
Abstract
Purpose
There is a need for biomarkers of drug efficacy for targeted therapies in triple-negative breast cancer (TNBC). As a step toward this, we identify multi-omic molecular determinants of anti-TNBC efficacy in cell lines for a panel of oncology drugs.
Methods
Using 23 TNBC cell lines, drug sensitivity scores (DSS3) were determined using a panel of investigational drugs and drugs approved for other indications. Molecular readouts were generated for each cell line using RNA sequencing, RNA targeted panels, DNA sequencing, and functional proteomics. DSS3 values were correlated with molecular readouts using a FDR-corrected significance cutoff of p* < 0.05 and yielded molecular determinant panels that predict anti-TNBC efficacy.
Results
Six molecular determinant panels were obtained from 12 drugs we prioritized based on their efficacy. Determinant panels were largely devoid of DNA mutations of the targeted pathway. Molecular determinants were obtained by correlating DSS3 with molecular readouts. We found that co-inhibiting molecular correlate pathways leads to robust synergy across many cell lines.
Conclusions
These findings demonstrate an integrated method to identify biomarkers of drug efficacy in TNBC where DNA predictions correlate poorly with drug response. Our work outlines a framework for the identification of novel molecular determinants and optimal companion drugs for combination therapy based on these correlates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Triple-negative breast cancer (TNBC) is a subtype of breast cancer defined by the absence of estrogen receptor (ER) and progesterone receptor (PR) expression and no overexpression of HER2/neu. TNBC is more likely to recur and metastasize, and have a higher grade than other subtypes of breast cancer [1, 2]. Moreover, it has few targeted approved treatments; thus surgery, radiation, and chemotherapy remain the mainstays of therapy for TNBC. This is largely due to TNBC being defined by what it lacks (ER, PR, Her2), while other forms of breast cancer are defined by the molecular features/targets they possess [3]. Thus far, many targeted therapies tested in traditionally-designed clinical trials largely without any biomarker-based patient selection have not fared well in TNBC [3,4,5]. Even a well-recognized marker such as homologous recombination deficiency, that has been advanced as promising for predicting response to checkpoint inhibitors, is under scrutiny in recent studies [6, 7]. Conversely, biomarker-guided selection for olaparib (using BRCA1 mutation or downregulation) and atezolizumab (using PD-L1 expression of immune cells) have led to FDA approvals of both drugs for TNBC, reflecting the observed improved patient outcomes [8,9,10,11]. A major reason proposed for the disappointing performance of targeted therapeutics in TNBC is the biological heterogeneity of the disease [12, 13]. Drugs approved on the basis of their biomarkers in TNBC, such as olaparib and atezolizumab, have shown promise in clinical trials for the small subset of patients matching the biomarker. There is however, a paucity of markers to predict the degree of clinical efficacy for drugs in breast cancer beyond ER or HER2 status. Broad efforts to identify biomarkers could enable larger numbers of TNBC patients to positively respond to targeted or conventional therapies [14]. These therapeutic challenges highlight the pressing clinical need for identifying robust biomarkers of drug efficacy in TNBC.
Using genetic information to inform drug discovery (and to “match” a drug to a patient) has been a significant focus since the sequencing of the human genome [15, 16]. While this has revolutionized how we treat specific tumors, such as EGFR mutant lung cancer [17], cancer-agnostic baskettrials have led to mixed results in selecting treatments for patients in the clinic [18,19,20]. It is now recognized that every occurrence of cancer is specific to each individual patient, even among patients who share certain mutations in notable cancer genes, such as TP53, RB1, BRCA1/2, PIK3CA, and others [21, 22]. While there is a push towards a true precision medicine where each patient’s tumor is evaluated and treated in a tumor-specific manner [23, 24], a significant gap remains in how to best match the therapeutic treatments to an individual tumor profile.
In an effort to identify molecular correlates to predict drug response in TNBC, we tested a library of 78 clinically approved and investigational new drugs (INDs) against a collection of 23 highly diverse TNBC cell lines. We measured the dose–response activity for each drug in each of 23 TNBC cell lines. Next, we determined the drug sensitivity score (DSS3), a drug response metric that incorporates both potency and efficacy, to facilitate quantitative correlations across drug types in each cell line [25]. We find that DNA mutational status alone generally does a poor job of predicting drug response in TNBC cell lines. However, using a combination of DNA sequencing, RNA sequencing, targeted RNA expression panels, and reverse-phase protein arrays (RPPA), we identify molecular features that correlate with DSS3 values. Further, co-inhibition that targets correlating molecular features identifies novel synergistic combinations.
Materials and methods
Cell culture
Cells used in this manuscript, including MUM51 Cell Line, BT20, BT549, CAL148, CAL51, DU4475, HCC1143, HCC1187, HCC1599, HCC1806, HCC1937, HCC2157, HCC38, HCC70, HS578t, MDA-MB-157, MDA-MB-231, MDA-MB-436, MDA-MB-453, MDA-MB-468, MFM223, SUM102, SUM149, SUM159, SUM185, SUM52, and VARI068 Cell Line, were cultured according to Supplemental Table 1 following dispersion with 0.05% Trypsin–EDTA (Invitrogen). All cell lines were appropriately validated or found to be unique by DNA short tandem repeat analysis (ATCC, Manassas, VA) within 6 months of use. All cell lines were tested and cleared from mycoplasma.
Chemicals and reagents
Chemicals were purchased from Selleckchem (Houston, TX), Sigma-Aldrich (St. Louis, MO), and Med Chem Express (Monmouth Junction, NJ). Compounds were diluted in DMSO (Sigma-Aldrich, D2650), except for copanlisib which was diluted in 10% Trifluoroacetic acid in DMSO due to solubility constraints (Sigma-Aldrich, T6508).
Primary antibodies used include Phospho AKT T308 Rabbit mAb (4056L, Cell Signaling Technology (CST)), Phospho AKT S473 Rabbit mAb (4058S, CST), pan-AKT Rabbit mAb (4691S, CST), PI3 Kinase p85 Rabbit Ab (4292S, CST), and Monoclonal Anti-β-Actin–Peroxidase (3854, Sigma-Aldrich). Secondary antibody used was anti-rabbit IgG-HRP (sc-2357, Santa Cruz Biotechnology).
Drug screening
Cells were screened in 96-well (Corning, 353072 or Costar, REF3610) format in triplicate. Cells were plated on day 0 at 3000 cells per well. On day 1, drugs were added at 1:1000, resulting in 0.1% final DMSO concentration per well. On day 4, viability was measured using cell proliferation reagent WST-1 (Sigma, 5015944001) followed by CellTiter-Glo (Promega, G9242) on a Synergy 4 plate reader (Bio Tek) or Envision plate reader (Perkin Elmer). For Chou–Talalay synergy calculations, cells were dosed at 1:500 DMSO (1:1000 of each drug) [26, 27]. IC50 values were determined for each drug and this IC50 value was used as the middle dose, with 3 doses twofold higher and 3 doses twofold lower, capturing points along the IC50 curve for synergy calculations.
Drug sensitivity score calculation
Drug dose response data were fit to the equation \(Y = {\text{bottom}} + ({\text{top}} - {\text{bottom}})/(1 + 10^{{({\text{Log}}10{\text{IC}}_{50} - X)\; \times \;{\text{HillSlope}}}} )\) where X = Log10 (concentration, M) and Y = % inhibition (vs. vehicle) using Graphpad Prism 7. Constraints used were bottom = 0 and top ≤ 100. DSS3 values were calculated as described by Yadav et al. [25]. Dose response curve readouts used for DSS3 calculation are listed in Supporting Information. The IC50 values, hillslope, maximum inhibition, and drug ranges were entered into the DSS package for Rstudio and DSS3 values (ranging 0 to 100) were calculated.
Molecular characterization
200,000 cells were plated in 6 cm dishes and grown for 24 h. DNA, RNA, and protein were collected from each of the 23 cell lines using the Qiagen AllPrep mini kit (Qiagen, 80204) or RPPA lysis buffer. DNA samples were sequenced using the Roche Human Oncology Panel. RNA was sequenced using QuantSeq 3′ (Lexogen) on an Illumina NovaSeq at TIGEM (Naples, Italy) and using Nanostring targeted panels. Protein was sent to MD Anderson for RPPA analysis. For full details, see Supplemental Materials and Methods.
Correlations and statistics
Molecular determinants were identified by calculating Pearson correlations between DSS3 values and molecular readouts including DNA variants, log2 normalized RNA expression levels from RNA-seq, log2 Nanostring array counts, normalized log2 Nanostring signature scores, and normalized log2 RPPA protein expression levels. To reduce problems associated with multiple testing and to identify significant correlations across 23 TNBC cell lines, we reduced the number of tests needed by eliminating low-count or low variability genes. From the nanostring and RPPA data, the bottom 15% of total counts were excluded. From the DNA sequencing, variants with VAFs of 0.0 or 1.0 were eliminated from analysis. Additionally, a second table was constructed by generating a roll-up of mutations to the same gene. From RNA sequencing, the top 5000 genes were selected based on the highest variance-mean ratio. Using R, Pearson correlations p-values were adjusted for multiple testing using the false discovery rate (FDR) procedure for each dataset. Significant molecular determinants were identified as those with an FDR-adjusted p*-value < 0.05. To visualize molecular correlates, signatures were ordered by DSS3 and hierarchically clustered before visualization with Morpheus (Broad Institute) [28]. Mann–Whitney and AUC-ROC testing was calculated using GraphPad Prism 7. Synergy of companion drugs with copanlisib was calculated using CompuSyn software (ComboSyn, Inc., Paramus, NJ) [26, 27, 29].
Results
Identifying drugs with anti-proliferative activity in TNBC cell lines
To identify molecular correlates of drug efficacy in TNBC, we developed a platform to correlate drug responses with molecular features. Seventy-eight FDA approved or investigational new drugs (IND) drugs were screened against a molecularly and ethnically diverse collection of 23 TNBC cell lines, in dose–response format. Drugs were chosen based on their efficacy in other solid tumors. From the 78 compounds, 61 compounds with discernable EC50 curves were identified (17 compounds were inactive across all cell lines tested). Drug sensitivity score 3 (DSS3) values were calculated for each the 61 drugs and 23 cell lines in an effort to compare potency and efficacy across the TNBC cell lines (Supplemental Table 2) [25]. The spread of DSS3 values was visualized by plotting median DSS3 value vs. interquartile range (IQR) of DSS3 values (Fig. 1). We reasoned that drugs displaying a high dispersion in their response across cell lines are more informative for deriving correlations. Drugs with an IQR greater than 15 and/or a median DSS3 over 20 were prioritized for further analysis as they exhibit sufficient range of activity and sensitivity, and thus were amenable to the analyses; the resulting top 12 compounds were advanced for molecular correlate identification [25].

Selection of molecular correlate candidate drugs. Twenty-three TNBC cell lines were treated with a panel of drugs yielding EC50 curves. The EC50 curves were used to derive DSS3 values. The IQR and Median DSS3 was plotted. Compounds with an IQR above 15 or median above 20 were prioritized, resulting in 12 drugs prioritized as molecular correlate targets. Correlations with molecular profiling yielded 6 sets of candidate molecular correlates (red)
Drug response predictions based on mutation status alone broadly fail in TNBC cell lines
Basket clinical trials use DNA mutational state of specific genes to attempt to match the patient tumor profiles to prospective treatments in advanced solid tumors, including TNBC [30]. To better understand how well basket-trial criteria predict drug response in our TNBC cell line collection, we grouped DSS3 values on the basis of their mutation status for drugs: copanlisib (PI3 K inhibitor), dasatinib (Src/Abl inhibitor), trametinib (MEK1/2 inhibitor), and TAK228 (mTOR inhibitor) (Fig. 2). PI3 K mutation status predicts copanlisib-sensitive cells with an average DSS of 54.6 and insensitive average of 16.9 (Fig. 2a). This prediction was significant via a Mann–Whitney test with a p < 0.01 and an area under the receiver operating characteristic curve (AUC-ROC) value of 0.89 (AUC = 0.5 denotes a poor classifier and AUC = 1 indicates a perfect classifier) [31]. PTEN mutations and PTEN or PI3 K mutations yield similar predictions for copanlisib with AUC values of 0.84 and 0.92, respectively. In basket-trials (NCT02465060), mutations in DDR2 are used to predict dasatinib sensitivity [32]; however, in our TNBC cell line collection there is almost no statistical difference between sensitive and insensitive groups via a Mann–Whitney test using this criterion, with a poor AUC value of 0.46 (Fig. 2b). Similarly, predictions for trametinib (BRAF, GNA11, and NF1 [33]) and TAK228 (TSC and mTOR [34, 35]) on the basis of mutation status (NCT02465060) fail to correctly predict differences in groups, with AUC values ranging from 0.11–0.59 (Fig. 2c, d).

DNA mutations alone poorly predict drug response in TNBC. Observing mutation status alone from basket-trial criteria, retrospective predictions of drug response are poor in TNBC cell lines. Average and spread of DSS3 compared in 23 TNBC cell lines. AUC values calculated for each paired comparison. a Mutation-based predictions for copanlisib sensitivity rely on PIK3CA and PTEN mutations. b Mutation-based predictions for dasatinib sensitivity rely on DDR2 mutations. c Mutation-based predictions for trametinib sensitivity rely on BRAF, GNA11, and NF1 mutations. d Mutation-based predictions for TAK228 rely on TSC1/2 and mTOR
Identifying molecular correlates for drug efficacy in TNBC cell lines using a multi-omic approach
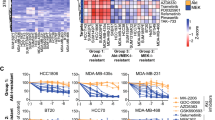
In an effort to improve on the current poor TNBC efficacy predictions based on DNA mutations alone, we elected to take an integrated multi-omics approach to identify novel molecular correlates of drug efficacy in TNBC cell lines. Twenty-three TNBC cell lines were molecularly characterized using RPPA, DNA sequencing, RNA sequencing, and targeted RNA profiling arrays. DSS3 was linearly correlated with each set of molecular features for the 12 prioritized drugs. The correlations resulted in molecular correlates of drug efficacy with FDR-adjusted p < 0.05 for 6 out of the 12 prioritized drugs (Fig. 3 and Supplemental Fig. 1). Six prioritized drugs (bortezomib, romidepsin, AZD-1775, paclitaxel, eribulin, and mubritinib) had no molecular correlates that met our selection criteria. Copanlisib DSS3 values were found to correlate with the PI3 K pathway as pathway members were identified via DNA mutation (PIK3CA), RNA expression (PIK3CD), and functional proteomics (AKT, GSKA, Tuberin) (Fig. 3a). In addition to the PI3 K pathway, we found that the RNA expression levels of other genes, as well as breast cancer subtype signatures, correlated with copanlisib efficacy meeting our selection criteria and FDR threshold. Dasatinib DSS3 values correlated with a novel panel of RNA gene hits, as well as three breast cancer subtype signatures (Fig. 3b). Trametinib DSS3 values gave correlations with both DNA mutations and RNA expression levels (Fig. 3c). Previous reports have demonstrated that ETS2 is a downstream transcription factor of the MAPK pathway [36]. CCND2 levels have been shown to correspond to MAPK signaling in other forms of cancer [37]. Additionally, TIAM1 is a parallel downstream Ras pathway member that has previously been reported to correspond to trametinib sensitivity [38]. TAK228 DSS3 values correlated with RNA markers as well as the Luminal A breast cancer subtype (Fig. 3d). TAK228 and Aurora inhibitors have previously been shown to synergize in TNBC [39]. Temsirolimus DSS3 values correlate with expression levels of TNFSF8 and SMAD4 (Supplemental Fig. 1a). Finally, we found that docetaxel DSS3 values correlate with a SMAD7 missense mutation (Supplemental Table 3), with non-responsive cell lines harboring SMAD7 mutations (Supplemental Fig. 1b).

Molecular correlates in TNBC cell lines. Examples of compounds with significant correlates: a copanlisib, b dasatinib, c trametinib, and d TAK228. All molecular marker readouts have a FDR-adjusted p*-value < 0.05. Top bars depict DSS3, with dark green representing the most sensitive lines and white representing the least sensitive. Orange labeled readouts are derived from DNA variants. Blue labeled readouts derived from functional proteomics. Black labeled readouts are derived from targeted RNA expression levels. Gray italicized labeled readouts are derived from global RNA expression levels. Pink labeled readouts are derived from breast cancer subtyping scores
Identifying synergistic combinations of targeted therapies using molecular correlates
We next wanted to determine whether we could leverage our molecular correlates of drug efficacy to identify synergistic drug combinations. Here, we focused on copanlisib due to its excellent anti-TNBC activity and robust selection of correlates available to discern companion drugs. We chose to target two molecular correlates of copanlisib drug response (EPHA2 and CENPE), in which their RNA expression levels are elevated in copanlisib-resistant cell lines and decreased in copanlisib-sensitive cell lines (Fig. 3a). Importantly, there are inhibitors available to target both EPHA2 and CENPE. We inhibited CENPE using the selective inhibitor, GSK923295, and inhibited EPHA2 using the selective inhibitor, ALW-II-41-27, and also the non-selective kinase inhibitor, dasatinib [40, 41]. We found that there a was significant synergy for each of the three combinations (GSK923295 + copanlisib, ALW-II-41-27 + copanlisib, and dasatinib + copanlisib) using Chou–Talalay analysis (Table 1). Briefly, Chou–Talalay analysis quantifies synergy at multiple therapeutically relevant effect levels (e.g., EC50, EC75-) where the Chou–Talalay combination index (CI) defines synergy, additivity, or antagonism (0 < CI < 1 is synergism, CI = 1 is additivity, CI > 1 is antagonism). CENPE chemical inhibition was found to have a synergistic effect with copanlisib in 5 of 7 cell lines tested at an effective dose (ED) of 50% (0.5) and 6 of 7 lines at an ED of 75% (0.75). EPHA2 chemical inhibition had a synergistic effect with copanlisib in all 7 cell lines tested at an ED of 50 and 75%.
Copanlisib anti-TNBC efficacy is not dependent on downstream AKT signaling
To examine the target engagement of copanlisib in TNBC cell lines, we collected cell lysates from three TNBC cell lines with a broad range of DSS3 values (MDA-MB-453 = 68.8; MDA-MB-436 = 8.9; and Cal51 = 62.0) treated with increasing concentrations of copanlisib. Western Blot analysis demonstrates that although there is a 20-fold difference in viability IC50 across these three cell lines, PI3 K signaling is reduced over a similar concentration range as measured by downstream AKT activation (Fig. 4a). Indeed, the least growth-sensitive cell line (MDA-MB-436, DSS3 = 8.9) has PI3 K-AKT pathway inhibition at 2 nM copanlisib (Fig. 4b, e, f). Meanwhile, PI3 K-mutated MDA-MB-453 is growth-sensitive to copanlisib (DSS3 = 68.8) and has a similar PI3 K-AKT pathway IC50 to the insensitive cell line MDA-MB-436. PI3 K-mutated Cal51 (DSS3 = 62.0) exhibited pathway inhibition at 8 nM and cell viability IC50 at 31 nM (Fig. 4c–f). Taken together, these results demonstrate that TNBC cell lines have a spectrum of sensitivity to PI3 K pathway inhibition that is not explained by the signaling of the PI3 K-AKT pathway alone.

Downstream changes in PI3 K signaling do not explain differences in DSS3 value. Signaling was measured following 2 h treatment with copanlisib at 5 × increasing doses. Cell lines measured were a MDA-MB-453, b MDA-MB-436, c and Cal51. Key for d–f located in e. d Cell viability curves for 72 h copanlisib treatment shows differential IC50 and cell death in the 3 cell lines. AKT activation is plotted for e. AKT pT308 and f AKT pS473, calculated by densitometry for pAKT/Total AKT from a–c
Discussion
Here, our work is focused on TNBC, while large pan-cancer resources, including the Genomics of Drug Sensitivity in Cancer (GDSC), have provided databases of cell line characterization and drug sensitivity data [42]. GDSC includes 22 TNBC cell lines, a similar number to the 23 cell lines in our study. In addition to a focused collection of TNBC-directed drugs that were examined in our study, we also utilized matched samples (identical material) for molecular characterization and drug sensitivity studies. It has recently been reported that individual cell lines undergo molecular diversification in culture, resulting in passage-specific changes at both the genetic and drug response level [43]. We utilize comprehensive molecular characterization of a diverse TNBC cell line collection and correlate drug responses to molecular readouts. From these data, we assemble panels of molecular correlates of therapeutic efficacy for the most promising anti-TNBC drugs. Current standards for prediction of drug response rely almost entirely on mutational changes within tumor cell DNA. Indeed, DNA-based biomarkers are prevalent in today’s large personalized medicine trials, including NCI-MATCH, TAPUR, and LOTUS [30, 44]. Though these trials are considered an important step toward true precision medicine, a significant shortcoming in these trials is that mutations alone are not generally robustly predictive of therapeutic response, even often in the case of therapies that directly target a mutated pathway. We found this discordance when comparing basket-trial enrollment criteria to drug response in TNBC cell lines (Fig. 2). While mutations in PIK3CA generate stratification of cell line response to copanlisib, mutation-based predictions alone for dasatinib, trametinib, and TAK228 fail in TNBC cell lines. In fact, the mutation-based criteria for trametinib and TAK228 predict the inverse of the experimental drug response where selection on the basis of basket-trial criteria incorrectly predicts high drug efficacy for a resistant group of cell lines. These results highlight the need for more robust biomarkers of drug efficacy.
Biomarkers of drug efficacy for cell lines have been successfully developed for other subtypes of cancer but are lacking for TNBC [45,46,47]. In an effort to identify highly robust molecular correlates of drug efficacy, we screened a collection of 23 TNBC cell lines. Our collection of TNBC cell lines was designed to maximally represent the molecular and ethnic diversity of the disease. While we found 23 cell lines to provide robust molecular correlates for drug response, additional TNBC cell lines would increase our predictive power and ease future application of molecular correlations to the clinic. We thus encourage the continual development of additional cell line models of TNBC and other cancers. Our goal was to correlate drug efficacy with a variety of molecular markers (DNA mutation, RNA expression, and functional proteomics). Correlates for copanlisib were heavily focused around the PI3 K pathway (Fig. 3a). However, it is striking that most of the derived molecular correlates for other targeted therapies did not directly include the drug-targeted pathway. For example, the drug efficacy of TAK228, an TORC1/2 inhibitor, is not correlated with genes or proteins from the mTOR pathway (Fig. 3d). Instead, TAK228 correlates strongly with AURKA, consistent with previously published results that demonstrated that the combination of TAK228 and Aurora inhibition is highly efficacious in TNBC [39]. Functional proteomic studies (RPPA) yielded significant correlations only for copanlisib, while global RNA expression and targeted RNA expression panels yielded significant molecular correlates for five of twelve drugs tested. Four of the newly identified panels contain correlations from DNA sequencing, though copanlisib is the only drug wherein the mutation in PIK3CA is within the drug-targeted pathway. Three of the six molecular correlate panels were also aligned with breast cancer subtyping scores (determined by nanoString PAM50 analysis [48]). While we did run a nanostring miRNA array containing nearly 800 well-studied miRNAs, we found no correlations that met our FDR-adjusted significance threshold for any of the prioritized compounds.
Copanlisib, a PI3 K inhibitor, was one of the most promising compounds that we identified in TNBC cell lines. Notably, the PI3 K pathway has long been of great interest for the treatment of TNBC, in both pre-clinical models and clinical trials [49,50,51]. TNBC presents with PIK3CA mutations in 20–30% of cases and low PTEN expression in 48% of cases [44, 52]. Together, these findings result in perturbation of PI3 K signaling in a significant number of TNBC patients. The frequent dysregulation of the PI3 K signaling pathway in TNBC is indicative of its importance in the pathogenesis of the disease. While PI3 K pathway inhibitors have shown some limited success in clinical trials, and especially so in subgroups of patients with aberrant mTOR/PI3 K/PTEN signaling [53, 54], the altered signaling in these cancers is not entirely explained by mutations. For example, the LOTUS trial identified that 48% of TNBC patients presented with decreased PTEN expression; however, only 29% of patients with low PTEN expression had corresponding PTEN genetic mutations [44]. This highlights a major challenge when selecting patients for clinical trials: mutations are not uniquely and robustly predictive of response to various therapies that directly or indirectly target the mutations or their downstream pathways [55]. Together, our findings emphasize the importance of considering other molecular profiling methodologies, such as gene and protein expression levels, in the design of clinical trials, if robust biomarker signatures can be deduced for the individual compounds.
Single agent targeted therapies are known to be prone to resistance [56,57,58]. Combining multiple efficacious targeted agents has emerged as a strategy to prevent or delay resistance (and to deal with resistance that has already emerged) [59,60,61,62]. It remains however a significant challenge to identify which drug(s) to combine with a specific targeted therapy. Most studies aimed at identifying companion drugs are performed using laborious and costly, seemingly “brute force”-like efforts, that explore all combinations without any basis for prioritization. We hypothesized that our molecular correlates could be used to identify optimal compounds to partner with a targeted agent. Notably, previous published work has described TAK228 and Aurora inhibitors as synergistic when administered in combination [39]. We found that AURKA was a correlate for TAK228, which lends support to using molecular correlates to identify drugs combinations. Thus, we hypothesized that we could identify novel synergistic combinations on the basis of our molecular correlate panels. As proof of principle for this approach, we selected copanlisib as the primary agent due to its excellent anti-TNBC efficacy. On the basis of copanlisib’s molecular correlate profile, EPHA2 and CENPE were prioritized correlates due to their high expression in less responsive cell lines. We measured significant anti-TNBC efficacy between copanlisib and EPHA2/CENPE inhibitors. Both combinations of inhibitors resulted in synergistic responses when administered at therapeutically relevant levels in six out of seven cell lines tested (Table 1). The cell line that did not respond to the combination (MDA-MB-436) is likely due to very high initial sensitivity to both of the companion drugs, resulting in difficulty to interpret curves and imperfect synergy analysis. Cell lines were chosen to be representative of the original data set, with a wide range of initial DSS3 values for copanlisib, as well as varied starting expression levels of both EPHA2 and CENPE (Supplemental Table 2). These results demonstrate that our molecular correlates cannot only be used to predict drug response, but because of their robustness, also prove valuable in the rational identification of companion drugs and thus, also provide a method for prioritizing the testing of drug combinations.
Our molecular correlates are notable because they did not generally include the direct pathway impacted by compound treatment. To better understand the relationship of downstream signaling to cellular viability, we measured downstream AKT signaling, following copanlisib treatment. We then compared inhibition of the PI3 K-AKT pathway to the reduced viability from copanlisib treatment (Fig. 4). We measured two copanlisib-sensitive cell lines (MDA-MB-453 and Cal51) and one growth-insensitive cell line (MDA-MB-436). Though MDA-MB-453 and MDA-MB-436 have a 100-fold difference in cell viability EC50, the suppression of downstream AKT signaling with copanlisib treatment is nearly identical. Cal51 has a fivefold differential in both cell viability EC50 and downstream signaling relative to MDA-MB-453. Thus, we observed that viability EC50 values between cell lines is not dependent on or correlated with differences in signaling suppression. Mutation of PI3 K thus predicts copanlisib sensitivity not because of elevated signaling, but likely because of a cellular addiction to PI3 K signaling or to signaling through other compensatory pathways.
In summary, our findings propose a potential solution for the pressing clinical need for robust biomarkers of drug efficacy in TNBC. Existing biomarkers for anti-TNBC drugs are scarce and generally rely on DNA alterations that poorly predict cellular response. Herein, we identified six molecular correlate panels for relevant clinical compounds using TNBC cell lines. Our work suggests that these drugs could be effective clinically when paired with the correct molecular correlate panels to guide patient selection. Modern instrumentation, such as the nanoString nCounter, which enables simultaneous analysis of DNA, RNA, and protein in rapid manner (2–3 days), could thus be used clinically to obtain a multi-omics panel for each putative patient. This work outlines a framework for the identification of biomarkers and has further utility in the identification of synergistic target pathways based on these correlate readouts. While expansion to the clinic would require extensive validation in ex vivo patient models, we believe that a multi-omic approach utilizing DNA, RNA, and protein has the potential to yield more robust biomarkers than examining DNA mutations alone. Together our work provides key insights into the interplay between drug sensitivity and molecular signaling in TNBC.
References
Pareja F, Reis-Filho JS (2018) Triple-negative breast cancers—a panoply of cancer types. Nat Rev Clin Oncol 15(6):347–348
Wahba HA, El-Hadaad HA (2015) Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med 12(2):106–116
Kalimutho M, Parsons K, Mittal D, Lopez JA, Srihari S, Khanna KK (2015) Targeted therapies for triple-negative breast cancer: combating a stubborn disease. Trends Pharmacol Sci 36(12):822–846
Le Du F, Ueno NT (2015) Targeted therapies in triple-negative breast cancer: failure and future. Womens Health (Lond) 11(1):1–5
Yam C, Mani SA, Moulder SL (2017) Targeting the molecular subtypes of triple negative breast cancer: understanding the diversity to progress the field. Oncologist 22(9):1086–1093
Pellegrino B, Mateo J, Serra V, Balmana J (2019) Controversies in oncology: are genomic tests quantifying homologous recombination repair deficiency (HRD) useful for treatment decision making? Esmo Open. https://doi.org/10.1136/esmoopen-2018-000480
Mayer EL, Abramson VG, Jankowitz RC, Falkson CI, Marcom PK, Traina TA, Carey LA, Rimawi MF, Specht JM, Miller K et al (2019) TBCRC 030: a randomized phase II study of preoperative cisplatin versus paclitaxel in TNBC—evaluating the homologous recombination deficiency (HRD) biomarker. J Clin Oncol 37(15_suppl):507
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G et al (2018) Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 379(22):2108–2121
Heimes AS, Schmidt M (2019) Atezolizumab for the treatment of triple-negative breast cancer. Expert Opin Investig Drugs 28(1):1–5
Griguolo G, Dieci MV, Guarneri V, Conte P (2018) Olaparib for the treatment of breast cancer. Expert Rev Anticancer Ther 18(6):519–530
Romero D (2019) Benefit in patients with PD-L1-positive TNBC. Nat Rev Clin Oncol 16(1):6
Bareche Y, Venet D, Ignatiadis M, Aftimos P, Piccart M, Rothe F, Sotiriou C (2018) Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol 29(4):895–902
Chiu AM, Mitra M, Boymoushakian L, Coller HA (2018) Integrative analysis of the inter-tumoral heterogeneity of triple-negative breast cancer. Sci Rep 8(1):11807
Davis SL, Eckhardt SG, Tentler JJ, Diamond JR (2014) Triple-negative breast cancer: bridging the gap from cancer genomics to predictive biomarkers. Ther Adv Med Oncol 6(3):88–100
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al (2001) Initial sequencing and analysis of the human genome. Nature 409(6822):860–921
Saito M, Momma T, Kono K (2018) Targeted therapy according to next generation sequencing-based panel sequencing. Fukushima J Med Sci 64(1):9–14
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350(21):2129–2139
Garrido-Castro AC, Lin NU, Polyak K (2019) Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 9(2):176–198
Mardis E (2018) Many mutations in one clinical-trial basket. Nature 554(7691):173–175
Renfro LA, Sargent DJ (2017) Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol 28(1):34–43
Wheler J, Lee JJ, Kurzrock R (2014) Unique molecular landscapes in cancer: implications for individualized, curated drug combinations. Cancer Res 74(24):7181–7184
Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S, Mitchell TJ, Grossmann S, Lightfoot H, Egan DA et al (2018) Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556(7702):457–462
Hodson R (2016) Precision medicine. Nature 537(7619):S49
Dugger SA, Platt A, Goldstein DB (2018) Drug development in the era of precision medicine. Nat Rev Drug Discov 17(3):183–196
Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumagi A, Knowles J et al (2014) Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 4:5193
Chou TC (2010) Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res 70(2):440–446
Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58(3):621–681
Morpheus
Chou TC, Talalay P (1984) Quantitative analysis of dose–effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Brower V (2015) NCI-MATCH pairs tumor mutations with matching drugs. Nat Biotechnol 33(8):790–791
Fan J, Upadhye S, Worster A (2006) Understanding receiver operating characteristic (ROC) curves. CJEM 8(1):19–20
Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, Brace LE, Woods BA, Lin W, Zhang J et al (2011) Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov 1(1):78–89
Thota R, Johnson DB, Sosman JA (2015) Trametinib in the treatment of melanoma. Expert Opin Biol Ther 15(5):735–747
Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK, Zhao H, Chen K, Chen H, Do KA et al (2014) Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget 5(18):8544–8557
Guo Y, Kwiatkowski DJ (2013) Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Mol Cancer Res 11(5):467–473
Tetsu O, McCormick F (2017) ETS-targeted therapy: can it substitute for MEK inhibitors? Clin Transl Med 6(1):16
Rai K, Akdemir KC, Kwong LN, Fiziev P, Wu CJ, Keung EZ, Sharma S, Samant NS, Williams M, Axelrad JB et al (2015) Dual roles of RNF2 in melanoma progression. Cancer Discov 5(12):1314–1327
Xu J, Pfarr N, Endris V, Mai EK, Md Hanafiah NH, Lehners N, Penzel R, Weichert W, Ho AD, Schirmacher P et al (2017) Molecular signaling in multiple myeloma: association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 6(5):e337
Diamond JR, Eckhardt SG, Pitts TM, van Bokhoven A, Aisner D, Gustafson DL, Capasso A, Sams S, Kabos P, Zolman K et al (2018) A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res 20(1):82
Chang Q, Jorgensen C, Pawson T, Hedley DW (2008) Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br J Cancer 99(7):1074–1082
Shi H, Zhang CJ, Chen GY, Yao SQ (2012) Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J Am Chem Soc 134(6):3001–3014
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR et al (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41(Database issue):D955–D961
Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R et al (2018) Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560(7718):325–330
Kim SB, Dent R, Im SA, Espie M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ et al (2017) Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 18(10):1360–1372
Tamura S, Wang Y, Veeneman B, Hovelson D, Bankhead A 3rd, Broses LJ, Lorenzatti Hiles G, Liebert M, Rubin JR, Day KC et al (2018) molecular correlates of in vitro responses to dacomitinib and afatinib in bladder cancer. Bladder Cancer 4(1):77–90
Nagamine A, Araki T, Nagano D, Miyazaki M, Yamamoto K (2019) l-Lactate dehydrogenase B may be a predictive marker for sensitivity to anti-EGFR monoclonal antibodies in colorectal cancer cell lines. Oncol Lett 17(5):4710–4716
Oztop S, Isik A, Guner G, Gurdal H, Karabulut E, Yilmaz E, Akyol A (2019) Class III beta-tubulin expression in colorectal neoplasms is a potential predictive biomarker for paclitaxel response. Anticancer Res 39(2):655–662
Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, Liu S, Leung S, Geiss G, Snider J et al (2015) Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 8:54
Costa RLB, Han HS, Gradishar WJ (2018) Targeting the PI3 K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat 169(3):397–406
Ganesan P, Moulder S, Lee JJ, Janku F, Valero V, Zinner RG, Naing A, Fu S, Tsimberidou AM, Hong D et al (2014) Triple-negative breast cancer patients treated at MD Anderson Cancer Center in phase I trials: improved outcomes with combination chemotherapy and targeted agents. Mol Cancer Ther 13(12):3175–3184
Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J et al (2012) PI3 K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2(11):1036–1047
Shimoi T, Hamada A, Yamagishi M, Hirai M, Yoshida M, Nishikawa T, Sudo K, Shimomura A, Noguchi E, Yunokawa M et al (2018) PIK3CA mutation profiling in patients with breast cancer, using a highly sensitive detection system. Cancer Sci 109(8):2558–2566
Basho RK, Gilcrease M, Murthy RK, Helgason T, Karp DD, Meric-Bernstam F, Hess KR, Herbrich SM, Valero V, Albarracin C et al (2017) Targeting the PI3 K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol 3(4):509–515
Fouque A, Jean M, Weghe P, Legembre P (2016) Review of PI3 K/mTOR inhibitors entering clinical trials to treat triple negative breast cancers. Recent Pat Anticancer Drug Discov 11(3):283–296
Stanfield Z, Coskun M, Koyuturk M (2017) Corrigendum: drug response prediction as a link prediction problem. Sci Rep 7:44961
Groenendijk FH, Bernards R (2014) Drug resistance to targeted therapies: deja vu all over again. Mol Oncol 8(6):1067–1083
Wein L, Loi S (2017) Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast 34(Suppl 1):S27–S30
Mundt F, Rajput S, Li S, Ruggles KV, Mooradian AD, Mertins P, Gillette MA, Krug K, Guo Z, Hoog J et al (2018) Mass spectrometry-based proteomics reveals potential roles of NEK9 and MAP2K4 in resistance to PI3 K inhibition in triple-negative breast cancers. Cancer Res 78(10):2732–2746
Wu ZH, Lin C, Liu MM, Zhang J, Tao ZH, Hu XC (2016) Src inhibition can synergize with gemcitabine and reverse resistance in triple negative breast cancer cells via the AKT/c-Jun pathway. PLoS ONE 11(12):e0169230
Mumin NH, Drobnitzky N, Patel A, Lourenco LM, Cahill FF, Jiang Y, Kong A, Ryan AJ (2019) Overcoming acquired resistance to HSP90 inhibition by targeting JAK-STAT signalling in triple-negative breast cancer. BMC Cancer 19(1):102
Miles D, von Minckwitz G, Seidman AD (2002) Combination versus sequential single-agent therapy in metastatic breast cancer. Oncologist 7(Suppl 6):13–19
Lee J, Yesilkanal AE, Wynne JP, Frankenberger C, Liu J, Yan J, Elbaz M, Rabe DC, Rustandy FD, Tiwari P et al (2019) Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 568(7751):254–258
Acknowledgements
We kindly thank Rork Kuick (University of Michigan) for his insight on statistical analysis and careful editing of this manuscript. We thank the MD Anderson RPPA core for protein analysis, TIGEM in Naples, Italy for RNA sequencing, and the University of Michigan sequencing core for DNA sequencing.
Funding
This research was supported by the National Institutes of Health (1R21CA218498 to M.B.S. and S.D.M.), the Breast Cancer Research Foundation (to S.D.M.), Tempting Tables (to S.D.M.), The Rose Run (to S.D.M.), and the Kathy Bruk Pearce Research Fund of the University of Michigan Rogel Cancer Center (to M.B.S.).
Author information
Authors and Affiliations
Contributions
Study concept and design: M.B.S., N.M.M. Acquisition of data: N.M.M., N.M.V., E.J.L., L.W.B. Drafting the manuscript: N.M.M., J.A.Y., M.B.S., S.D.M. Analysis and interpretation of data: N.M.M., M.B.S., P.J.L, J.P.L., S.D.M. Experimentation: N.M.M., N.M.V., E.J.L., L.W.B. Statistical analysis: N.M.M., P.U.L., J.P.L. Administrative, technical, or material support: J.A.Y., A.M. Study supervision: M.B.S., S.D.M. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
All authors declare that they have no conflicts of interest related to this work.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2019_5473_MOESM2_ESM.eps
Supplementary material 2 Candidate molecular correlates in TNBC cell lines. Examples of compounds with significant correlates include: A. temsirolimus and B. docetaxel. All molecular marker readouts have a FDR-adjusted p*-value < 0.05. Top bars depict DSS3, with dark green representing the most sensitive lines and white representing the least sensitive. Orange labeled readouts are derived from DNA variants. Black labeled readouts are derived from targeted RNA expression levels (EPS 656 kb)
10549_2019_5473_MOESM4_ESM.docx
Supplementary material 4 Cell culture conditions. When comparing the Nanostring readouts with RNA sequencing, we observed strong correlations for A. IL7R, B. ERBB2, C. KIF2C, D. CD164, E. PRR15L. F. CACNB3, G. MYCL, H. EPHA2, I. PIK3CD, J. IGFBP4, and K. IRAK2. We observed poor correlations for L. FOSL1 (DOCX 314 kb)
10549_2019_5473_MOESM5_ESM.pdf
Supplementary material 5 Table showing IC50 values, hill-slopes, max response, dose range, and DSS3 values in individual cell lines. (PDF 312 kb)
10549_2019_5473_MOESM6_ESM.pdf
Supplementary material 6 Individual correlating DNA non-synonymous mutations in the molecular correlate panel. Genes marked with “*” indicate a roll-up of mutations was used for correlations. SIFT, Polyphen2, MutationTaster, MutationAssessor, and FATHMM were used to generate functional predictions. (PDF 25 kb)
Rights and permissions
About this article

Cite this article
Merrill, N.M., Lachacz, E.J., Vandecan, N.M. et al. Molecular determinants of drug response in TNBC cell lines. Breast Cancer Res Treat 179, 337–347 (2020). https://doi.org/10.1007/s10549-019-05473-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-019-05473-9