenomics of cancer. Cell, 128(4), 683–692.
Chuang, J. C., & Jones, P. A. (2007). Epigenetics and microRNAs. Pediatric Research, 61(5 Pt 2), 24R–29R.
Dawson, M. A., & Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell, 150(1), 12–27.
Keohane, C., Mesa, R., & Harrison, C. (2013). The role of JAK1/2 inhibitors in the treatment of chronic myeloproliferative neoplasms. American Society of Clinical Oncology Educational Book, 301–305.
Liu, S. V., et al. (2013). Epigenetic therapy in lung cancer. Frontiers in Oncology, 3, 135.
Lyko, F., & Brown, R. (2005). DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. Journal of the National Cancer Institute, 97(20), 1498–1506.
Tang, M., et al. (2009). Potential of DNMT and its epigenetic regulation for lung cancer therapy. Current Genomics, 10(5), 336–352.
Christman, J. K. (2002). 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene, 21(35), 5483–5495.
Komashko, V. M., & Farnham, P. J. (2010). 5-azacytidine treatment reorganizes genomic histone modification patterns. Epigenetics, 5(3), 229–240.
Kim, H. J., & Bae, S. C. (2011). Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. American Journal of Translational Research, 3(2), 166–179.
Brazelle, W., et al. (2010). Histone deacetylase inhibitors downregulate checkpoint kinase 1 expression to induce cell death in non-small cell lung cancer cells. PLoS One, 5(12), e14335.
Tang, Y. A., et al. (2010). A novel histone deacetylase inhibitor exhibits antitumor activity via apoptosis induction, F-actin disruption and gene acetylation in lung cancer. PLoS One, 5(9), e12417.
Perrino, E., et al. (2008). New sulfurated derivatives of valproic acid with enhanced histone deacetylase inhibitory activity. Bioorganic and Medicinal Chemistry Letters, 18(6), 1893–1897.
Tesei, A., et al. (2012). Organosulfur derivatives of the HDAC inhibitor valproic acid sensitize human lung cancer cell lines to apoptosis and to cisplatin cytotoxicity. Journal of Cellular Physiology, 227(10), 3389–3396.
Zhang, W., et al. (2009). Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. Journal of Thoracic Oncology, 4(2), 161–166.
Karthik, S., et al. (2014). Romidepsin induces cell cycle arrest, apoptosis, histone hyperacetylation and reduces matrix metalloproteinases 2 and 9 expression in bortezomib sensitized non-small cell lung cancer cells. Biomedicine and Pharmacotherapy, 68(3), 327–334.
Papeleu, P., et al. (2003). Trichostatin A induces differential cell cycle arrests but does not induce apoptosis in primary cultures of mitogen-stimulated rat hepatocytes. Journal of Hepatology, 39(3), 374–382.
Mukhopadhyay, N. K., et al. (2006). Effectiveness of trichostatin A as a potential candidate for anticancer therapy in non-small-cell lung cancer. Annals of Thoracic Surgery, 81(3), 1034–1042.
Platta, C. S., et al. (2007). The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. Journal of Surgical Research, 142(2), 219–226.
Juergens, R.A., & Rudin, C.M. (2013). Aberrant epigenetic regulation: a central contributor to lung carcinogenesis and a new therapeutic target. American Society of Clinical Oncology Educational Book.
Petta, V., et al. (2013). Histones and lung cancer: are the histone deacetylases a promising therapeutic target? Cancer Chemotherapy and Pharmacology, 72(5), 935–952.
Li, C. T., et al. (2011). Vorinostat, SAHA, represses telomerase activity via epigenetic regulation of telomerase reverse transcriptase in non-small cell lung cancer cells. Journal of Cellular Biochemistry, 112(10), 3044–3053.
Forde, P. M., Brahmer, J. R., & Kelly, R. J. (2014). New strategies in lung cancer: epigenetic therapy for non-small cell lung cancer. Clinical Cancer Research, 20(9), 2244–2248.
Belinsky, S. A., et al. (2011). Combination therapy with Vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Research, 71(2), 454–462.
Noro, R., et al. (2007). PTEN inactivation in lung cancer cells and the effect of its recovery on treatment with epidermal growth factor receptor tyrosine kinase inhibitors. International Journal of Oncology, 31(5), 1157–1163.
Peipp, M., et al. (2002). A recombinant CD7-specific single-chain immunotoxin is a potent inducer of apoptosis in acute leukemic T cells. Cancer Research, 62(10), 2848–2855.
Deng, X. K., Nesbit, L. A., & Morrow, K. J., Jr. (2003). Recombinant single-chain variable fragment antibodies directed against Clostridium difficile toxin B produced by use of an optimized phage display system. Clinical and Diagnostic Laboratory Immunology, 10(4), 587–595.
Frenzel, A., Hust, M., & Schirrmann, T. (2013). Expression of recombinant antibodies. Frontiers in Immunology, 4, 217.
Wang, R., et al. (2013). Engineering production of functional scFv antibody in E. coli by co-expressing the molecule chaperone Skp. Frontiers in Cellular and Infection Microbiology, 3, 72.
Kumar, P., et al. (2008). T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell, 134(4), 577–586.
Xu, X., et al. (2012). Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America, 109(13), 4910–4915.
Oh, Y. K., & Park, T. G. (2009). siRNA delivery systems for cancer treatment. Advanced Drug Delivery Reviews, 61(10), 850–862.
Acknowledgments
We thank Indrabahadur Singh and Julio Cordero for helpful discussions. This work was done according to the program for competitive growth of the Kazan Federal University and the Russian Government. Addi J. Romero-Olmedo received a fellowship from CONACyT - COCyT (CVU 510283). Guillermo Barreto is funded by the „LOEWE-Initiative der Landesförderung“ (III L4–18/15.004 2009) and the DFG grant BA 4036/1-1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Affiliations in Germany are members of the Universities of Giessen and Marburg Lung Center (UGMLC) and the German Center of Lung Research (DZL).
Rights and permissions
About this article

Cite this article
Mehta, A., Dobersch, S., Romero-Olmedo, A.J. et al. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev 34, 229–241 (2015). https://doi.org/10.1007/s10555-015-9563-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-015-9563-3