
Abstract
PRKAG2 encodes the γ2 subunit of AMP-activated protein kinase (AMPK), which is an important regulator of cardiac metabolism. Mutations in PRKAG2 cause a cardiac syndrome comprising ventricular hypertrophy, pre-excitation, and progressive conduction-system disease, which is typically not diagnosed until adolescence or young adulthood. However, significant variability exists in the presentation and outcomes of patients with PRKAG2 mutations, with presentation in infancy being underrecognized. The diagnosis of PRKAG2 can be challenging in infants, and we describe our experience with three patients who were initially suspected to have Pompe disease yet ultimately diagnosed with mutations in PRKAG2. A disease-causing PRKAG2 mutation was identified in each case, with a novel missense mutation described in one patient. We highlight the potential for patients with PRKAG2 mutations to mimic Pompe disease in infancy and the need for confirmatory testing when diagnosing Pompe disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The unique association of hypertrophic cardiomyopathy (HCM) and ventricular pre-excitation in certain individuals prompted the search for a specific unifying genetic mutation and the discovery of PRKAG2 (MacRae et al. 1995; Blair et al. 2001; Gollob et al. 2001). PRKAG2 encodes the γ2 subunit of AMP-activated protein kinase (AMPK), which is an important regulator of cardiac metabolism (Kemp et al. 1999; Hardie 2003). Mutations in PRKAG2 follow an autosomal dominant inheritance pattern and are now known to cause a cardiac syndrome comprising ventricular hypertrophy, pre-excitation, and progressive conduction-system disease (Arad et al. 2003). Cardiac hypertrophy and conduction abnormalities have been postulated to be due to cytoplasmic glycogen accumulation in cardiomyocytes and the conduction system (Arad et al. 2002). However, recent PRKAG2 mouse models suggests that cardiac hypertrophy may be independent of increased glycogen, leaving the underlying cause unknown (Kim et al. 2014). Patients with PRKAG2 cardiac syndrome typically present in late adolescence to early adulthood, with the most common symptom being palpitations. Conduction abnormalities may progress to complete heart block, requiring pacemaker placement, often in the fourth to fifth decade of life (Gollob et al. 2002; Murphy et al. 2005). However, some heterogeneity has been described in the presentation of patients with PRKAG2 mutations, with five previously described cases in infancy outside of our group (Burwinkel et al. 2005; Akman et al. 2007; Kelly et al. 2009; Austin et al. 2017). Additionally, skeletal myopathy and seizures have been noted in a minority of patients with PRKAG2 mutations, broadening the previously described phenotype of these patients (Murphy et al. 2005; Laforet et al. 2006).
Heterogeneity in disease presentation and severity is also a significant feature in Pompe disease. Also known as glycogen storage disease type II, Pompe disease is a rare, autosomal recessive disease caused by a deficiency of acid-α-glucosidase (GAA), which leads to glycogen accumulation in lysosomes. Infantile- (IOPD) and late- (LOPD) onset Pompe disease represent a continuum of the disease spectrum. Its severity typically correlates with the level of GAA enzyme activity, with IOPD being the most severe form (Kishnani et al. 2006). Patients with IOPD present with HCM, generalized weakness, hypotonia, feeding difficulty, failure to thrive, and respiratory insufficiency—typically within the first days to weeks of life. In some cases, HCM can be detected on fetal echocardiogram (ECG), and prompt early postnatal evaluation for possible IOPD (Hamdan et al. 2010). IOPD progresses rapidly, and patients typically succumb to cardiopulmonary failure around 1 or 2 years of age without intervention (Kishnani et al. 2006). Enzyme replacement therapy (ERT) with recombinant human acid-α-glucosidase (rhGAA, alglucosidase alfa) is currently the only treatment for IOPD, which significantly improves survival, reduces cardiac hypertrophy, and improves cardiac and skeletal muscle function (Kishnani et al. 2006).
Here we discuss three infants whose initial presentations were suspicious for Pompe disease but were subsequently found to have PRKAG2 mutations (Table 1), with a novel mutation described in case 1. These cases emphasize the importance of considering PRKAG2 mutation in the differential diagnosis of patients undergoing evaluation for IOPD and the value of performing confirmatory testing via gene sequencing prior to diagnosing and treating IOPD. Our cases not only highlight the potential for misdiagnosis of Pompe disease, but also demonstrate the variability in patient presentation and outcomes of patients with PRKAG2 mutations.
Case presentations
Case 1
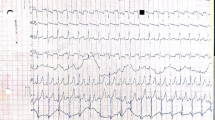
A full term, female infant was born to a G1P0 mother whose fetal ultrasound was concerning for HCM at 27 weeks gestation. The newborn had a normal clinical exam with normal muscular tone, and postnatal ECG showed mild hypertrophy of the interventricular septum (Fig. 1a). Electrocardiogram (EKG) was notable for a very short PR interval, ventricular pre-excitation, and high ventricular voltages (Fig. 1b). At 2 days of life, GAA enzyme level was low at 1.5 nmol/mg protein (normal 6–37 nmol/mg protein) on leukocyte-based assayat a local lab, urine (E)-hex-4-en-1-yl butyrate (HEX4) was elevated at 34 nmol/mol (normal <19 nmol/mol creatinine), and creatinine kinase was elevated at 2261 IU/L (normal 38–174 IU/L). Given this presentation, suspicion for IOPD was high. Biopsies of skeletal muscle and skin were obtained, and ERT with alglucosidase alfa was promptly started while waiting for biopsy results to determine CRIM status.

Cardiovascular evaluation in case 1: a Parasternal long-axis echocardiogram view from initial study showing mild intraventricular septal hypertrophy. b Initial electrocardiogram showing a very short PR interval, ventricular pre-excitation, and high ventricular voltages
Echocardiography at age 3 months no longer showed hypertrophy of the intraventricular septum, but the mitral valve was thickened and tethered by shortened chordae. There was moderate mitral regurgitation and subsequent moderate left ventricular dilation, for which captopril was started. The patient remained on ERT, with normalization of creatinine kinase and urine HEX4 but demonstrating failure to thrive and mild developmental delay. As the clinical picture and cardiac findings did not fit IOPD skin fibroblast GAA level using maltose as substrate was performed, which measured at 1.8 μmol/min/mg protein, which was 8.82% of the normal control (control mean = 20.4 ± 2.3 μmol/min/mg protein) and not consistent with IOPD. GAA sequencing subsequently identified a single common late-onset heterozygous mutation (c. -32-13 T > G). DNA microarray analysis was negative for a large deletion of the GAA gene in trans. Biochemical analysis of the skeletal muscle biopsy showed normal glycogen content of 0.49% (control 0.94 ± 0.55%) and low–normal GAA activity of 0.2 μmol/min/g tissue (control 0.42 ± 0.20 μmol/min/g tissue). Subsequently, blood GAA enzyme activity was tested after stopping ERT for a few months; AS it measured in the low–normal range (6.13pmo/punch/h; control = 10.88 pmol/punch/h), it further ruled out the diagnosis of Pompe disease. Light microscopic examination of skeletal muscle showed mild myopathic changes, including increased variation in fiber diameter, a mild increase in lipid, and slightly increased amounts of cytoplasmic glycogen. No rimmed vacuoles were seen, and electron microscopy found no membrane-bound glycogen, so there was no support for a lysosomal glycogen storage disorder.
Given these results, the diagnosis of Pompe disease was no longer supported. Blood was sent for a commercially available HCM panel, and a heterozygous mutation in PRKAG2 (c.1423A > G p. Lys475Glu) was identified. This was a novel missense mutation predicted to be disease causing for PRKAG2 cardiac syndrome, as it leads to a nonconservative amino acid substitution, occurs at a highly conserved position in the gene, is not a known benign polymorphism, and mutations affecting nearby codons (Tyr487His and Asn488Ile) have been described as disease causing. Echocardiogram (ECG), electrocardiogram (EKG), and PRKAG2 genetic testing were normal in both parents, indicating a de novo mutation in their daughter.
The patient was diagnosed with PRKAG2 cardiac syndrome with a carrier status for LOPD, and ERT was stopped at 8 months of age. Creatinine kinase and urine HEX4 levels were followed for 1 year after stopping ERT and remained normal. Follow-up ECGs continued to show thickened mitral valve leaflets with shortened chordae and moderate mitral valve regurgitation with left ventricular dilation but no ventricular hypertrophy. At 5 years of age, the child continues on captopril for afterload reduction, but she remains asymptomatic from a cardiovascular standpoint and has no overt musculoskeletal weakness per report from the treating physician.
Case 2
A full-term female infant had wheezing at 5 weeks of age and failed bronchodilator therapy for presumed reactive airway disease. At 11 weeks of age, she presented with more severe wheezing at her pediatrician’s office, which prompted referral to the emergency room. Chest X-ray showed severe cardiomegaly, and an ECG demonstrated severe biventricular hypertrophy, severe mitral regurgitation, and moderate tricuspid regurgitation (Fig. 2a). EKG showed biventricular hypertrophy, a short PR interval, and interventricular conduction delay (Fig. 2b). She was transferred to the cardiac intensive care unit where she was started on milrinone, oxygen, and furosemide for heart failure. She developed persistent ventricular ectopy, which required a lidocaine infusion. Suspicion for IOPD was high, but blood GAA enzyme activity level was normal at 15.8 pmol/punch/h (normal ≥ 10.88 pmol/punch/h), and GAA gene sequencing and urine HEX4 were also normal. Because of the care team’s experience with PRKAG2 cardiac syndrome in the patient described in case 1, this was high in the differential diagnosis. Blood was sent for a commercially available familial HCM panel, which showed a disease-causing mutation in the PRKAG2 gene (c.1592G > A p. Arg531Gln). The patient had persistent arrhythmias and poor cardiac function requiring high-dose inotropy and was therefore listed for cardiac transplant at 3 months of age. She remained in the hospital on milrinone infusion awaiting transplant, but her clinical status continued to decline. At 4 months of age, she had an episode of cardiac arrest requiring extracorporeal membrane oxygenation (ECMO) cannulation. She then developed severe renal dysfunction, a large right intracerebral hemorrhage presumably from anticoagulation required for ECMO, and was unable to recover any meaningful cardiac function. The decision was made by the family and healthcare team to withdraw support, and the patient died at 4 months of age. No muscle tissue biopsy or autopsy was performed.

Cardiovascular evaluation in case 2. a Parasternal long-axis echocardiogram view showing severe biventricular hypertrophy in . b Initial electrocardiogram showing biventricular hypertrophy, a short PR interval, and interventricular conduction delay
Case 3
A previously healthy, full-term, male infant was noted to have hypotonia, areflexia, and generalized weakness at 2 months of age. Over the next several months, he continued to show generalized weakness and developed recurrent respiratory infections with frequent hospital admissions. Evaluation during these admissions showed creatinine kinase mildly elevated at 197 IU/L (normal 38–174 IU/L) and blood GAA enzyme level (done at the local facility) was low at 4.81 U (18.66-U control sample). ECG showed mild hypertrophy of the interventricular septum but an otherwise normal left ventricular mass. EKG showed a normal sinus rhythm without evidence of pre-excitation or hypertrophy. Due to his persistent hypotonia, muscle weakness, and low GAA enzyme level, he was diagnosed with nonclassic IOPD at 11 months of age and started on ERT with alglucosidase alfa
After just 3 months of ERT, the patient showed significant improvement in motor skills; he could crawl well, sit unassisted, and pull to a standing position—none of which he could do prior to starting ERT. At 2.5 years of age, he had a normal gait and could jump and climb stairs. The previously noted hypertrophy of the interventricular septum resolved, and he had no abnormalities on ECG or EKG.
Given the patient’s unusual course and atypical presentation for IOPD, additional testing was performed at 2.5 years of age. GAA enzyme testing on skin fibroblasts was normal at 50.7 nmol/h/mg (control 45–180 nmol/h/mg). GAA gene sequencing identified a heterozygous mutation (c. -32-13 T > G), consistent with being a carrier of LOPD. Parental testing showed the same heterozygous mutation in his mother and no abnormalities in his father. These new data no longer supported a diagnosis of IOPD, and ERT was discontinued.
To identify the true cause of his myopathy, further testing was performed. At age 3, he underwent a quadriceps muscle biopsy that showed mild myopathic features on light microscopy, including increased variation in fiber size, focal myocyte degeneration, a mild increase in cytoplasmic glycogen and other intermyofibrillar material, and subsarcolemmal blebs of uncertain composition. No rimmed vacuoles were seen, and electron microscopy showed no membrane-bound glycogen, which argued against a lysosomal glycogen storage disease. Finally, a glycogen storage disease sequencing panel showed a pathogenic mutation in PRKAG2 (c. 298G > A p. Gly100Ser), a known disease-causing mutation.
The patient, now 7 years old, was restarted on ERT just before his fourth birthday due to worsening muscle weakness when off ERT. He continues to make progress in gross motor skills and has some fine motor delays, but there is no further evidence of HCM or pre-excitation.
Discussion
Patients with PRKAG2 mutations typically present in adolescence or young adulthood, but five cases presenting in infancy have been described outside of our group (Table 2). Three of those patients had a p.Arg531Gln mutation, which was also found in our case 2 patient. All three of those previously reported cases had similar presentations of fetal bradycardia, severe HCM, and a short PR interval on EKG as seen in case 2. They were acutely ill shortly after birth and had a relatively rapid cardiopulmonary decline, succumbing between 2 and 11 weeks of age. Phosphorylase b kinase (PHK) level was low in all three patients, and PHK mutations were assumed to cause this severe phenotype. However, further study showed no PHK mutations and, instead, identified a p. Arg531Gln PRKAG2 mutation in each case (Burwinkel et al. 2005). In another report of a PRKAG2 mutation presenting in infancy, a baby girl presented at 10 weeks of age with severe HCM, left ventricular outflow obstruction, and pre-excitation on electrocardiogram. Analysis of cultured skin fibroblasts did not support a diagnosis of Pompe disease. Due to progressive cardiac hypertrophy and failure to thrive, she was listed for heart transplant but died of aspiration pneumonia at 5 months of age. PHK level was undetectable, and the infant was initially diagnosed with PHK-deficient cardiomyopathy. Due to the aforementioned PRKAG2 mutations found in infantile presentations of HCM and the low PHK, postmortem PRKAG2 sequencing was performed and showed a p. Arg384Thr mutation (Akman et al. 2007). Third, a 6-month-old boy presented to a pediatric cardiologist for a murmur and was found to have HCM with moderate left ventricular outflow tract obstruction and no pre-excitation on EKG. By 10 months of age, he had developed pre-excitation and severely obstructive HCM requiring surgical septal myomectomy. His father had undergone heart transplant at age 29 for cardiomyopathy, and his sister was had high–normal left ventricular mass on ECG. All three family members carried a heterozygous p. Glu506Gln mutation in PRKAG2.
Our case series highlights the challenges of diagnosing PRKAG2 in infancy due to varying degrees of disease severity and progression. Similar to the three previously described cases with p. Arg531Gln mutation in PRKAG2 (Burwinkel et al. 2005), the patient in case 2 had severe HCM and heart failure at 11 weeks of age and succumbed at 4 months of age, which supports the idea that this particular mutation leads to a severe phenotype. Case 1 was unique in that HCM was noted prenatally, but postnatal ECG showed only mild hypertrophy of the interventricular septum (Fig. 1a), which resolved months later. She has thickened mitral valve leaflets with shortened chordae and moderate mitral regurgitation and remains clinically stable on captopril, with no overt muscle weakness and experiencing normal development. While this cardiac phenotype has not been described in other PRKAG2 patients, interestingly, in vitro work studying this novel PRKAG2 mutation (c.1423A > G p. Lys475Glu) suggests an alteration in mammalian target of rapamycin (mTOR) signaling that results in cellular hypertrophy, which can be reversed with rapamycin (Xu et al. 2017). The patient in case 3 presented with significant myopathy and mild hypertrophy of the interventricular septum that resolved while on ERT with alglucosidase alfa. Whereas myalgia during or immediately after exercise has been described in eight patients with PRKAG2 mutations (Murphy et al. 2005; Laforet et al. 2006), our case 3 is the only description of clinically significant muscle weakness and an essentially isolated skeletal myopathy without an overt cardiac phenotype. This patient improved on alglucosidase alfa ERT, and we recently described this patient’s presentation to prompt further study as to whether ERT may benefit other patients with PRKAG2 mutations (Austin et al. 2017). The use of ERT for ventricular hypertrophy in patients with PRKAG2 mutations and comparison of cardiac biopsy specimens in PRKAG2 patients with and without a cardiac phenotype could be revealing.
All of our cases presented in infancy but had quite different clinical courses, and none shared the same PRKAG2 mutations. These examples support the notion that a wide range of phenotypic variability exists among patients with PRKAG2 mutations (Gollob 2008), making the initial diagnosis challenging. Furthermore, our cases demonstrate the potential for a misdiagnosis of IOPD in these patients. In combination with the five previously reported patients, our cases demonstrate that some PRKAG2 patients present very early in life with HCM and/or skeletal myopathy, making IOPD high on the differential diagnosis among other causes of HCM that may present in infancy (Table 3). With typical mortality around 1–2 years of life without intervention, there is strong motivation to promptly diagnose IOPD and begin ERT. In two of our cases, GAA enzyme activity was low on blood-based assays tested locally, suggesting a diagnosis of IOPD. Both patients were started on ERT due to concern for IOPD, but subsequent testing of GAA activity in skin fibroblasts and skeletal muscle tissue showed levels inconsistent with IOPD. Both patients were subsequently found heterozygous for the common late-onset mutation c. -32-13 T > G, making them carriers for LOPD (Huie et al. 1994; de Vries et al. 2012). The estimated allele frequency for the c. -32-13 T > G GAA mutation is 0.3% in the general population (National Center for Biotechnology Information 2017). It is possible that there is interaction between the c. -32-13 T > G GAA and PRKAG2 mutations, contributing to the unique and early presentation of these two cases, but it is also possible that this is merely an association. Further assessment of the interaction of these mutations should be considered. Additionally, in case 1, the skin fibroblast GAA level being 8% of control is lower than expected with a heterozygous c.-32-13 T > G mutation. It is possible that altered kinase activity in patients with PRKAG2 mutations affects in vitro measurement of skin fibroblast GAA activity. In contrast, when measuring GAA activity on muscle biopsy, the muscle tissue itself serves as the substrate, without the need for in vitro cell culture (Kishnani et al. 2006), and may have been more accurate in this patient. In our laboratory, a similar phenomenon has been encountered when measuring phosphorylase kinase activity in patients with PRKAG2 mutations.
Methods for IOPD diagnosis have evolved. Initially, the presence of membrane bound glycogen on ultrastructural analysis of skeletal muscle biopsy tissue was used to make the diagnosis. Then, measuring GAA enzyme activity in muscle or cultured skin fibroblasts became the method of diagnosing Pompe disease. Currently, GAA enzyme activity is typically measured using simple blood-based assays (purified lymphocytes and mixed leukocytes) and by noninvasive dried blood spot assays. While blood-based tests are less invasive and allow more rapid diagnosis, in less experienced laboratories, false-positives can occur (Kishnani et al. 2014). The three cases presented here demonstrate the similarities that may exist between patients with IOPD and PRKAG2 mutations and underscores the importance of the most recent American College of Medical Genetics recommendation that more than one diagnostic tool be used to confirm the diagnosis of Pompe disease (Kishnani et al. 2006). Furthermore, a PRKAG2 mutation should be in the differential diagnosis of any patient being evaluated for IOPD, and our three cases demonstrate the utility of genetic sequencing to differentiate these diagnoses.
Conclusion
Classically, patients with mutations in PRKAG2 had HCM, ventricular pre-excitation, and progressive conduction delay, presenting initially in adolescence or young adulthood. Our case series combined with those previously published demonstrates a cohort of patients with PRKAG2 mutations presenting in infancy. These patients have varying presentations, ranging from severe cardiopulmonary failure and death within weeks, to HCM requiring surgical septal myectomy, and even to skeletal myopathy with little or no cardiac phenotype. Considerable overlap exists in the presentation of IOPD and patients with PRKAG2 mutations, making the risk of a misdiagnosis of IOPD high. We emphasize the importance of considering the diagnosis of PRKAG2 mutation in patients suspected of having IOPD and the need for confirmatory testing before diagnosing IOPD.
References
Akman HO et al (2007) Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res 62(4):499–504
Arad M et al (2002) Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 109(3):357–362
Arad M et al (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107(22):2850–2856
Austin SL et al (2017) Alglucosidase alfa enzyme replacement therapy as a therapeutic approach for a patient presenting with a PRKAG2 mutation. Mol Genet Metab 120(1–2):96–100
Blair E et al (2001) Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10(11):1215–1220
Bobele GB et al (1993) Hypertrophic cardiomyopathy during corticotropin therapy for infantile spasms. A clinical and echocardiographic study. Am J Dis Child 147(2):223–225
Burwinkel B et al (2005) Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 76(6):1034–1049
Colan SD (2010) Hypertrophic cardiomyopathy in childhood. Heart Fail Clin 6(4):433–444 vii-iii
de Vries JM et al (2012) Effect of enzyme therapy and prognostic factors in 69 adults with Pompe disease: an open-label single-center study. Orphanet J Rare Dis 7:73
Doyle LW, et al (2014) Early (<8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database Syst Rev (5): CD001146
Fu L et al (2016) Identification of LAMP2 mutations in early-onset Danon disease with hypertrophic cardiomyopathy by targeted next-generation sequencing. Am J Cardiol 118(6):888–894
Gollob MH (2008) Modulating phenotypic expression of the PRKAG2 cardiac syndrome. Circulation 117(2):134–135
Gollob MH et al (2001) Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 344(24):1823–1831
Gollob MH et al (2002) PRKAG2 cardiac syndrome: familial ventricular pre-excitation, conduction system disease, and cardiac hypertrophy. Curr Opin Cardiol 17(3):229–234
Hamdan MA et al (2010) Antenatal diagnosis of pompe disease by fetal echocardiography: impact on outcome after early initiation of enzyme replacement therapy. J Inherit Metab Dis 33(Suppl 3):S333–S339
Hardie DG (2003) Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144(12):5179–5183
Hay WW Jr (2012) Care of the infant of the diabetic mother. Curr Diab Rep 12(1):4–15
Huie ML et al (1994) Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (−13T-->G) mutation in a majority of patients and a novel IVS10 (+1GT-->CT) mutation. Hum Mol Genet 3(12):2231–2236
Kelly BP et al (2009) Severe hypertrophic cardiomyopathy in an infant with a novel PRKAG2 gene mutation: potential differences between infantile and adult onset presentation. Pediatr Cardiol 30(8):1176–1179
Kemp BE et al (1999) Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci 24(1):22–25
Kim M et al (2014) Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res 114(6):966–975
Kishnani PS et al (2006) Pompe disease diagnosis and management guideline. Genet Med 8(5):267–288
Kishnani PS et al (2014) Methods of diagnosis of patients with Pompe disease: data from the Pompe registry. Mol Genet Metab 113(1–2):84–91
Laforet P et al (2006) A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis. Neuromuscul Disord 16(3):178–182
MacRae CA et al (1995) Familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome maps to a locus on chromosome 7q3. J Clin Invest 96(3):1216–1220
Murphy RT et al (2005) Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol 45(6):922–930
National Center for Biotechnology Information: ClinVar (2017). Retrieved from https://www.ncbi.nlm.nih.gov/clinvar/variation/4027
Roberts AE et al (2013) Noonan syndrome. Lancet 381(9863):333–342
Xu Y, et al (2017) A novel, de novo mutation in PRKAG2 gene: infantile-onset phenotype and signaling pathway involved. Am J Physiol Heart Circ Physiol: ajpheart 00813 02016
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
R. D. Torok, S. Austin, C. Phornphutkul, K. Rotondo, D. Bali, G. Tatum, S. Wechsler, A. Buckley, and P. Kishnani declare that they have no conflict of interest.
Additional information
Responsible Editor: Olaf Bodamer, MD PhD
Rights and permissions
About this article

Cite this article
Torok, R.D., Austin, S.L., Phornphutkul, C. et al. PRKAG2 mutations presenting in infancy. J Inherit Metab Dis 40, 823–830 (2017). https://doi.org/10.1007/s10545-017-0072-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-017-0072-0