
Abstract
The insulin-like growth factor I receptor (IGF1R) has been linked to resistance to HER2-directed therapy with trastuzumab (Herceptin). We examined the anti-tumor activity of figitumumab (CP-751,871), a human monoclonal antibody that blocks IGF1R ligand binding, alone and in combination with the therapeutic anti-HER2 antibody trastuzumab and the pan-HER family tyrosine kinase inhibitor neratinib, using in vitro and in vivo breast cancer model systems. In vitro assays of proliferation, apoptosis, and signaling, and in vivo anti-tumor experiments were conducted in HER2-overexpressing (BT474) and HER2-normal (MCF7) models. We find single-agent activity of the HER2-targeting drugs but not figitumumab in the BT474 model, while the reverse is true in the MCF7 model. However, in both models, combining figitumumab with HER2-targeting drugs shows synergistic anti-proliferative and apoptosis-inducing effects, and optimum inhibition of downstream signaling. In murine xenograft models, synergistic anti-tumor effects were observed in the HER2-normal MCF7 model for the combination of figitumumab with trastuzumab, and, in the HER2-overexpressing BT474 model, enhanced anti-tumor effects were observed for the combination of figitumumab with either trastuzumab or neratinib. Analysis of tumor extracts from the in vivo experiments showed evidence of the most optimal inhibition of downstream signaling for the drug combinations over the single-agent therapies. These results suggest promise for such combinations in treating patients with breast cancer, and that, unlike the case for single-agent therapy, the therapeutic effects of such combinations may be independent of expression levels of the individual receptors or the single-agent activity profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
HER2 overexpression occurs approximately in 15–20 % of human breast tumors, and the therapeutic anti-HER2 antibody, trastuzumab (Herceptin), is the cornerstone of treatment of such disease. Potential mechanisms of intrinsic and acquired resistance to trastuzumab that have been discovered, and include the presence of truncated or cleaved forms of constitutively activated HER2 lacking the trastuzumab-binding extracellular domain [1, 2], a splice variant eliminating a 16 amino acid exon from the extracellular domain [3, 4], constitutive activation of the PI3 K pathway [5–10], MUC4 [11, 12], signaling via src [3, 13, 14], enhanced activity of other EGFR family receptors and expression of their ligands [15–17], and signaling by the insulin-like growth factor I receptor (IGF1R) [18–21].
The IGF1R is a heterotetrameric transmembrane receptor tyrosine kinase that is widely expressed in normal human tissues [22, 23]. The IGF1R is a receptor tyrosine kinase that, like HER2, can signal via the anti-apoptotic phosphatidylinositol-3-kinase (PI3 K)-AKT and proliferation-driving Ras/Raf/ERK (MAPK) pathways [24]. After binding to the ligand IGF-I, the receptor phosphorylates the adaptor protein insulin receptor substrate 1 (IRS-1) that is responsible for mediating the activation of multiple downstream signaling networks, including the PI3 K or the ERK pathways. Activation of these signaling pathways leads to cell cycle progression and resistance to cell death. In addition, a second adaptor protein, termed IRS-2, is also activated by the IGF1R and participates in mediating motility signals. IGF1R is believed to have a central role in malignant transformation, and deregulated IGF1R signaling appears common to many solid tumors including breast cancer [22, 25–27], making it a target of therapeutic interest.
IGF1R signaling is necessary for cellular transformation by a variety of agents. In fact, IGF1R-deficient cells are resistant to transformation by a wide variety of oncogenes [28]. In cells already transformed, inhibition of IGF1R signaling typically only produces mild growth inhibition of cells growing in monolayer, but can induce cell death under anchorage-independent conditions, such as in soft agar or xenograft experiments [28, 29]. The IGFIR also appears to have a role in the metastatic process [30, 31]. These critical roles of IGF signaling in malignancy make it a promising therapeutic target for the treatment of cancer. As a mono-therapy, IGF1R inhibition does not uniformly display in vivo anti-tumor activity in model systems [32]. However, its critical anti-apoptotic role in established tumors makes IGF1R targeting a promising strategy to combine with other therapies.
Signaling cross-talk and physical interactions of IGF1R with HER2 have been discovered [33, 34]. Forced overexpression of IGF1R results in IGF-I-mediated trastuzumab resistance [20, 35]; furthermore, cells cultured long term in trastuzumab to induce acquired resistance up-regulated endogenous IGF1R, developed a physical association between HER2 and IGF1R in co-immunoprecipitation (co-IP) experiments, and IGF became capable of inducing HER2 cross phosphorylation [34]. Association of IGF1R with clinical resistance to trastuzumab has also been reported [18].
Given the potential role of IGF1R in trastuzumab resistance, co-targeting of HER2 and IGF1R in HER2-overexpressing breast cancer is a rational therapeutic strategy. We previously reported on the anti-tumor effects of reagent-grade IGF1R-inhibiting agents in combination with trastuzumab in cell culture models and found that such dual targeting resulted in disruption of HER2/IGF1R cross-talk, synergistic inhibition of cell proliferation, and induction of cellular apoptosis in the HER2-overexpressing BT474 cell line model; most remarkably, we also found that the killing effects of the HER2/IGF1R combination targeting occurred even in a HER2-normal cell line that does not have amplification or overexpression of HER2; HER2 overexpression was not required for the synergistic killing effects of HER2/IGF1R co-targeting [36]. Targeting IGF1R was able to bring about a dramatic anti-tumor effect of trastuzumab in a HER2-normal tumor model.
Here we describe the anti-tumor activity of figitumumab (CP-751,871), a human therapeutic anti-IGF1R antibody, alone and in combination with trastuzumab, using in vitro and in vivo models. Figitumumab is a fully human IgG2 specific for IGF1R, that blocks IGF1R ligand binding, is a potent inhibitor of tumor growth as a single agent, and has been shown to enhance the efficacy of other anticancer agents in human tumor xenograft models [37]. Since trastuzumab resistance may also arise from excess signaling by HER family receptors, another approach to overcoming trastuzumab resistance is the simultaneous inhibition of multiple HER family receptors. Therefore, we also explored the anti-tumor activity of figitumumab in combination with a pan-HER family receptor tyrosine kinase inhibitor (TKI), Neratinib (HKI-272). Neratinib is a potent low molecular weight, orally administered, irreversible pan-HER family receptor TKI [38, 39]. We report on the activity of these drug combinations in HER2-overexpressing and HER2-normal breast cancer models in vitro and in vivo.
Materials and methods
Drugs
Trastuzumab (Herceptin), a humanized therapeutic monoclonal antibody against HER2, was obtained from the Yale Cancer Center Medical Oncology pharmacy; a 20-mg/ml stock preparation was kept at 4 °C and was further diluted in sterile PBS before addition to cells in culture. The human monoclonal antibody to IGF1R, figitumumab (CP-751,871), (20 mg/ml stock in sterile PBS), and the pan-HER inhibitor neratinib were kindly provided by Pfizer Global Research & Development (Groton, CT).
Cell culture
BT474 cells (HER2-overexpressing and IGF1R low), obtained from ATCC, and MCF7 cells (HER2-normal and IGF1R high), a gift from Dr. Marc Lippman (University of Michigan), were cultured as described previously [40].
Assays of proliferation
Proliferation/viability of cells was detected using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI). For in vitro growth assays, cells (1 × 104/well for BT474, and 1 × 103/well for MCF7) were plated in 96-well plates and treated with the indicated concentrations of the inhibitors for 5 days. Thereafter, luminescence was read using the Envision plate reader. Results were expressed as percentage of control (vehicle DMSO-treated) cells. Results presented are mean ± SD from three separate experiments done in triplicate.
Apoptosis determination by Annexin-V assay
For apoptosis analyses by Annexin-V assay, after 5 days of drug exposure, both adherent and nonadherent cells were harvested, washed twice with cold phosphate-buffered saline (PBS) and then resuspended in binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) at a concentration of 1x106 cells/ml. In total, 100,000 cells in 100 μl binding buffer were incubated for 15 min in the dark with 5 μl of Annexin-V–fluorescein isothiocyanate (FITC) (BD Biosciences) and 1 μl of propidium iodide (1 μg/ml, final concentration). Finally, 300 μl of binding buffer was added before apoptosis analyses on a FACSCalibur machine using the CellQuest software (BD Biosciences, Franklin Lakes, NJ) as described previously [36].
Apoptosis determination by poly(ADP-ribose) polymerase (PARP) cleavage analysis
PARP cleavage assays were performed after 4 days of drug exposure as described previously [36].
Immunoblot analysis for HER2, phospho-HER2, IGF1R, phospho-IGF1R, AKT, phospho-AKT, ERK-1/2, and phospho-ERK-1/2
Anti-HER2 polyclonal antibody sc-284 and anti-IGFIR polyclonal antibody (clone C-20) were from Santa Cruz Biotechnology, Inc. (Santa Cruz CA); anti-phospho-HER2 monoclonal antibody (Tyr-1248; clone PN2A) and anti-phospho-IGFIR were from NeoMarkers (Fremont CA) and Biosource International, Inc. (Camarillo CA), respectively. Rabbit polyclonal antibodies against phospho-AKT, total AKT, phospho-extracellular signal-regulated kinase ERK1/2, and total ERK1/2 were all from Cell Signaling Technology (Danvers MA). Mouse monoclonal β-actin antibody (Sigma, St. Louis MO) was used as an internal loading control. All the secondary antibodies, horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG or goat anti-mouse IgG, were purchased from Santa Cruz Biotechnology (Santa Cruz CA). Immunoblot analysis was performed as described previously [36]. Bands were detected by chemiluminescence (Perkin-Elmer Life Sciences, Boston, MA).
Mouse xenograft studies
Mice were maintained and handled in accordance with Yale Institutional Animal Care and Use Committee protocols and regulations. Six- to eight-week-old female BALB/c athymic, ovariectomized nude mice were purchased from Harlan Laboratories (South Easton, MA). Since the BT474 and MCF7 models are estrogen dependent, a 0.72 mg, 60-day release, 17β-estradiol pellet (Innovative Research of America, Sarasota, FL) was implanted subcutaneously on the back of each mouse one day before tumor inoculation. The following day, 2 × 107 BT474 cells or 2 × 106 MCF cells suspended in 100 μl of PBS were injected s.c. into the right flank of each mouse after mixing with an equal volume of Matrigel (BD Biosciences, Bedford, MA). When mice were bearing tumors of approximately 200–250 mm3 in volume (approximately day 10), they were divided into treatment groups consisting of approximately 7 mice, and drug treatment was initiated.
Treatment groups consisted of vehicle control (PBS), figitumumab, trastuzumab, neratinib, and figitumumab plus either trastuzumab or neratinib. Based on previous experiments [41] and preliminary dose–response experiments (data not shown), the following doses and frequency of administration were chosen. Trastuzumab was administered every other day by i.p injection at 2 and 4 μg/mouse for BT474 and MCF7, respectively. Figitumumab was administered by i.p. injection once weekly at a dose of 500 μg/mouse bearing BT474 tumor and 250 μg/mouse bearing MCF7 tumor. Neratinib was administered every other day by i.p. injection at a dosage of 50 μg/mouse (in 0.1 ml PBS) bearing BT474. Treatment was continued for up to 8 weeks.
Tumors were measured three times per week using digital calipers (Chicago Brand Industrial, Inc., Fremont, CA), and tumor volume was calculated as d 2 × D × π/6 where d represented the smaller diameter and D the larger diameter [42]. Data were expressed as mean ± SD, and differences were considered statistically significant at p < 0.05 by Student t test.
Mice were sacrificed by CO2 asphyxiation 59 days after tumor inoculation. Tumors were excised and bisected. Half of each tumor was frozen immediately using liquid nitrogen and stored at 80 °C for preparation of extracts. Tumor specimen(s) and extracts were also prepared from one or two mice from each group at short time points (e.g., 4 days) after initiation of drug treatment for studies of downstream signaling events.
Results
Anti-proliferative effects
The anti-proliferative effects of figitumumab, trastuzumab, and neratinib were analyzed in two breast cancer cell lines representative of distinct molecular pathologic tumor models, BT474, a model cell line with HER2 overexpression and gene amplification, and MCF7, a model cell line with low/normal HER2 level. Both cell lines express IGF1R, with relatively higher levels observed in MCF cells and comparatively low levels in BT474 cells [36, 43].
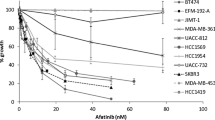
In MCF7 cells, treatment with figitumumab resulted in a small degree of inhibition of proliferation in a dose-dependent manner, while no growth inhibition was observed with single-agent trastuzumab or neratinib. BT474 cells, unlike MCF7 cells, were unaffected by figitumumab, but were found, as expected, to be very sensitive to trastuzumab or neratinib (Fig. 1).

Dose–response experiments of anti-proliferative effects of trastuzumab, neratinib, and figitumumab on BT474 and MCF7 cells. BT474 (panels A, C, E) and MCF7 (panels B, D, F) cells in exponential growth phase were seeded on day-1 in 96-well plates at 1 × 104 cells/well and 1 × 103 cells/well, respectively. They were allowed to adhere overnight and then treated with drugs the following day (day 0) at the indicated doses. After 5 days of incubation, the CellTiter-Glo proliferation assay was performed as described in Materials and Methods. Results were expressed as percentage of control (vehicle-treated cells). The mean and SD values of four wells from at least three experiments for each point are shown
We next evaluated the effect of the combination of figitumumab with trastuzumab or neratinib on each cell line. In both cell lines, treatment with the drug combinations resulted in a significant inhibitory effect compared with the single agents (Fig. 2). Hence, in BT474 cells, the IGF1R inhibitor figitumumab potentiates the anti-proliferative effect of the HER2-targeting drugs, despite the fact that figitumumab as a single agent is inactive, and in MCF7 cells the HER2-targeting drugs potentiate the anti-proliferative effect of figitumumab, despite the HER2-targeting drugs being inactive as single agents in this cell line. Hence, synergistic anti-proliferative effects of the drug combinations are observed.

Anti-proliferative effects of the combination of figitumumab with trastuzumab and with neratinib on BT474 and MCF7 cells. Figitumumab (10 μg/ml) was combined with either trastuzumab or neratinib at the indicated doses in BT474 (panel A) and MCF7 (panel B) cells and proliferation measured as in Fig. 1. Results are expressed as percentage of control (vehicle-treated cells). V Vehicle, CP CP-751,871/figitumumab. The mean and SD values of four wells from at least three experiments for each point are shown. Symbols Asterisk and Double Asterisk indicate p < 0.05 and p < 0.01, respectively
Apoptosis-inducing effects
Apoptosis was assayed after 5 days of drug treatment by analyzing the percentage of cells that were Annexin-V positive (Fig. 3), and after 4 days of drug treatment by PARP cleavage assay (Fig. 4). Both assays revealed that the concomitant inhibition of IGF1R and HER2 signaling in both BT474 and MCF7 cells enhanced apoptotic cell death compared to inhibition of one signaling pathway.

Apoptosis-inducing effects of the combination of figitumumab with trastuzumab and with neratinib on BT474 and MCF7 cells measured by Annexin-V assay. Figitumumab was combined with either trastuzumab or neratinib at the indicated doses in BT474 (panel A) and MCF7 (panel B) cells and apoptosis measured after 5 days of drug exposure by Annexin-V staining. The collected cells were washed and treated with propidium iodide (PI) and Annexin-V Alexa Fluor 488. Fluorescence-activated cell-sorting analysis was used to quantify Annexin-V positive (apoptotic) cells. V Vehicle, Ne neratinib, CP CP-751,871/figitumumab, Trastu trastuzumab. Results are mean ± SD from 3 different experiments. Symbols Asterisk and Double Asterisk indicate p < 0.05 and p < 0.01, respectively, when compared with vehicle control

Apoptosis-inducing effects of the combination of figitumumab with trastuzumab and with neratinib on BT474 and MCF7 cells measured by PARP cleavage assay. Figitumumab was combined with either trastuzumab or neratinib at the indicated doses in BT474 (panel A) and MCF7 (panel B) cells and apoptosis measured after 4 days of drug exposure by the PARP cleavage assay. Adherent and floating cells were harvested, and whole cell lysates were prepared as described in “Materials and methods” section. Immunoblotting was carried out using an anti-PARP antibody, and also with β-actin, as a loading control. Detection was carried out using ECL reagent. V Vehicle, Ne neratinib, CP CP-751,871/figitumumab, Trastu trastuzumab
Signal transduction effects in vitro
Immunoblot analyses of cell extracts after drug treatment for 2 days were performed to analyze drug effects on receptor common downstream signaling pathways (Fig. 5). In BT474 cells, which express high levels of HER2 but low levels of IGF1R, both HER2-targeting drugs trastuzumab and neratinib caused a decrease in the phosphorylation level of HER2, while figitumumab had no direct effect on levels or phosphorylation of HER2. In MCF7 cells, which express high levels of IGF1R but low levels of HER2, figitumumab caused a decrease in the phosphorylation level of IGF1R; the HER2-targeting drugs trasuzumab and neratinib as single agents did not effect IGF1R level or phosphorylation, however, when added to figitumumab, resulted in an additional decrease in phosphorylation of IGF1R, likely reflecting cross-talk between the two receptors, as our previous work has also suggested [36]. Analysis of downstream signaling pathways revealed that the combination of figitumumab with the HER2-targeting drugs trastuzumab and neratinib maximally inhibited receptor downstream signaling, as indicated by effects on levels of phosphorylated AKT and phosphorylated ERK (Fig. 5).

Signal transduction effects in vitro of the combination of figitumumab with trastuzumab and with neratinib on BT474 and MCF7 cells. Cells (BT474, panel A; MCF7, panel B) were treated with single drugs or with figitumumab combined with either trastuzumab or neratinib at the indicated concentrations for 2 days. Cell lysates were prepared and 40 μg of protein per lane was loaded. Membranes were immunoblotted for phospho-HER2 (PHER2), total HER2 (THER2), phospho-IGF1R (PIGF1R), total IGF1R (TIGF1R), phospho-AKT (PAKT), total AKT (TAKT), phospho-ERK (PERK), total ERK (TERK), and β-actin as a loading control. Detection of bands was carried out using ECL reagent. V Vehicle, Ne neratinib, CP CP-751,871/figitumumab, Trastu trastuzumab
In vivo anti-tumor activity
The in vivo anti-tumor activity of the individual agents and the combination of figitumumab with either trastuzumab or neratinib was examined in xenograft experiments.
Combination of figitumumab with trastuzumab in BT474 xenograft model
Animals bearing BT474 xenografts were treated with trastuzumab at 2 μg every other day, figitumumab at 500 µg weekly, both, or neither. When plotted as the average tumor volume of each group over the time period, there were no significant effects of the drugs when used as single agents; however, inhibition of tumor growth was observed in those animals treated with the combination (Fig. 6).

Growth of BT474 tumor xenografts in BALB/c nude mice treated with trastuzumab, figitumumab, or the combination. Estrogen supplemented, overiectomized, athymic nude mice bearing s.q. BT474 xenografts were randomly allocated to treatment with trastuzumab (2 μg/mouse, every alternative day), figitumumab (500 μg/mouse, once a week), the combination, or vehicle for consecutive 8 weeks. Treatment began when the average tumor size was approximately 200 mm3. Drugs were administered for 48 days by i.p. injection. Tumor volumes were measured twice weekly. Each data point represents the mean tumor volume from five mice. Bars represent standard deviation. Asterisk indicates statistically significant difference from the vehicle and/or single-drug treatment at p < 0.05
Combination of figitumumab with trastuzumab in the MCF7 xenograft model
Animals bearing MCF7 xenografts were treated with trastuzumab at 4 μg every other day, figitumumab at 250 µg weekly, both, or neither. Single-agent trastuzumab or figitumumab showed minimal tumor growth inhibition, but the drug combination resulted in significant tumor growth inhibition (Fig. 7).

Growth of MCF7 tumor xenografts in BALB/c nude mice treated with trastuzumab, figitumumab, or the combination. Estrogen supplemented, overiectomized, athymic nude mice bearing s.q. MCF7 xenografts were randomly allocated to treatment with trastuzumab (4 μg/mouse, every alternative day), figitumumab (250 μg/mouse, once a week), the combination, or vehicle. Treatment began when the average tumor size was approximately 200 mm3. Drugs were administered for 48 days by i.p. injection. Tumor volumes were measured twice weekly. Each data point represents the mean tumor volume from five mice. Bars represent standard deviation. Trastu trastuzumab. Asterisk indicates statistically significant difference from the vehicle and/or single-drug treatment at p < 0.05
Combination of figitumumab with neratinib in the BT474 xenograft model
Animals bearing BT474 xenografts were treated with neratinib at 50 μg every other day, figitumumab at 500 µg weekly, both, or neither. There was moderate tumor growth inhibition with neratinib; however, the greatest degree of inhibition of tumor growth was observed in the animals treated with the combination (Fig. 8).

Growth of BT474 tumor xenografts in BALB/c nude mice treated with neratinib, figitumumab, or the combination. Estrogen supplemented, overiectomized, athymic nude mice bearing s.q. BT474 xenografts were randomly allocated to treatment with neratinib (50 μg/mouse, every alternative day), figitumumab (500 μg/mouse, once a week), the combination, or vehicle. Treatment began when the average tumor size was approximately 200 mm3. Drugs were administered for 48 days by i.p. injection. Tumor volumes were measured twice weekly. Each data point represents the mean tumor volume from five mice. Bars represent standard deviation. Asterisk indicates statistically significant difference from the vehicle and/or single-drug treatment at p < 0.05
In vivo pharmacodynamic studies
As indicators of downstream signaling from IGF1R and HER2, levels of phosphorylated HER2, phosphorylated IGF1R, phosphorylated AKT [pAKT(Ser473)], and phosphorylated ERK [pERK(Thr202/Tyr204)] were examined in tumor tissues harvested from animals sacrificed at 96 and 169 h after initiating drug therapy with figitumumab, trastuzumab, neratinib, or their combinations. Preliminary experiments showed that no apparent trends for impact on signaling were evident in the tumor extracts prepared after long-term drug treatment; however, in tumor extracts prepared after just a short period of treatment (e.g., 4 days), signaling effects were apparent (data not shown).
In the BT474 tumor models, either trastuzumab or neratinib caused a decrease in levels of HER2 phosphorylation (Figs. 9, 10). Likewise, in the MCF7 tumor model, figitumumab caused a decrease in level of IGF1R phosphorylation (Fig. 9). In all three animal experiments described above, signaling studies on tumor extracts from mice sacrificed 96 h after first drug treatment showed minimal effects of single agents, but the combinations resulted in enhanced inhibition of signaling as evidenced by decreased levels of phospho-AKT and phospho-ERK (Figs. 9, 10).

Immunoblot analysis of downstream signaling in mouse xenograft tumor samples after treatment with trastuzumab, figitumumab, or the combination. Frozen tumor tissue from mice bearing xenograft tumors of BT474 (panel A) and MCF7 (panel B) tumors after 4 days of drug treatment was used to prepare tumor extracts. Protein samples were electrophoresed and immunoblotted with antibodies to phospho-HER2 (PHER2), total HER2 (THER2), phospho-IGF1R (PIGF1R), total IGF1R (TIGF1R), phospho-AKT (PAKT), total AKT (TAKT), phospho-ERK (PERK), total ERK (TERK). Immunoblot with human pan-cytokeratin antibody was performed to demonstrate the relative amount of human epithelial cells in tumor samples. Each lane represents an individual mouse’s tumor from within each group. Detection of bands was carried out using ECL reagent. V Vehicle, CP CP-751,871/figitumumab, Trastu trastuzumab

Immunoblot analysis of downstream signaling in BT474 mouse xenograft tumor samples after treatment with neratinib, figitumumab, or the combination. Frozen tumor tissue from mice bearing xenograft tumors of BT474 after 4 days of drug treatment was used to prepare tumor extracts. Protein samples were electrophoresed and immunoblotted with antibodies to phospho-HER2 (PHER2), total HER2 (THER2), phospho-IGF1R (PIGF1R), total IGF1R (TIGF1R), phospho-AKT (PAKT), total AKT (TAKT), phospho-ERK (PERK), total ERK (TERK). Immunoblot with human pan-cytokeratin antibody was performed to demonstrate the relative amount of human epithelial cells in tumor samples. Each lane represents an individual mouse’s tumor from within each group. Detection of bands was carried out using ECL reagent. V Vehicle, CP CP-751,871/figitumumab, Ne neratinib
Discussion
In breast cancer models, cross-talk and physical interactions of IGF1R with HER2 [33, 34] implicated the IGF1R in trastuzumab resistance, and clinical trial results have reported this association as well [18]. In other work, it was found that the EGFR also interacts with IGF1R in a human breast cancer cell line [44]. One group reported that an IGF1R TKI (AG1024) and the EGFR TKI gefitinib gave additive-to-synergistic growth inhibition of several breast carcinoma cell lines, and the combination gave greater apoptosis than either single agent [45].
HER2 is the only member of the EGFR/HER family that has no ligand, and therefore, HER2 signaling requires heterodimerization with another HER family receptor (HER1/EGFR, HER3, or HER4). The HER2/HER3 heterodimer is implicated as the most potent with regard to mitogenic signaling, transforming ability, and the malignant phenotype [46, 47]. The presence of HER family ligands, and activation of other HER family receptors, can reduce the efficacy of HER2-directed therapies [15, 16]. Furthermore, overactive signaling via HER3 has been implicated in resistance to cancer therapies that target the other receptor tyrosine kinases, including drugs that target EGFR [48] and HER2 [16, 17].
These considerations suggest potential limitations of a therapy that targets solely HER2, since optimal anti-tumor effects may require silencing of the function of all of the HER family receptors. Recent clinical success from adding the therapeutic anti-HER2 antibody pertuzumab to trastuzumab [49, 50] supports this notion, as pertuzumab binds to the dimerization arm of HER2 and prevents dimerization with other receptors in the HER family. These considerations also suggest that agents such as neratinib may add benefit in treating HER2-overexpressing breast cancer. Furthermore, some breast cancers are known to express constitutively activated truncated forms of HER2 that lack the extracellular domain, and to which trastuzumab cannot bind; such tumors are resistant to trastuzumab [1], but sensitive to HER2 TKIs [1, 51].
Given these considerations, and the ability of IGF1R to interact with both HER2 and EGFR (HER1) and act as an escape mechanism from trastuzumab effect, we were interested in combining neratinib with IGF1R inhibitors. In accordance with our previous observations [36], we find that combining figitumumab with trastuzumab or neratinib in representative human breast cancer cell lines enhances growth inhibition and apoptosis in vitro, in models where drugs employed as single agents were incapable of inducing such an effect, similar to results we previously reported with reagent grade IGF1R inhibitors in combination with trastuzumab. We also find such enhanced effects of the drug combinations using in vivo xenograft models. Furthermore, biochemical analysis of downstream signaling in the tumors from these animals demonstrates enhanced anti-signaling effects of the drug combinations.
Clinical trial results showed that a HER2/EGFR dual TKI, lapatinib, has clinical activity in HER2-positive metastatic breast cancer after progression on trastuzumab-based therapy [52]. One group reported that lapatinib could kill trastuzumab-resistant SKBR3 cells, and that an IGF1R antagonistic antibody potentiated this effect [21]. Using the BT474 and MCF7 cell lines, we found that combining IGF1R inhibitors with a lapatinib analog (GW2974) induces high levels of apoptosis, when again single agents could not (unpublished data).
Neratinib has demonstrated activity in HER2-positive breast cancer patients, both in the setting of no prior trastuzumab treatment (response rate 56 %, progression-free survival 39.6 weeks) and with prior trastuzumab treatment (response rate 24 %, progression-free survival 22.3 weeks) [53]. Patients with trastuzumab pretreated HER2-positive metastatic breast cancer receiving neratinib plus capecitabine had an overall response rate of 64 % for those with no prior lapatinib therapy and 57 % for those previously treated with labtinib [54]. A recent news release from Puma Biotechnology, currently developing neratinib, states a positive effect in a randomized phase III trial of using neratinib in extended adjuvant therapy after adjuvant therapy with trastuzumab (http://www.pumabiotechnology.com/pr2014072202.html).
Figitumumab has been tested as a cancer therapeutic in a number of clinical trials. In a randomized phase II trial of conventional cytotoxic chemotherapy with or without figitumumab for advanced non-small cell lung cancer, initial results (subsequently retracted) suggested improved outcomes when added to conventional chemotherapy, especially for squamous cell histology. This led to a randomized phase III trial in this setting for non-small cell lung cancer patients with non-adenocarcinoma histology; however, the phase III trial was terminated early due to lack of improved effect and concern about excess toxicity [55]. Furthermore, additional analysis of the randomized phase II trial called into question the initial encouraging results and led to a retraction of the initial publication. The clinical development of figitumumab has been discontinued [55]. However, other drugs to target IGF1R are still in clinical development and in clinical trials. Relevant to HER2-positive breast cancer, a clinical trial of lapatinib/capecitabine with or without the anti-IGF1R therapeutic antibody cixutumumab (IMC-A12) was recently reported in abstract form, and reported that the addition of cixutumamb did not meet the primary endpoint of improved progression-free survival in unselected patients with HER2-positive metastatic breast cancer, though subsetting patients by IGFBP levels might help predict patients who may benefit [56]. The highly related insulin receptor also has tumor-promoting properties, and hybrid IGF1R/insulin receptor signaling is believed to be important in cancer. Pre-clinical research has suggested that co-targeting both IGF1R and insulin receptor may be required for optimal anti-tumor effects [57], and dual IGF1R/insulin receptor inhibitors are also in clinical development.
In conclusion, combining IGF1R inhibitors with HER2-targeting agents is a promising strategy to treat breast cancer, potentially irrespective of the tumor HER2 expression level, as demonstrated here using the HER2-normal MCF7 model. This work suggests that selected targeted therapy drug combinations may have dramatic synergy, even in settings where single drugs are inactive. Co-targeting of the IGF-signaling axis and HER family axis may hold promise in treating breast cancers of various subtypes.
References
Scaltriti M et al (2007) Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst 99(8):628–638
Christianson TA et al (1998) NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res 58:5123–5129
Mitra D et al (2009) An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther 8(8):2152–2162
Kwong KY, Hung MC (1998) Identification of a novel alternative splicing form of human HER2/neu proto-oncogene (ABSTRACT). Proc Am Assoc Cancer Res 39:205
Berns K et al (2007) A functional genetic approach identifies the PI3 K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12(4):395–402
Chakrabarty A et al (2010) H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene 29(37):5193–5203
Dave B et al (2011) Loss of phasphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol 29(2):166–173
Esteva FJ et al (2010) PTEN, PIK3CA, p-AKT, and p-p70S6 K status - Association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol 177(4):1647–1656
Majewski IJ et al (2015) PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol 56:2439
Nagata Y et al (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6(2):117–127
Nagy P et al (2005) Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res 65(2):473–482
Price-Schiavi SA et al (2002) Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer 99(6):783–791
Rexer BN et al (2011) Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene 30(40):4163–4174
Zhang S et al (2011) Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 17(4):461–469
Diermeier S et al (2005) Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp Cell Res 304:604–619
Motoyama AB, Hynes NE, Lane HA (2002) The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res 62(11):3151–3158
Sergina NV et al (2007) Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445(7126):437–441
Harris LN et al (2007) Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res 13(4):1198–1207
Huang X et al (2010) Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-I receptor in breast cancer cells resistant to Herceptin. Cancer Res 70(3):1204–1214
Lu YH et al (2001) Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst 93(4):1852–1857
Nahta R et al (2007) Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther 6(2):667–674
Surmacz E (2000) Function of the IGF-I receptor in breast cancer. J Mammary Gland Biol Neoplasia 5(1):95–105
Ibrahim YH, Yee D (2005) Insulin-like growth factor-I and breast cancer therapy. Clin Cancer Res 11:944s–950s
Surmacz E (2003) Growth factor receptors as therapeutic targets: strategies to inhibit the insulin-like growth factor I receptor. Oncogene 22:6589–6597
Carboni JM et al (2005) Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res 65(9):3781–3787
Hartog H et al (2007) The insulin-like growth factor 1 receptor in cancer: old focus, new future. Eur J Cancer 43:1895–1904
Jerome L, Shiry L, Leyland-Jones B (2003) Deregulation of the IGF axis in cancer: epidemiological evidence and potential therapeutic interventions. Endocr Relat Cancer 10:561–578
Baserga R (2004) Targeting the IGF-1 receptor: from rags to riches. Eur J Cancer 40(14):2013–2015
Resnicoff M et al (1995) The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res 55:2463–2469
Dunn SE et al (1998) A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res 58(15):3353–3361
Sachdve D et al (2004) A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem 279(6):5017–5024
Arteaga CL (1992) Interference of the IGF system as a strategy to inhibit breast cancer growth. Breast Cancer Res Treat 22(1):101–106
Balañá ME et al (2001) Activation of ErbB-2 via a hierarchical interaction between ErbB-2 and type I insulin-like growth factor receptor in mammary tumor cells. Oncogene 19(1):34–47
Nahta R et al (2005) Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 65(23):11118–11128
Camirand A, Lu Y, Pollak M (2002) Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit 8(12):BR521–BR526
Chakraborty AK, Liang K, DiGiovanna MP (2008) Co-targeting insulin-like growth factor I receptor and HER2: dramatic effects of HER2 inhibitors on nonoverexpressing breast cancer. Cancer Res 68(5):1538–1545
Cohen BD et al (2005) Combination therapy enhances the inhibition of tumor growth with the fully human anti–type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res 11:2063–2073
Canonici A et al (2013) Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 4(10):1592–1605
Wissner A, Mansour TS (2008) The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm Chem Life Sci 341:465–477
Chakraborty AK, Welsh A, DiGiovanna MP (2010) Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and pro-apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast Cancer Res Treat 120:327–335
Wang C-X et al (2005) In vitro and in vivo effects of combination of trastuzumab (Herceptin) and tamoxifen in breast cancer. Breast Cancer Res Treat 92:251–263
Euhus DM et al (1986) Tumor measurement in the nude mouse. J Surg Oncol 31(4):229–234
Neve RM et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10(6):515–527
Riedemann J et al (2007) The EGF receptor interacts with the type 1 IGF receptor and regulates its stability. Biochem Biophys Res Commun 355(3):707–714
Camirand A et al (2005) Inhibition of insulin-like growth factor-1 receptor signaling enhances growth-inhibitory and proapoptotic effects of gefitinib (Iressa) in human breast cancer cells. Breast Cancer Res 7(4):R570–R579
Alimandi M et al (1995) Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 10(9):1813–1821
Pinkas-Kramarski R et al (1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO 15(10):2452–2467
Engelman JA et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316(5827):1039–1043
Baselga J et al (2012) Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 336(2):109–119
Gianni L et al (2012) Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol 13:25–32
Scaltriti M et al (2010) Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2–positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res 16(9):2688–2695
Geyer CE et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355(26):2733–2743
Burstein HJ et al (2010) Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol 28(8):1301–1307
Saura C et al (2014) Safety and efficacy of neratinib in combination with capecitabine in patients with metastatic human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol 32(32):3626–3633
Langer CJ et al (2014) Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients With advanced non–small-cell lung cancer. J Clin Oncol 32(19):2059–2066
Haluska P et al. (2014) Randomized phase II trial of capecitabine and lapatinib with or without cixutumumab in patients with HER2 + breast cancer previously treated with trastuzumab and an anthracycline and/or a taxane: NCCTG N0733 (Alliance). J Clin Oncol. 32: supplement(5s) (abstract p 632)
Buck E et al (2010) Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther 9(10):2652–2664
Acknowledgments
Supported by grants from Pfizer and Connecticut Breast Health Initiative, Inc, to MPD. We thank Dr. Mark Lippman for MCF7 cells and advice on their use. We also thank the services of the animal facility of the Yale School of Medicine, and the Yale Cancer Center Flow Cytometry shared resource (supported by US Public Health Service Grant CA-16359 from the National Cancer institute) for assistance with flow cytometry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
This work was supported in part by a research grant from Pfizer to MPD. MPD has received royalties from DAKO and NeoMarkers, and consulting fees from Merck. CZ and AKC declare that they do not have any financial conflicts of interest.
Ethical Approval
The experiments conducted herein comply with the current laws of the United States of America. Mice were maintained and handled in accordance with Yale Institutional Animal Care and Use Committee protocols and regulations, which also approved the research protocol. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
Rights and permissions
About this article

Cite this article
Chakraborty, A.K., Zerillo, C. & DiGiovanna, M.P. In vitro and in vivo studies of the combination of IGF1R inhibitor figitumumab (CP-751,871) with HER2 inhibitors trastuzumab and neratinib. Breast Cancer Res Treat 152, 533–544 (2015). https://doi.org/10.1007/s10549-015-3504-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3504-2