
Abstract
Background
Multiple common variants identified by genome-wide association studies showed limited evidence of the risk of breast cancer in Taiwan. In this study, we analyzed the breast cancer risk in relation to 13 individual single-nucleotide polymorphisms (SNPs) identified by a GWAS in an Asian population.
Methods
In total, 446 breast cancer patients and 514 healthy controls were recruited for this case–control study. In addition, we developed a polygenic risk score (PRS) including those variants significantly associated with breast cancer risk, and also evaluated the contribution of PRS and clinical risk factors to breast cancer using receiver operating characteristic curve (AUC).
Results
Logistic regression results showed that nine individual SNPs were significantly associated with breast cancer risk after multiple testing. Among all SNPs, six variants, namely FGFR2 (rs2981582), HCN1 (rs981782), MAP3K1 (rs889312), TOX3 (rs3803662), ZNF365 (rs10822013), and RAD51B (rs3784099), were selected to create PRS model. A dose–response association was observed between breast cancer risk and the PRS. Women in the highest quartile of PRS had a significantly increased risk compared to women in the lowest quartile (odds ratio 2.26; 95% confidence interval 1.51–3.38). The AUC for a model which contained the PRS in addition to clinical risk factors was 66.52%, whereas that for a model which with established risk factors only was 63.38%.
Conclusions
Our data identified a genetic risk predictor of breast cancer in Taiwanese population and suggest that risk models including PRS and clinical risk factors are useful in discriminating women at high risk of breast cancer from those at low risk.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the most common cancers for women in the world, including Taiwan, is breast cancer. Although rates of new cases of breast cancer in Western countries are higher than those in Taiwan, there has been a sharp sixfold rise in the breast cancer incidence over the past three decades here [1]. Breast cancer is known to be a complex disease which is determined by both genetic and environmental factors. Recently, the estimated heritability of breast cancer was 31%, while the common environmental component was 16% [2], showing that the diversity of disease-associated genes is a major issue of current scientific inquiry into breast cancer.
Several previous genome-wide association studies (GWASs) identified numerous susceptible common variants associated with breast cancer among Caucasian or Asian populations [3–10]. However, most of them were low-penetrance risk variants in addition to BRCA1 or BRCA2 [11], which contributed 25% to the familial risk and <5% to the total breast cancer incidence due to rare mutation frequencies [12, 13]. Researchers have tended to estimate the polygenic manner of single-nucleotide polymorphism (SNP) variants through developing composite genetic risk scores [14–16]. Most of these developed polygenic risk scores (PRSs) were derived from Caucasian population databases, and there is limited evidence on how the risk of breast cancer depend on PRSs which the SNPs were constructed from Asian populations.
In this study, we systematically reviewed GWAS-identified susceptibility loci in an Asian population and investigated the associations between established breast cancer susceptibility loci and breast cancer risks in Taiwanese. Further, we developed a composite risk score which was used to examine the relationship with breast cancer risk by tumor subtypes.
Materials and methods
Study population
This was a population-based breast cancer case–control study. Cases were identified as women aged 20–90 years with a histologically confirmed diagnosis of first primary in situ or invasive breast cancer from Taipei Medical University Hospital, Wan Fang Hospital, Shuang Ho Hospital and Cathy General Hospital between 2010 and 2014 (n = 446). Population-based healthy controls were randomly selected from a physical examination program at Taipei Medical University Hospital or a community-based prospective study of a nutrition health education program in Taipei City [17]. Finally, 514 controls without a cancer history were enrolled in this study and were frequency matched with an approximate age distribution of cases within 5-year age strata. Participants signed informed consent during study enrollment. This study was conducted under approval of the Ethics Committee of the Joint Institutional Review Board of Taipei Medical University and Cathay General Hospital.
Risk factor information
Structured questionnaires including lifestyle risk factors, menstrual history, reproductive history, and disease history were administered by well-trained nurses. The ages of menarche and menopause were retrospectively estimated by considering the first and last menstrual cycles. The duration of lifetime estrogen exposure was calculated for postmenopausal women as the number of years between the ages of menarche and the ages of menopause. Parity was determined as nulliparous, 1–2, 3–4, and ≥5 live births. The body mass index (BMI) was calculated as the weight in kilograms divided by the height in meters squared. The waist-to-hip ratio (WHR) is the ratio of the circumference of the waist to that of the hips.
DNA extraction, SNP selection, and genotyping
Genomic DNA was extracted from peripheral blood according to the phenol chloroform protocol and stored at −80 °C until being used for further analysis. SNP selection was identified by a review of GWASs or candidate-gene association studies for Asian populations based on the available literature at the time of the genotype analysis [3, 7, 8, 18–22]. Due to our sample size and limited budget, we finally selected 13 SNPS which were the most significantly associated with breast cancer, including FGFR2 (rs2981579, rs3750817, rs1219648, and rs2981582), HCN1 (rs981782), MAP3K1 (rs889312 and rs1686165), TOX3 (rs3803662 and rs4784227), C6orf97 (rs2046210), ZNF365 (rs10822013), RAD51B (rs3784099), and ERBB (rs13393577).
SNP genotyping was performed using custom-designed Illumina Golden Gate SNP Genotyping Arrays (Illumina, San Diego, CA) according to the manufacturer’s instructions. To assure genotyping quality, detailed quality control (QC) procedures, including the duplicate identification of genotypes, a Hardy–Weinberg equilibrium (HWE) test, and a call rate of >99%, were carried out. A total of 47 (5%) quality control samples were successfully genotyped, and the concordance rate was 100%.
Statistical analysis
HWE was examined among controls using a goodness-of-fit Chi squared test. Comparisons between cases and controls were conducted using Student’s t test and a Chi squared test. The odds ratio (OR) and 95% confidence interval (CI) were calculated using a logistic regression model to assess the association between the SNPs and risk of breast cancer. Adjustment for multiple testing was performed using the permutation test. All statistical analyses were performed using SAS vers. 9.4. (SAS Institute, Cary, NC) on two-sided probabilities.
A PRS was established to estimate the polygenic contribution of breast cancer susceptibility loci, which was created using marginally significant SNPs associated with breast cancer risk (p < 0.15) based on any one of the per-allele, dominant, or recessive logistic regression models. For strong linkage disequilibrium SNPs located on the same gene or chromosome (each D′ > 0.9), we choose the one variant with the lowest P value as candidate. Then, a weighted PRS was calculated as the sum of the product of the number of risk allele copies of the selected SNPs and the corresponding log odds estimate. A multivariate logistic regression model that comprised PRS, age, BMI, parity, and menopausal status was applied to investigate independent associations of PRS with risk of breast cancer. When stratified by menopausal status, a logistic regression model was composed of age, BMI, and parity to explore the association between PRS and breast cancer risk. To avoid potential overfitting, we took advantage of a bootstrap procedure with 1000 repetitions to adjust model regression coefficients. We applied cross-validation and split-sample design to increase the accuracy of prediction for internal validation [23, 24]. Briefly, bootstrapping resampled repeatedly and randomly by drawing samples with replacement from the original dataset. In cross-validation procedure, the dataset was divided into k subsets, which is used as the test set, and the holdout method was repeated k times. The other k−1 subsets were combined to form a training set. In addition, we separated the original dataset in development and validation samples, including 80 and 20% of the original dataset, respectively. The area under the receiver operating characteristic curve (AUC) was applied to evaluate the discriminative ability of the model.
Results
Distributions of the characteristic of breast cancer patients and healthy controls are shown in Table 1. The mean age of cases and controls were 51.3 ± 9.8 and 51.0 ± 10.1, respectively. The frequencies or distributions of educational level, cigarette smoking, alcohol consumption, age at menarche, age at menopause, age at first full-term birth, and lifetime estrogen exposure were not significantly different between cases and controls. However, the mean BMI and WHR were higher in cases than those in controls. In addition, breast cancer patients had higher percentages of parity number and premenopausal status than controls.
Table 2 indicates the association between the 13 SNPs previously reported in GWAS and breast cancer risk. For per-allele model, there were nine of these SNPs which revealed significant associations with risk of breast cancer. The four SNPs in FGFR gene, named rs2981579, rs3750817, rs1219648, and rs2981582, showed similar statistic tendency under dominant model and were in strong LD (each D′ > 0.7). HCN1 gene rs981782 were significantly associated with risk of breast cancer either in per-allele model or under dominant model. The two SNPs rs889312 and rs16886165 located in MAP3K1 gene demonstrated significant association with breast cancer risk under dominant model and were also in LD (D′ > 0.9). We also confirmed a significant association with risk of breast cancer for the remaining two SNPs, TOX3 rs3803662 under recessive model and RAD51B rs3784099 under per-allele model (each p < 0.05).
We selected six SNPs, namely rs2981582, rs981782, rs889312, rs3803662, rs10822013, and rs3784099 to create PRS model. Since four SNPs located on FGFR gene were in strong linkage disequilibrium, rs2981582 were chosen as a representative of FGFR due to lowest P value. The selection criteria were similar with rs889312 of MAP3K1 and rs3803662 of TOX3. The rest of other SNPs were picked up due to marginal significantly associated with breast cancer risk. According to the quartile distribution of the normal controls, women in the second (1.36–1.66), third (1.67–1.91), and fourth quartiles (≥1.92) had 1.66-, 1.83-, and 2.26-fold increased breast cancer risks compared to women in the lowest quartile (<1.36), showing a significant trend (p = 0.0001). When the same risk score was separately applied to premenopausal and postmenopausal women, the trend of the log odds was also significant (Fig. 1).

Odds ratio of breast cancer in groups defined by quartiles of the polygenic risk score among controls
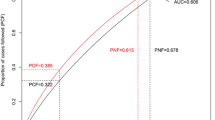
Finally, the AUC was calculated to evaluate how the risk models discriminated between women with and without breast cancer (Fig. 2). The AUC estimated for a model which contained the PRS in addition to established risk factors including age, BMI, age at menarche, parity, and menopausal status was 66.52%, whereas that for a model which with established risk factors only was 63.38%, while that for PRS model only was 59.79%.

AUC in the three risk models. The gray line with an AUC of 50% is reference. AUC of the upper blue line, which showed the combined PRS and clinical risk factors, is 66.52%. The green line with an AUC of 63.38% is clinical risk factors model, whereas AUC of the red line representing PRS is 59.79%
Discussion
In this study, we evaluated possible relationships between an increased breast cancer risk and 13 GWAS-identified SNPs in an Asian population. Among them, 9 SNPs were significantly associated with breast cancer risk, including FGFR2 (rs2981579, rs3750817, rs1219648, and rs2981582), HCN1 (rs981782), MAP3K1 (rs889312 and rs16886165), TOX3 (rs3803662), and RAD51B (rs3784099) after multiple testing. The findings provide extra benefit for the worth of genome-based studies associated with breast cancer, particularly for those carried out in Asian population. In addition, we constructed the PRS using six selected variants, which took into account the magnitudes of individual SNP effects as done in previous studies, and was most efficient when considering multiple loci in combination [14–16, 21, 25, 26]. Furthermore, we developed the risk model, combined the PRS and clinical risk factors to discriminate women with and without breast cancer, which provided adequate power with an AUC of approximately 70%.
In this study, breast cancer patients had higher value of BMI, WHR, and parity number, and higher frequency of premenopausal status than healthy controls. It is interesting that a greater proportion of cases were premenopausal despite similar ages between cases and controls. The reason might be due to early tumor onset, showing a relatively younger median age at diagnosis in Asian women [27].
Fibroblast growth factor (FGF) receptor 2 (FGFR2) is a transmembrane tyrosine kinase receptor that belongs to FGFR family, is able to integrate with FGF, and is involved in various biologic processes including cell migration, proliferation, survival, and differentiation [28]. The human FGFR2 gene, located on chromosome 10q26, was reported to be one of the first susceptible genes associated with breast cancer in GWASs [4, 6, 10]. Subsequent replication studies were widely conducted, and a meta-analysis indicated that the association between FGFR2 variants and breast cancer risk was more notable in Caucasians and Asians than Africans [29]. In our study, the single-locus analysis showed that the ORs for rs2981579, rs3750817, rs1219648, and rs2981582 were 1.37 (95% CI 1.03–1.83), 1.39 (95% CI 1.04–1.87), 1.39 (95% CI 1.06–1.83), and 1.47 (95% CI 1.13–1.91), respectively, under dominant model. The mitogen-activated protein kinase kinase kinase 1 (MAP3K1) gene was identified in GWASs of breast cancer [4, 6], and it acts in the mitogen-activated protein kinase (MAPK) signaling pathway. MAPK signal transduction is a major pathway in the cellular response to mitogens and is responsible for regulating transcription of important cancer genes which could influence susceptibility to breast cancer [30]. A recent meta-analysis suggested that MAP3K1 rs889312 and rs16886165 might be risk factors for breast cancer, especially in Europeans and Asians [31]. Consistent findings were also observed in our study, which showed significant associations of MAP3K1 rs889312 and rs16886156 with the breast cancer risk. The universal significance of FGFR and MAP3K1 for Europeans and Asians implies that the MAPK pathway may play an important role in the etiology of breast cancer.
GWASs identified the intergenic SNP rs981782 on the chromosome 5p12 region as a hotspot for breast cancer susceptibility [4, 10]. However, subsequent studies showed inconsistent results between rs981782 and breast cancer risk [25, 32, 33]. Our findings indicated that subjects with the risk allele of SNP rs981782 had an increased risk of breast cancer. The SNP rs3803662 at 16q12, close to the TOX3 gene, represented one of the susceptibility loci identified by GWAS [18] and was also observed in women of Asian descent [34, 35]. This study showed a significant association between rs3803662 and breast cancer under recessive model. SNP rs3784099, located in the RAD51B gene, was found to influence breast cancer prognosis in a GWAS [20]. The RAD51B gene belongs to RAD51 paralogs, which play an important role in the DNA repair system. A previous study also indicated that inactivation of RAD51B is involved in tumorigenesis [36]. In the case of the RAD51B gene, our data showed the OR per allele for rs3784099 was a 1.37-fold (95% CI 1.02–1.83) increased risk of breast cancer.
Previous studies investigated associations between the PRS of multiple SNPs and breast cancer risk. Our PRS results showed a linear association with an increased risk of breast cancer, indicating a greater than twofold risk for women in the highest quartile compared to those in the lowest. Reeves et al. used PRS based on 4, 7, or 10 SNPs to study their effects on the risk of breast cancer and found that the effect of the PRS with 7 SNPs was more prominent for estrogen receptor positive than for estrogen receptor negative cancers [16]. Harlid et al. investigated the combined effect of low-penetrant SNPs on breast cancer including ten SNPs [25]. Although they used a simple addition risk allele method, their results were still almost the same as ours. Two PRS studies in Asian population, a Japanese study conducted by Sueta et al. [26] and a Chinese study carried out by Chan et al. [37] also developed their own model to show a dose-dependent association between the risk of breast cancer and a genetic risk score. Anderson et al. created a PRS composed of seven SNPs and found that the OR was around twofold for women in the highest quintile compared to those in the lowest quintile of the score [15]. Mavaddat et al. constructed a 77-SNP PRS for breast cancer and found a threefold increase in risk when comparing the polygenic scores of the highest 1% and the middle quintiles [14].
In addition, we also constructed the risk prediction model including PRS and clinical risk factors for discrimination of breast cancer with an improved accuracy with AUC of 66.52%. This moderate discriminatory accuracy is nevertheless relatively high compared with the risk assessment model reported by Wacholder et al., Zheng et al., and Hüsing et al. with AUC of 61.8, 63.0, and 60.5%, respectively [21, 38, 39]. A risk assessment model in a Japanese population conducted by Sueta et al. that included both genetic markers and established risk models gave a higher AUC of 69.33%, which is similar with our present findings [26]. Interestingly, only one SNP rs3803662 on TOX3 gene was chosen simultaneously in genetic risk model developed by our study and in other Asian population [21, 26].
The strength of this study was the collection of tumor pathology data which were all assessed using the same processing protocols and criteria. Nevertheless, there are certain issues that needed to be discussed for a proper interpretation of this study. First, this study had nearly 80% power to detect a log-additive OR of 1.3 with allelic frequencies of >35%; however, other SNPs with ORs of <1.3 may need larger sample size to increase statistical power. Larger sample sizes could be considered to help improve the power in future studies. Second, our current analysis was limited to 13 common risk variants for breast cancer identified by a GWAS in an Asian population. In the near future, larger effect sizes of sequence variants with intermediate frequencies are likely to be uncovered; therefore, our PRS results should be interpreted carefully. In addition, although this study included comprehensive information on reproductive and menstrual factors, no interactions were found between the effects of the genes investigated in this study and hormone risk factors for breast cancer. Further studies should take other environmental risk factors into account to investigate gene–environment interactions on breast cancer risk.
In conclusion, our data evaluated the association of 13 genetic variants identified in previous GWASs in Asian populations as risk factors for breast cancer in a Taiwanese population. The PRS model, a combination of six selected variants, with clinical risk factors are useful in discriminating women at high risk of breast cancer from those at low risk. Future comprehensive evaluations of the genetic risk variants in a larger population are warranted.
References
Health Promotion Administration MOHAW. Health indicator 123. https://olap.hpa.gov.tw/en_US/index.aspx. Accessed May 5 2016
Moller S, Mucci LA, Harris JR et al (2016) The heritability of breast cancer among women in the nordic twin study of cancer. Cancer Epidemiol Biomark Prev 25:145–150
Cai Q, Long J, Lu W et al (2011) Genome-wide association study identifies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium. Hum Mol Genet 20:4991–4999
Easton DF, Pooley KA, Dunning AM et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093
Gold B, Kirchhoff T, Stefanov S et al (2008) Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci USA 105:4340–4345
Hunter DJ, Kraft P, Jacobs KB et al (2007) A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39:870–874
Kim HC, Lee JY, Sung H et al (2012) A genome-wide association study identifies a breast cancer risk variant in ERBB4 at 2q34: results from the Seoul Breast Cancer Study. Breast Cancer Res 14:R56
Long J, Cai Q, Sung H et al (2012) Genome-wide association study in East Asians identifies novel susceptibility loci for breast cancer. PLoS Genet 8:e1002532
Stacey SN, Manolescu A, Sulem P et al (2007) Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39:865–869
Stacey SN, Manolescu A, Sulem P et al (2008) Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 40:703–706
Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M (2010) Genetic susceptibility to breast cancer. Mol Oncol 4:174–191
Peto J, Collins N, Barfoot R et al (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 91:943–949
Pharoah PD, Dunning AM, Ponder BA, Easton DF (2004) Association studies for finding cancer-susceptibility genetic variants. Nat Rev Cancer 4:850–860
Mavaddat N, Pharoah PD, Michailidou K et al (2015) Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst 107:djv036
Warren Andersen S, Trentham-Dietz A, Gangnon RE et al (2013) The associations between a polygenic score, reproductive and menstrual risk factors and breast cancer risk. Breast Cancer Res Treat 140:427–434
Reeves GK, Travis RC, Green J et al (2010) Incidence of breast cancer and its subtypes in relation to individual and multiple low-penetrance genetic susceptibility loci. JAMA 304:426–434
Hsieh YC, Hung CT, Lien LM et al (2009) A significant decrease in blood pressure through a family-based nutrition health education programme among community residents in Taiwan. Public Health Nutr 12(4):570–577
Long J, Cai Q, Shu XO et al (2010) Identification of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium. PLoS Genet 6:e1001002
Hein R, Maranian M, Hopper JL et al (2012) Comparison of 6q25 breast cancer hits from Asian and European Genome Wide Association Studies in the Breast Cancer Association Consortium (BCAC). PLoS ONE 7:e42380
Shu XO, Long J, Lu W et al (2012) Novel genetic markers of breast cancer survival identified by a genome-wide association study. Cancer Res 72:1182–1189
Zheng W, Wen W, Gao YT et al (2010) Genetic and clinical predictors for breast cancer risk assessment and stratification among Chinese women. J Natl Cancer Inst 102(13):972–981
Zheng W, Zhang B, Cai Q et al (2013) Common genetic determinants of breast-cancer risk in East Asian women: a collaborative study of 23 637 breast cancer cases and 25 579 controls. Hum Mol Genet 22(12):2539–2550
Steyerberg EW, Harrell FE Jr, Borsboom GJ et al (2001) Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol 54(8):774–781
Efron B, Tibshirani R (1997) Improvements on cross-validation: the 632+ bootstrap method. J Am Stat Assoc 92:548–560
Harlid S, Ivarsson MI, Butt S et al (2012) Combined effect of low-penetrant SNPs on breast cancer risk. Br J Cancer 106:389–396
Sueta A, Ito H, Kawase T et al (2012) A genetic risk predictor for breast cancer using a combination of low-penetrance polymorphisms in a Japanese population. Breast Cancer Res Treat 132:711–721
Huang CS, Lin CH, Lu YS, Shen CY (2010) Unique features of breast cancer in Asian women–breast cancer in Taiwan as an example. J Steroid Biochem Mol Biol 118:300–303
Turner N, Grose R (2010) Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10:116–129
Cui F, Wu D, Wang W, He X, Wang M (2016) Variants of FGFR2 and their associations with breast cancer risk: a HUGE systematic review and meta-analysis. Breast Cancer Res Treat 155:313–335
Klinge CM, Blankenship KA, Risinger KE et al (2005) Resveratrol and estradiol rapidly activate MAPK signaling through estrogen receptors alpha and beta in endothelial cells. J Biol Chem 280:7460–7468
Zheng Q, Ye J, Wu H, Yu Q, Cao J (2014) Association between mitogen-activated protein kinase kinase kinase 1 polymorphisms and breast cancer susceptibility: a meta-analysis of 20 case-control studies. PLoS ONE 9:e90771
Yu Y, Chen Z, Wang H, Zhang Y (2013) Quantitative assessment of common genetic variants on chromosome 5p12 and hormone receptor status with breast cancer risk. PLoS ONE 8:e72154
Liang H, Li H, Yang X et al (2016) Associations of genetic variants at nongenic susceptibility loci with breast cancer risk and heterogeneity by tumor subtype in Southern Han Chinese Women. Biomed Res Int 2016:3065493
Han W, Woo JH, Yu JH et al (2011) Common genetic variants associated with breast cancer in Korean women and differential susceptibility according to intrinsic subtype. Cancer Epidemiol Biomark Prev 20:793–798
Long J, Shu XO, Cai Q et al (2010) Evaluation of breast cancer susceptibility loci in Chinese women. Cancer Epidemiol Biomark Prev 19:2357–2365
Wadt KA, Aoude LG, Golmard L et al (2015) Germline RAD51B truncating mutation in a family with cutaneous melanoma. Fam Cancer 14:337–340
Chan M, Ji SM, Liaw CS et al (2012) Association of common genetic variants with breast cancer risk and clinicopathological characteristics in a Chinese population. Breast Cancer Res Treat 136:209–220
Wacholder S, Hartge P, Prentice R et al (2010) Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362(11):986–993
Hüsing A, Canzian F, Beckmann L et al (2012) Prediction of breast cancer risk by genetic risk factors, overall and by hormone receptor status. J Med Genet 49:601–608
Acknowledgements
This study was supported by the Health and Welfare Surcharge of Tobacco Products, Ministry of Health and Welfare to the Comprehensive Cancer Center of Taipei Medical University (MOHW105-TDU-B-212-134001), Taipei Medical University Hospital (103TMU-TMUH-09), Taipei Medical University (TMU102-AE1-B03), and the Top University Project- Cancer Translational Center of Taipei Medical University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Chin-Sheng Hung and Hung-Yi Chiou contributed equally to this study.
Rights and permissions
About this article

Cite this article
Hsieh, YC., Tu, SH., Su, CT. et al. A polygenic risk score for breast cancer risk in a Taiwanese population. Breast Cancer Res Treat 163, 131–138 (2017). https://doi.org/10.1007/s10549-017-4144-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4144-5