
Abstract
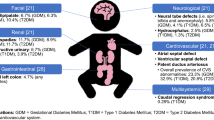
Gestational diabetes mellitus (GDM) from all causes of diabetes is the most common medical complication of pregnancy and is increasing in incidence, particularly as type 2 diabetes continues to increase worldwide. Despite advances in perinatal care, infants of diabetic mothers (IDMs) remain at risk for a multitude of physiologic, metabolic, and congenital complications such as preterm birth, macrosomia, asphyxia, respiratory distress, hypoglycemia, hypocalcemia, hyperbilirubinemia, polycythemia and hyperviscosity, hypertrophic cardiomyopathy, and congenital anomalies, particularly of the central nervous system. Overt type 1 diabetes around conception produces marked risk of embryopathy (neural tube defects, cardiac defects, caudal regression syndrome), whereas later in gestation, severe and unstable type 1 maternal diabetes carries a higher risk of intrauterine growth restriction, asphyxia, and fetal death. IDMs born to mothers with type 2 diabetes are more commonly obese (macrosomic) with milder conditions of the common problems found in IDMs. IDMs from all causes of GDM also are predisposed to later-life risk of obesity, diabetes, and cardiovascular disease. Care of the IDM neonate needs to focus on ensuring adequate cardiorespiratory adaptation at birth, possible birth injuries, maintenance of normal glucose metabolism, and close observation for polycythemia, hyperbilirubinemia, and feeding intolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gestational diabetes mellitus (GDM) from all causes of diabetes is the most common medical complication of pregnancy and is increasing in incidence, particularly as type 2 diabetes continues to increase worldwide. Recent literature documents the same problems noted for infants of diabetic mothers (IDMs) in textbooks over the past 40 years, with little change in management [1–9]. Most of the advances in the care of IDMs have come from improved management of the pregnant diabetic mother, as well as the increased understanding of how maternal obesity contributes to development of GDM. Despite advances in perinatal care, however, IDMs remain at risk for preterm birth and many of the problems that occur in all late preterm infants, as well as a multitude of physiologic, metabolic, and congenital complications unique to fetal adaptation to maternal diabetes before and during pregnancy. Perinatal mortality rates in IDMs from type 1 diabetic mothers are increased three- to 10-fold and congenital malformation rates are four- to 10-fold higher than in normal infants of the same gestational age, although there is considerable overlap in clinical problems of IDMs from mothers with both type 1 and type 2 diabetes. For example, in a recent survey, Cordero et al. [10] noted the following in a series of 530 infants born to 322 women with GDM and 177 women with insulin-dependent (type 1) diabetes mellitus: 36% were large for gestational age (LGA); 2% were small for gestational age; 36% were born preterm (14% before 34 weeks gestation and 22% between 34 and 37 weeks gestation); 47% were admitted to a neonatal intensive care unit because of respiratory distress, preterm birth, hypoglycemia, or congenital malformation; 5% had polycythemia; 25% had hyperbilirubinemia; and 4% had hypocalcemia. Induced delivery rates, either for unstable diabetes with fetal growth restriction and other fetal complications or from fetal macrosomia, often are two- to threefold greater than normal and cesarean section rates are threefold higher than normal (67% vs 22%) [11–13]. Therefore, successful management of IDMs requires close consultation between maternal (obestetricians, maternal-fetal medicine specialists) and infant (pediatricians, neonatologists) caregivers, during pregnancy and at the time of labor and delivery [11, 12]. Anticipation of potential problems and early evaluation and diagnosis of disorders help preempt many if not all of these disorders (Table 1) and prevent their complications during the newborn period. This anticipatory approach also is cost effective in reducing the financial burden of diabetes in pregnancy, for the mother and the infant [13].
Large for Gestational Age, Macrosomic IDMs
LGA IDMs develop their excess size in pregnancies from both type 1 and type 2 diabetes, particularly when the maternal glucose concentrations are generally high but unstable. They have anthropometric indices of weight, length, and head circumference above the 90th percentile for the normal population of newborn infants [14]. Many of these LGA IDMs are macrosomic, usually defined as a birth weight greater than 4 kg, although the exact definition of macrosomia is debatable [15]. Skinfold thickness measurements and other indices of body composition indicate that most of the excess weight in macrosomic IDMs is due to excess body fat. The risk for macrosomia increases in later gestation, the period of fat deposition in fetal life, and is increased in those fetuses whose mothers’ diabetes is poorly controlled. The excess adiposity develops in IDMs as maternal hyperglycemia produces fetal hyperglycemia followed by increased fetal insulin secretion and the production of fat from glucose, glycerol, fatty acids, and triglycerides. The risk for having a fetus with macrosomia increases as the mean maternal glucose concentration chronically exceeds 130 mg/dL, but the greatest degree of macrosomia occurs when maternal glucose concentrations are episodically increased, particularly following a meal (meal-associated or pulsatile hyperglycemia), as this form of hyperglycemia, when translated into surges of hyperglycemia in the fetus, has the greatest effect on promoting fetal insulin secretion [16]. IDMs also have increased glycogen content in the liver, kidney, skeletal muscle, and heart, which contributes to their characteristic organomegaly [17]. Although many have considered glucose to be the primary source of fat production in such infants, most of the fetal fat gain that produces the macrosomia (or fetal obesity) is due to maternal hyperlipidemia and increased lipid transfer to the fetus. In recent multivariate models, increased maternal glucose concentrations accounted for less than 25% of the variability in birth weight percentiles in IDMs, whereas maternal free fatty acids and triglycerides were better predictors of birth weight and the degree of macrosomia [18–24].
The principal risk of being LGA or macrosomic is injury to the head and major nerves of the upper extremities from stretching of the neck during vaginal birth (cephalopelvic disproportion) [1, 14, 25]. Shoulder dystocia is the most common of these problems, producing a 10-fold increase in the incidence of Erb’s palsy (brachial nerve palsy, roots C5-7), as well as Klumpke’s paralysis (roots C7-8), diaphragmatic nerve paralysis (roots C3-5), and recurrent laryngeal nerve damage (roots T1-2). Fractures of the humerus and clavicle also are common. Although not often diagnosed, subarachnoid hemorrhage with vaginal birth of LGA infants is common, especially when forceps are used, which also often cause facial nerve palsies.
Perinatal Asphyxia
Perinatal asphyxia can occur in up to 25% of IDMs and correlates with maternal atherosclerotic vascular disease, hyperglycemia (before delivery but particularly during labor), maternal nephropathy, and recurrent preterm labor [11, 26]. Such problems are almost exclusively confined to type 1 diabetic mothers whose diabetes is chronically unstable. In such cases, acute, severe maternal diabetic hyperglycemia with ketoacidosis is inversely related to uterine and placental blood flow, most likely due to the simultaneous increase in maternal catecholamine concentrations, which may increase the risk of fetal hypoxia. Glycated hemoglobin A1c (HbA1c) concentrations are increased in the mother with long-standing diabetes and this glycated hemoglobin (Hgb) increases the affinity of maternal Hgb for oxygen [27, 28]. This increase in Hgb-O2 affinity will decrease the uterine venous PO2 (PutVO2), as this Hgb will tend to bind oxygen until the PO2 is much lower than with lower Hgb-O2 affinity. A lower PutVO2 will lead to reduced transfer of oxygen to the fetus, which is directly dependent on the PO2 gradient across the placenta. Furthermore, chronic decreases in maternal red blood cell 2,3-diphosphoglycerate (2,3-DPG) concentration also can increase maternal Hgb-O2 affinity and thereby further decrease the PutVO2 and the potential transfer of oxygen to the fetus. Further potential reduction in oxygen transfer to the fetus may be caused by increased diffusion distance in the thickened basement membranes or edematous trophoblast villi in diabetic placentas and by increased placental glucose consumption. The latter is the result of increased fetal glucose concentration, which reduces transport of glucose from mother to fetus and in turn promotes uteroplacental net glucose uptake and metabolism (oxidation, lactate production, glycogen deposition) [29, 30].
Metabolic Characteristics
At birth, the neonate is separated from the constant supply of glucose from the maternal circulation. Although macrosomic IDMs have abundant adipose tissue and glycogen stores, they can be less able than normal infants to mobilize these substrate stores. Insulin is an anabolic hormone that promotes growth and the movement of glucose, fatty acids, and amino acids into tissues in the late stages of gestation and in the neonate [31]. Normal neonates rapidly mobilize glycogen from the liver and other sites to maintain plasma glucose concentration [32]. However, high plasma insulin concentrations inhibit glycogenolysis by specifically blocking glycogen phosphorylase enzyme activity. Insulin also inhibits the normal postnatal activation of phosphoenolpyruvate carboxykinase (PEPCK), the rate-limiting enzyme of gluconeogenesis, by glucagon and cortisol as plasma glucose concentrations decrease. Fatty acid release from fat cells and subsequent oxidative metabolism and ketone body oxidation also are important for the maintenance of normal glucose concentrations. These processes can be impaired in IDMs by high insulin concentrations. Insulin acts to increase transport of fatty acids into adipocytes for fat synthesis. It also promotes the production of malonyl-coenzyme A (CoA) and acetyl CoA carboxylase activity, which promotes glucose oxidation, inhibits fatty acid oxidation, and re-directs fatty acids into synthetic pathways. Previous studies indicated that hyperinsulinism in IDMs inhibits free fatty acid release from adipose tissue and limits the availability of fatty acids as an alternative substrate for oxidative metabolism. Such studies have noted that ketone body production from fatty acids also is decreased for the same reasons, decreasing the availability of this important alternate energy source. Thus, hyperinsulinemia in the IDM, whether from type 1 or type 2 diabetic mothers, can lead to increased glucose utilization, decreased glucose production, and decreased availability of alternate substrates. In recent studies, however, despite increased insulin concentrations, lipolysis (as measured by glycerol production) in a small group of IDMs was unimpaired and the gluconeogenic contribution from glycerol was higher than in healthy newborns [33–35]. This may represent a mechanism to compensate for the reduced rate of glucose production in these infants and should be studied in considerable more detail. However, one should not assume that IDMs can mount adequate alternative energy substrate production (free fatty acids, ketoacids) during episodes of low glucose concentrations.
Blood and/or plasma glucose concentrations should be tested using a glucose monitoring device or a glucose oxidase strip by at least 2 hours of life in known IDMs. Earlier measurements often show low glucose values; therefore, the advantage of waiting until 2 hours is to ensure that the infant has upregulated gluose production and normalized glucose concentrations. Although obese non-IDMs do not have the same frequency or degree of hypoglycemia as do obese IDMs, those infants weighing more than 4 kg and/or exhibiting the physical characteristics of IDMs also should be carefully observed for signs of hypoglycemia and frequently monitored for initial and subsequent glucose concentrations as if they were IDMs. Many of these infants actually are IDMs and this condition was missed in prenatal care or they are the offspring of obese mothers who had meal-associated hyperglycemia. Non-IDM LGA infants should be measured carefully for body fat content, most simply by determining whether they have a disproportionately high ponderal Index (higher weight percentile than their length and head circumference percentiles).
When treating IDMs with intravenous (IV) glucose, one must be careful not to administer too much glucose too fast to prevent stimulation of insulin secretion. Because fat has a relatively low metabolic rate, basing nutrition and treatments such as IV glucose infusion rates on a weight that matches the head circumference percentile will help to limit excessive glucose infusion rates, rebound hyperinsulinemia, and, in turn, further hypoglycemia. Glucose concentrations should be maintained as constant as possible to prevent acute hyperglycemia-induced insulin secretion.
Low Glucose Concentrations (“Hypoglycemia”)
Transient low glucose concentrations in the immediate postnatal period are the norm in all infants, but tend to occur sooner, more frequently, and to lower levels in IDMs, especially those that had evidence of unstable maternal glucose concentrations during later gestation and particularly during labor [32]. The phrase “low glucose concentrations” is preferable to the common term “hypoglycemia,” which has been variably defined, lacks current universally accepted definition, and has legal implications that usually are unwarranted in most IDMs [36]. Current working definitions of the acceptable lower limit for plasma glucose concentration (no urgent treatment required, but evaluation should occur and potential treatment should be considered) are statistically based, not outcomes related, and range from 30 to 45 mg/dL (25–40 in blood) in term infants [37••, 38, 39]. The best treatment of moderately low, transient neonatal glucose concentrations is early feeding, whether the neonate is an IDM or not. A plasma glucose concentration less than 30 mg/dL and associated with significant signs of low glucose concentrations should be treated with IV glucose administration unless the infant readily takes a good feeding, corrects its glucose concentration on its own, and remains normoglycemic. With persistent low glucose concentrations (more than 30–60 minutes following an initial low glucose concentration), IV glucose administration should be started, especially if the infant does not feed and/or has emesis [40].
If the infant is mildly to moderately symptomatic (tremulousness, mildly to moderately hypotonic, unable to feed after initially feeding well), a constant rate IV infusion of glucose should be started at 4 to 6 mg/min/kg body weight. A constant infusion of glucose is almost always preferable in IDMs because this approach helps prevent sudden hyperglycemia-induced insulin secretion. This regimen will rapidly correct very low glucose concentrations (usually within 10–15 minutes) and helps to minimize the risk of rebound hypoglycemia. However, if the glucose concentration is extremely low (unmeasurable to < 10–20 mg/dL) and accompanied by prolonged, recurrent, and marked clinical signs consistent with severe hypoglycemia (marked hypotonia, reduced alertness, hypothermia, seizures, and/or coma), the infant should receive a minibolus of IV glucose followed by continued IV glucose infusion as the preferred therapy [41]. With this regimen, the infant should receive 200 mg/kg of glucose (2 mL of 10% glucose per kilogram of body weight) over 2 to 4 minutes, followed by a constant infusion of 6 mg/kg/min (D10W at 80 mL/kg/day) [42]. This rate should be adjusted in response to frequent glucose concentration measurements and reduced when possible to avoid further hyperglycemia and insulin secretion. Such infants, when stable, can be started again on enteral feedings; sometimes smaller and more frequent feedings or even constant gavage infusion of milk will help stabilize glucose concentrations. Milk is preferred for enteral feeding rather than glucose-containing solutions as milk sugar, lactose, contains 50% galactose. Galactose is first pass cleared by the liver after absorption from the intestine into the portal vein for glycogen production and does not stimulate insulin secretion by the pancreas.
Continuous Glucose Concentration Monitoring
Recent reports document the use of continuous subcutaneous glucose concentration monitoring in neonates at risk of hypoglycemia [43••, 44–46]. Continuous glucose monitoring in IDMs would provide much-needed data to determine the incidence, severity, and duration of low glucose concentrations, their relationship with symptoms, and their correlation with concurrent morbidities and neurodevelopmental outcomes. In IDMs requiring treatment for low glucose concentrations, continuous glucose monitoring could help determine when low glucose concentrations had been corrected, how stable (or not) they remained during treatment, and when treatment could be safely and effectively discontinued. Such monitoring also would help determine when such infants had achieved glucose homeostasis sufficient for safe discharge. Such continuous monitoring also might uncover low glucose concentrations that tend to occur when IV glucose infusion rates are reduced as enteral feeding is advanced. Through such careful monitoring, aided by further research, continuous glucose monitoring in IDMs might help to determine relationships between very low glucose concentration and acute or longer-term evidence of neurologic injury, particularly in those infants whose glucose concentrations fall to extremely low values and/or stay very low for extended periods [47]. This value, or narrow range of values, probably differs among infants. It also is not known how low, how often if episodic, or for how long low plasma glucose concentrations must be present before irreversible neuronal damage occurs [46]. Relationship with other pathology also is confusing, with little data to determine at which values of cardiac output, blood pressure, brain blood/plasma flow, hematocrit, availability of alternative brain energy substrates such as ketones, and relationship to prior or concurrent hypoxic-ischemic conditions [48] a given low plasma glucose concentration becomes too low for normal brain glucose and energy needs [46, 48]. Furthermore, controversy continues about whether IDMs suffer injury while entirely asymptomatic, as low glucose concentrations in asymptomatic infants often are insufficiently documented [49–51]. Continuous glucose monitoring in recent clinical trials of insulin infusion in very low birth weight neonates detected many more episodes of low glucose concentrations than standard intermittent blood glucose measurements [52]. This might be particularly applicable to IDMs who often have quite variable plasma glucose concentrations; detecting these, but also learning how to treat them without stimulating insulin secretion, will be helpful.
Hypocalcemia
At birth, IDMs demonstrate a negligible rise in parathyroid hormone, resulting in a temporary hypoparathyroidism. This hypoparathyroidism is related to maternal and fetal hypomagnesemia, both of which then contribute to neonatal hypocalcemia [53]. Hypocalcemia occurs in a high percentage of IDMs, at rates up to 50% in the past, but much less frequently today [54]. Risks of developing hypocalcemia include acidosis, which releases intracellular phosphorus, use of sodium bicarbonate to buffer acidosis, stress-induced calcitonin release, preterm delivery with limited parathyroid hormone release, sustained calcitonin production despite hypocalcemia, and end-organ resistance to 1,25-(OH)2 cholecalciferol, the most active form of vitamin D [55, 56]. Maternal magnesium deficiency also occurs more commonly in mothers with diabetes who have renal insufficiency, which leads to fetal hypomagnesemia and impaired parathyroid hormone secretion and action via limited magnesium-dependent adenylate cyclase action. Hypocalcemia, defined as a calcium level of less than 7 mg/dL, can contribute to agitation, irritability, and, in occasional infants, decreased myocardial contractility [57]. Treatment rarely is necessary (other than normal enteral milk feeding) but, when indicated, consists of 1 to 2 mL/kg of 10% calcium gluconate given intravenously and 50 to 60 mg/kg/day given orally for maintenance. Hypomagnesemia (< 1.5 mg/dL) also can occur but rarely is of clinical significance. Magnesium treatment should be given to the IDM with documented clinical signs of hypocalcemia and measured low plasma calcium and magnesium concentrations.
Hyperbilirubinemia
Hyperbilirubinemia also is an index of immaturity and occurs in 20% to 30% of IDMs. Contributing factors include newborn polycythemia, which produces immature red cell precursors that break down more rapidly, hepatic immaturity that reduces bilirubin conjugation and excretion, and occasionally bruising in macrosomic infants who had a difficult vaginal birth with superficial head trauma. Serum bilirubin concentrations should be measured early and often and may need to be followed for up to 5 to 7 days. Hyperbilirubinemia is exacerbated by preterm delivery and the normal problems with reduced gut transit time (from developmental cause and delayed enteral feeding) and decreased bilirubin excretion found in preterm infants.
Polycythemia
IDMs may have symptomatic polycythemia [57]. As already noted, increased maternal Hgb-O2 affinity from increased glycation of Hgb and reduced 2,3-DPG concentrations can reduce the transplacental PO2 gradient, and the placenta can increase oxygen consumption, both of which can reduce oxygen transport to the fetus [58, 59]. Fetal hyperglycemia and hyperinsulinism result in increased fetal oxygen consumption. These conditions may lead to fetal hypoxia and increased levels of fetal erythropoietin. Increased insulin and insulin-like growth factors also can increase red blood cell production. High concentrations of β-hydroxybutyrate derived from mild maternal ketosis also independently stimulate erythropoiesis [60]. The fetal erythropoietic response to hypoxia may occur at the expense of other bone marrow cell lines, particularly platelets producing transient thrombocytopenia [57].
Polycythemia with hyperviscosity can be treated by partial exchange transfusion using saline to decrease the central hematocrit to less than 55%. Indications include respiratory distress with signs of vascular congestion on chest radiographs and heart failure, which represent complications of the hyperviscosity that results from the polycythemia and increases exponentially above hematocrit values of 60%, persistent hypoglycemia, and absolute hematocrits above 70% to 75%. Although IV hydration often is recommended, there is little evidence that this reduces the clinical complications associated with hyperviscosity, since water distributes rapidly into all tissues and cells; thus, even an excessive water load of 5% body weight (50 cc/kg) would only reduce the hematocrit by 5% (eg, from 75 to 71).
Thrombosis
IDMs are at increased risk for the development of deep vessel thromboses, most commonly recognized as renal vein thrombosis with hematuria often as the presenting sign [57, 61]. These problems are more common in IDMs from unstable type 1 diabetic mothers. The pathogenesis may relate to polycythemia with hyperviscosity or to decreased cardiac output secondary to cardiomyopathy. IDMs, like their mothers, also tend to have low blood concentrations of the anticoagulant proteins, protein C, protein S, and antithrombin—the production of these proteins is decreased in the fetus by hyperinsulinemia. Insulin also increases the production of procoagulants [61]. Treatment of deep vessel thromboses is supportive, including normalization of oxygenation and blood acid–base balance, but should include IV or low molecular weight heparin when significant deep vessel thromboses are documented by Doppler flow ultrasound studies. The overall propensity to thrombophilia extends to the placenta, where vascular thromboses can lead to placental insufficiency (even in the presence of a large placenta), and to fetal organs, particularly the brain, where infarcts may be seen, although microvascular thromboses may restrict neural development and brain growth without obvious strokes.
Respiratory Disorders
IDMs characteristically have worse respiratory distress for gestational age than non IDMs. The risk of surfactant deficiency with respiratory distress syndrome and hyaline membrane disease developing in these infants is six times that of normal infants until gestational week 38 and is more common in IDMs whose mothers had unstable type 1 diabetes. Late preterm (34–37 weeks) IDMs commonly have apparent surfactant deficiency respiratory distress syndrome despite their advanced gestational age. Fetal hyperglycemia and hyperinsulinism have been associated with pulmonary immaturity and decreased synthesis and secretion by type II pneumocytes of surfactant phospholipids and their associated proteins [62–66]. Earlier studies indicated that high glucose and insulin concentrations inhibit the production of surfactant by blocking key enzymes in the phosphatidylcholine pathway. Furthermore, there are fewer type II pneumocytes and alveolar lining cells and lamellar bodies/alveolar lining cells in high glucose conditions, suggesting a block in the trafficking of lipids from fibroblasts to the type II cells, along with reduced mRNA expression and protein concentrations of surfactant proteins B and C. More recent studies in mice genetically altered to overexpress Akt also have shown that increased insulin signaling through the insulin signal transduction pathway, specifically via increasing Akt-mTOR signaling in lung epithelial cells, reduces surfactant production and produces greater respiratory distress [67]. Akt overexpression also resulted in downregulated hypoxia-inducible factor 2α and its target, vascular endothelial growth factor. Such adverse changes could be attenuated, including the associated respiratory distress, with inhibitors of the Akt-mTOR protein activities and the overall insulin signal transduction pathway. Therefore, it is possible that although glucose and insulin might reduce surfactant production, other pulmonary vascular pathology could result from increased stimulation of key proteins in the insulin signal transduction pathway, including reduced vascular function and even angiogenesis.
Cesarean birth in the absence of labor is a significant risk factor for transient tachypnea of the newborn due to retained or slow clearance of fluid in the lungs. Hyperviscosity resulting from polycythemia, as well as the underlying chronic fetal hypoxia, may predispose the infant to persistent pulmonary hypertension of the newborn [57, 59]. Fortunately, better maternal diabetes control during pregnancy reduces the incidence and severity of all of these pulmonary and respiratory tract-related symptoms.
Cardiac Disorders
Congenital cardiac malformations and disorders are quite common in IDMs, but those with congenital malformations of the heart are almost exclusively from mothers with unstable type 1 diabetes [68•]. Thirty percent of infants born to mothers with GDM and pre-existing diabetes present with cardiac malformations. Approximately half the cardiovascular defects are conotruncal anomalies, including transposition of the great arteries, persistent truncus arteriosus, visceral heterotaxia, and single ventricle. Elevated HbA1c values during the first trimester are associated with these cardiovascular defects. This suggests a profound teratogenic effect at a very early stage in cardiogenesis. Other cardiac disorders, which can be seen in IDMs of both type 1 and type 2 diabetic mothers, include asymmetric septal hypertrophy, transient hypertrophic subaortic stenosis from ventricular septal defects, and/or a thickened myocardium. Septal hypertrophy occurs from 25% to 75% of IDMs (the high variability is probably related to how often and how carefully caregivers look for this disorder) [69]. The cardiomyopathy is attributed to neonatal hyperglycemia, hyperinsulinemia, hypertrophic myocardium with septal hypertrophy and outflow tract obstruction, or congestive failure. In the presence of cardiomegaly, poor perfusion, and hypotension, a differential diagnosis is important, as IDMs may present with heart failure and poor cardiac output, which worsens with increasing septal thickness and cardiomegaly. With supportive care, most symptoms resolve spontaneously within 2 weeks. Septal hypertrophy usually resolves by 4 months. All IDMs with septal hypertrophy should receive consultation and follow-up with a pediatric cardiologist.
Cardiomegaly also is common in IDMs, often associated with thickening of the interventricular septum and subaortic stenosis, representing a hypertrophic response to increased afterload [70]. This complication may be a cause of the sudden late gestational deaths and stillbirths that occur occasionally, although unexplained arrhythmias also should be considered. Septal hypertrophy is found by echocardiography in 30% to 40% of known IDMs and can reproducibly be detected as early as the 18th week of pregnancy, whereas progressive thickening can be documented up through at least the 33rd week of gestation, allowing considerable opportunity for producing tighter glucose control in the mother. Although septal hypertrophy is less common in well-controlled pregnant diabetic women, it remains uncertain whether producing tighter glucose control after the diagnosis of fetal ventricular septal hypertrophy will lead to regression of the hypertrophy and reduce the severity of this condition. Interestingly, this complication is observed also in low birth weight infants who are receiving long-term parenteral nutrition and become chronically hyperglycemic. In such infants, this postnatal, glucose infusion-related septal hypertrophy is reversible with normalization of the blood glucose concentration by decreasing IV glucose infusion rates.
Hypertrophic Cardiomyopathy
Fetuses exposed to hyperglycemic ± hyperinsulinemic conditions during diabetic pregnancies are prone to develop “hypertrophic” cardiomyopathy, primarily affecting the interventricular septum, but extending to the myocardium in more severe cases [69]. Even more concerning is that IDMs are at risk in later life for metabolic disorders that include insulin resistance and hypertension, which could be worsened by the prenatal development of hypertrophic cardiomyopathy, making this problem a public health issue. Despite such concerns, there is a large gap in the understanding of how hyperglycemia and hyperinsulinemia during fetal life in markedly diabetic pregnancies cause cardiomyocyte hypertrophy and dysfunction. It is not known whether increased fetal growth factors such as insulin and insulin-like growth factor-1 drive septal hypertrophy, to what extent hyperglycemia influences cardiomyocyte growth and function, or how these conditions are affected by hyperglycemia-induced oxidative injury [71]. The degree to which immature myocytes are more sensitive to hyperglycemic injury also is not known.
Supranormal levels of glucose can also inflict damage to cells including endothelial cells and cardiomyocytes [72, 73]. The common pathway of damage is thought to be the overproduction of superoxide by the mitochondrial electron transport chain [73]. Thus, high intracellular glucose levels force excessive amounts of electron donors into the electron transport chain; this increases the voltage across the mitochondrial membrane so that complex III of the chain is blocked. This allows coenzyme Q to donate the electrons to molecular oxygen and reactive oxygen species (ROS), such as superoxide, are generated [73]. These disordered processes lead to poor cell function and expression of inflammatory genes [74]. The generation of advanced glycation end products (AGE) is the inevitable result of elevated glucose levels in plasma and within the cytosol. In diabetes, the commonly found AGE species include carboxymethyl-lysine protein adducts, carboxyethyl-lysine protein adducts, and many others, which have been shown to stimulate AGE receptor-inflammatory pathways [75–77].
Most fetuses whose mothers have increased plasma glucose levels have abnormal cardiac function or growth ranging from asymmetrical septal hypertrophy in the mildest cases, dilated hypertrophic cardiomyopathy in moderate cases, or massive hypertrophy and lethal heart failure in extreme cases [78, 79]. Even in mild to moderate cases, the hypertrophic condition is accompanied of markers of cardiomyopathy (troponin T and pro-B-type natriuretic peptide) [80]. Common dogma says that hypertrophy “resolves” over the first 6 postnatal months after birth, meaning it can no longer be detected by ultrasound. But whether such hypertrophic growth results in persistent and indolent cardiomyopathy is unknown. A few studies have found that ejection fraction, estimated by echocardiography, is actually elevated in the diabetic fetal hypertrophic heart [81], whereas others have found that heart function is compromised [82]. Certainly diabetic cardiomyopathy in adults is characterized by depressed myocardial metabolism and reduced oxygen consumption [83]. The impact of cardiomyocyte enlargement (hypertrophy) or proliferation (hyperplasia) on the increase in myocardial mass and cardiac function remains to be elucidated.
Congenital Malformations
Congenital malformations are frequent in diabetic pregnancies, almost exclusive from mothers with unstable hyperglycemia prior to and around the time of conception. The incidence of congenital heart disease and other major malformations is two to three times greater than that of the general population (3% to 5%) and has been related to poor control of maternal diabetes during the periconceptional period and the embryonic period that encompasses organogenesis. The incidence of all congenital malformations may be in excess of 10% in poorly controlled diabetic pregnancies. Renal (hydronephrosis, renal agenesis, ureteral duplication), cardiovascular (single umbilical artery, ventral septal and atrial septal defects, coarctation of the aorta, cardiomegaly), and gastrointestinal (duodenal, anorectal atresia) anomalies are frequent in IDMs [84, 85].
Most major congenital malformations occur very early in gestation and, therefore, cannot be attributed to fetal hyperinsulinemia. The fetal pancreas does not make insulin until after day 100 of gestation, and the functional importance of insulin probably develops only after weeks 26 and 27 of pregnancy. Malformations in IDMs primarily are caused by hyperglycemia during critical periods of organogenesis in the early embryonic stage of development [86]. Several teratological mechanisms in the embryonic tissues have been suggested to be involved, including alterations in the metabolism of inositol, arachidonic acid/prostaglandins, formation of sorbitol, glycation of proteins, and generation of ROS. Optimal control of maternal diabetes should be instituted even before conception occurs, to be certain of good control during the period of organogenesis, when congenital malformations occur.
Neurologic Impairments
IDMs can present with acute neurologic abnormalities secondary to central nervous system dysfunction. This is more common in IDMs whose mothers had unstable type 1 diabetes. Central nervous system changes occur as a result of perinatal asphyxia, glucose and electrolyte abnormalities, polycythemic vascular sludging and stroke, and birth trauma [87]. The timing of the neurologic symptoms may provide clues as to the cause. Symptoms from perinatal depression or hypoglycemia typically have their onset in the first 24 hours postpartum, whereas symptoms from hypocalcemia or hypomagnesemia present between 24 and 72 hours of life. Cerebral symptoms may include seizures, jitteriness, lethargy, changes in tone, and movement disorders. The spinal cord is also vulnerable to birth trauma with symptoms related to palsies of the brachial plexus.
The macrosomic IDM is particularly vulnerable to birth asphyxia as a consequence of fetal macrosomia, which increases the risk of shoulder dystocia during delivery, and a compromised prenatal milieu characterized by fetal hypoxia, cardiomyopathy, and tissue iron deficiency. The latter three factors may decrease the IDM’s tolerance to the stress of delivery and may limit the response of the infant to respond optimally to resuscitation. Signs and symptoms of perinatal asphyxia include initial hypotonia or flaccidity followed by increasing tone, jitteriness, and seizure risk. The risk of seizures from birth asphyxia typically peaks at 24 hours of age [88]. Initial treatment consists of correcting any underlying metabolic or hematologic abnormality before the initiation of anticonvulsant agents. Treatment of the underlying disorder frequently aborts the seizures without recurrence.
The immature sucking patterns seen in infants born to insulin-managed mothers with diabetes represent neurologic immaturity [89]. Neurologic state changes are common in compromised IDMs. More commonly, symptoms include lethargy and hypotonia, but jitteriness and hypertonicity can occur. Potential factors that contribute to the classic picture of the plethoric, sluggish IDM are perinatal depression, glucose and electrolyte abnormalities, polycythemia, cardiomyopathy, and tissue (eg, brain) iron deficiency. These symptoms may persist for up to a week. The lethargic IDM tends to be a poor feeder.
Among IDMs, central nervous system malformations are 16 times more likely compared with nondiabetic mothers. The risk of anencephaly is 13 times higher, the risk of spina bifida is 20 times higher, and the risk of caudal dysplasia is up to 600 times higher in these infants [90]. The caudal regression syndrome, which demonstrates a general hypoplasia of the sacrum and lower extremities, is found almost exclusively in IDMs whose mothers had relatively unstable type 1 diabetes during the periconceptional and early embryonic period.
Central Nervous System Complications of Polycythemia and Redistribution of Iron
Increased red blood cell production can contribute to redistribution of iron from the developing neurons in the brain to the liver and bone marrow, producing neuronal iron deficiency, which has been correlated with limitations in cognitive development. Diabetic pregnancies are characterized by chronic metabolic insults, including iron deficiency, that place the developing brain at risk for memory impairment later in life. Electrophysiologic studies demonstrate that both encoding and retrieval processes are compromised, indicating that prenatal iron deficiency leads to changes in neural development that have lasting impact on memory ability, including specifically declarative memory [91].
Iron deficiency in IDMs increases the infant’s risk for neurodevelopmental abnormalities; increased hematopoiesis depletes hepatic, cardiac, and cerebral iron stores by 93.7%, 56.1%, and 40%, respectively, potentiating already compromised neurodevelopment [92]. Studies have shown that 65% of all IDMs and up to 95% of LGA IDMs demonstrate abnormalities of iron metabolism at birth. Most of these infants have low ferritin concentrations, but more severely affected infants have increased total iron binding capacity concentrations, decreased transferrin saturation, and increased free-erythrocyte protoporphyrin concentrations, which indicate accelerated erythropoiesis. Iron-deficient infants are at increased risk for neurodevelopmental and neurobehavioral abnormalities [93]. Animal models demonstrate that gestational and early postnatal iron deficiency adversely affects myelination [94], brain energy metabolism [95], and brain monoamine neurotransmitter metabolism [96]. Perinatal iron deficiency seems to increase the vulnerability of the neonatal brain, particularly the hippocampus, to hypoxic-ischemic insult [97]. Perinatal iron deficiency may place IDMs, who have an increased risk of acute and chronic hypoxemia, at even greater risk of perinatal brain injury. A recent study provided evidence that IDMs demonstrate abnormal cognitive processing in the newborn period [98] and that this effect may be related partly to iron status.
Small Left Colon Syndrome
IDMs also may exhibit the neonatal small left colon syndrome [99], which can present much like Hirschsprung disease (rectal/colonic aganglionosis). Neonatal small left colon syndrome is the most common cause of intestinal obstruction in IDMs. Maternal diabetes has been reported in 40% to 50% of the published cases of neonatal small left colon syndrome. Neonatal small left colon syndrome can be confidently diagnosed in this population based on the classic clinical and radiologic findings.
Intrauterine Growth Restriction
Intrauterine growth restriction can occur if maternal diabetes is associated with severe vascular disease, particularly when the mother is hypertensive and/or has preeclampsia. Such mothers usually have chronically unstable type 1 diabetes. Maternal renovascular disease is a particularly common cause of poor fetal growth in diabetic pregnancies, resulting in impaired fetal growth [8]. Pregnancies thus affected should be monitored carefully and may require early delivery if fetal well-being is jeopardized.
Predisposition to Later-Life Pathology
IDMs may not maintain their macrosomia in early childhood, but they tend to regain their obesity earlier in childhood and as adolescents tend to have increased adipose tissue mass [100]. This tendency has implications for the development of later-life obesity as well as insulin resistance and the development of type 2 diabetes. A recent study tested the hypothesis that intrauterine exposure to GDM predicts childhood growth independent of the effect on infant birth weight [101]. This study involved a prospective analysis of 28,358 mother-infant pairs who enrolled in the National Collaborative Perinatal Project between 1959 and 1965. As expected, compared with their nondiabetic counterparts, mothers with GDM gave birth to offspring that had higher weights at birth. The offspring of mothers with GDM were larger at age 7 years as indicated by greater weight, body mass index (BMI), and BMI z-score compared with the offspring of mothers without GDM at that age (all P < 0.05). These differences at age 7 years persisted even after adjusting for infant birth weight. Furthermore, the offspring of mothers with GDM had a 61% higher odds of being overweight at age 7 years compared with the offspring of mothers without GDM after adjusting for maternal BMI, pregnancy weight gain, family income, race, and birth weight (OR, 1.61; 95% CI,1.07, 1.28). These results indicate that maternal GDM status is associated with overweight offspring during childhood. This relationship is only partially mediated by effects of birth weight.
In GDM offspring of mothers with type 1 and type 2 diabetes, obesity leads to insulin resistance as early as childhood [102]. The insulin resistance and associated hyperglycemia initially increase pancreatic insulin production, which then is followed by failure of pancreatic insulin secretion resulting in persistant hyperglycemia from failure to promote normal glucose uptake and utilization rates in peripheral tissue, primarily muscle. This hyperglycemia also is augmented by hepatic insulin resistance leading to increased glucose production [103]. These observations indicate that fetal hyperinsulinism and hyperglycemia, as well as hyperlipidemia, contribute to an increase in the adult metabolic syndrome and may represent an example of environmental programming of fetal development with lifelong implications for later disease [104]. Exposure to GDM moderately increases the risk of subsequent metabolic complications [105••, 106], although fetal exposure to maternal blood glucose is only one of several risk factors of the metabolic syndrome. In particular, the role of maternal weight in the occurrence of the metabolic syndrome in the offspring is difficult to distinguish from that of GDM [105••].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Nold JL, Georgieff MK. Infants of diabetic mothers. Pediatr Clin North Am. 2004;51:619–37.
Schwartz R, Teramo KA. Effects of diabetic pregnancy on the fetus and newborn. Semin Perinatol. 2000;24:120–35.
American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2005;28 Suppl 1:S4–S36.
Chu SY, Callaghan WM, Kim SY, et al. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care. 2007;30:2070–6.
Cowett RM. The infant of the diabetic mother. Neo Rev. 2002;3:el73–89.
Crowther CA, Hiller E, Moss R, et al. Australian Carbohydrate intolerance Study in pregnant women (ACHOIS) Trial Group. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med. 2005;352:2477–86.
Yang JB, Cummings EA, O’Connell CP, Jangaard KMD. Fetal and neonatal outcomes of diabetic pregnancies. Obstet Gynecol. 2006;108:644–50.
Meur S, Mann NP. Infant outcomes following diabetic pregnancies. Paediatrics and Child Health. 2007;17:217–22.
Cowett RM, Schwartz R. The infant of the diabetic mother. Pediatr Clin North Am. 1982;29:1213–31.
Cordero L, Treuer SH, Landon MB, Gabbe SG. Management of infants of diabetic mothers. Arch Pediatr Adolesc Med. 1998;152:249–54.
Anonymous. The Confidential Enquiry Into Maternal and Child Health (CEMACH). Diabetes in pregnancy: caring for the baby after birth. Findings of a National Enquiry: England, Wales and Northern Ireland. London: CEMACH; 2007.
Coetzee EJ, Levitt NS. Maternal diabetes and neonatal outcome. Semin Neonatol. 2000;5:221–9.
Elixhauser A, Weschler JM, Kitzmiller JL, et al. Cost-benefit analysis of preconception care for women with established diabetes mellitus. Diabetes Care. 1993;16:1146–57.
Weindling MA. Offspring of diabetic pregnancy: short-term outcomes. Semin Fetal Neonatal Med. 2009;14:111–8.
Visser GHA, Evers IM, Giorgio Mello G. Management of the macrosomic fetus. In: Hod M, Jovanovic L, Di Renzo GC, De Leiva A, Langer O, editors. Textbook of Diabetes & Pregnancy. 2nd ed. London: Informa; 2008.
Carver TD, Anderson SM, Aldoretta PW, Hay Jr WW. Effect of low-level basal plus marked “pulsatile” hyperglycemia on insulin secretion in fetal sheep. Am J Physiol. 1996;271(Endo. Metab 34):E865–71.
Hay Jr WW. Nutrition and development of the fetus: carbohydrate and lipid metabolism (Chapter 28). In: Walker WA, Watkins JB, Duggan CP, editors. Nutrition in Pediatrics (Basic Science and Clinical Applications). 4th ed. BC Decker Inc Publisher, Hamilton, Ontario: Canada; 2008. p. 311–25.
Ouzilleau C, Roy MA, Leblanc L, et al. An observational study comparing 2-hour 75-g oral glucose tolerance with fasting plasma glucose in pregnant women: both poorly predictive of birth weight. CMAJ. 2003;168:403–9.
Knopp RH, Magee MS, Walden CE, et al. Prediction of infant birth weight by GDM screening tests. Importance of plasma triglyceride. Diabetes Care. 1992;11:1605–13.
Son GH, Kwon JY, Kim YH, Park YW. Maternal serum triglycerides as predictive factors for large-for-gestational age newborns in women with gestational diabetes mellitus. Acta Obstet Gynecol Scand. 2010;89:700–4.
Di Cianni G, Miccoli R, Volpe L, et al. Maternal triglyceride levels and newborn weight in pregnant women with normal glucose tolerance. Diabet Med. 2005;22:21–5.
Kitajima M, Oka S, Yasuhi I, et al. Maternal serum triglyceride at 24–32 weeks’ gestation and newborn weight in nondiabetic women with positive diabetic screens. Obstet Gynecol. 2001;97:776–80.
Catalano PM. Management of obesity in pregnancy. Obstet Gynecol. 2007;109:419–33.
Schaefer-Graf UM, Graf K, Kulbacka I, et al. Maternal lipids as strong determinants of fetal environment and growth in pregnancies with gestational diabetes mellitus. Diabetes Care. 2008;31:1858–63.
Wagner RK, Nielson PE, Gonik B. Controversies in labor management: shoulder dystocia. Obstet Gynecol Clin North Am. 1999;26:371–83.
Mimouni F, Miodovnik M, Siddiqi T, et al. Perinatal asphyxia in infants of insulin-dependent diabetic mothers. J Pediatr. 1988;113:345.
Bard H, Prosmanne J. Relative rates of fetal hemoglobin and adult hemoglobin synthesis in cord blood infants of insulin-dependent diabetic mothers. Pediatrics. 1987;75:1143–7.
Morris MA, Grandis AS, Litton JC. Glycosylated hemoglobin concentration in early gestation associated with neonatal outcome. Am J Obstet Gynecol. 1985;153:651–4.
Regnault TRH, Limesand SW, Hay Jr WW. Aspects of fetoplacental nutrition in intrauterine growth restriction and Macrosomia. In: Thureen PJ, Hay Jr WW, editors. Neonatal Nutrition and Metabolism. 2nd ed. Cambridge: Cambridge University Press; 2006. p. 32–46.
Hay Jr WW. Recent observations on the regulation of fetal metabolism by glucose. J Physiol. 2006;572:17–24.
Hay Jr WW. Nutrient delivery and metabolism in the fetus. In: Hod M, Jovanovic L, Di Renzo GC, de Leiva A, Langer O, editors. Textbook of Diabetes and Pregnancy. London: Martin Dunitz; 2003. p. 201–21.
Persson B. Neonatal glucose metabolism in offspring of mothers with varying degrees of hyperglycemia during pregnancy. Semin Fetal Neonatal Med. 2009;14:106–10.
Gustafsson J. Neonatal energy substrate production. Indian J Med Res. 2009;130:618–23.
Sunehag A, Ewald U, Larsson A, Gustafsson J. Attenuated hepatic glucose production but unimpaired lipolysis in newborn infants of mothers with diabetes. Pediatr Res. 1997;42:492–7.
Ahlsson FS, Diderholm B, Ewald U, Gustafsson J. Lipolysis and insulin sensitivity at birth in infants who are large for gestational age. Pediatrics. 2007;120:958–65.
Cornblath M, Hawdon JM, Williams AF, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105:1141–5.
•• American Academy of Pediatrics Committee on Fetus and Newborn. Screening and management of postnatal glucose homeostasis in late preterm and term SGA and LGA/IDM infants. Pediatrics 2011, 127:575–579. This article is the latest effort by the American Academy of Pediatrics (AAP) to provide rational guidelines for management of glucose metabolism and concentrations in newborn infants. Although the AAP recommended allowing asymptomatic infants, especially those breastfed, to have slightly lower glucose concentrations than previously preferred, the AAP continues to emphasize that IDMs and even LGA infants (many LGA infants are simply missed IDMs, usually from type 2 and/or obese mothers) should be monitored earlier and more frequently and treated earlier and with less bolus approaches.
Ward-Platt M, Deshpande S. Metabolic adaptations at birth. Semin Fetal Neonatal Med. 2005;10:341–50.
Heck IJ, Erenberg A. Serum glucose levels in term neonates during the first 48 hours of life. J Pediatr. 1987;110:119–22.
Rozance PJ, Hay Jr WW. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neonate. 2006;90:74–86.
Rozance PJ, Hay, WW, Jr. Describing hypoglycemia - definition or operational threshold? Invited Review in: Hawdon. J., Guest Editor, Neonatal Hypoglycaemia. Best Practice Guidelines. Early Human Development 2010, 86:275–280.
Lilien LD, Pildes RS, Srinivasan G, et al. Treatment of neonatal hypoglycemia with minibolus and intravenous glucose infusion. J Pediatr. 1980;97:295–8.
•• Harris DL, Battin MR, Weston PJ, Harding JE. Continuous glucose monitoring in newborn babies at risk of neonatal hypoglycaemia. Journal of Pediatrics 2010, 157:198–202. Continuous subcutaneous glucose monitoring could help define glucose concentrations previously unrecognized that contribute to complications, it could exonerate glucose concentrations previously thought to cause complications, or it could lead to excessive and unnecessary treatments. Time and experience will tell. This article provides novel data establishing the validity of this new technology and the potential to be a useful and perhaps important addition to monitoring neonatal glycemia and treatments for both high and low glucose concentrations.
Platas II, Lluch MT, Alminana NP, et al. Continuous glucose monitoring in infants of very low birth weight. Neonatology. 2009;95:217–23.
Beardsall K, Ogilvy-Stuart A, Ahluwalia J, et al. The continuous glucose monitoring sensor in neonatal intensive care. Arch Dis Child Fetal Neonatal Ed. 2005;90:F307–10.
Hay Jr WW, Rozance PJ. Continuous glucose monitoring for diagnosis and treatment of neonatal hypoglycemia. Invited Editorial Commentary regarding Harris DL, Battin MR, Weston PJ, Harding JE. Continuous glucose monitoring in newborn babies at risk of hypoglycaemia. J Pediatr. 2010;157:180–2.
Boluyt N, van Kempen A, Offringa M. Neurodevelopment after neonatal hypoglycaemia: a systematic review and design of an optimal future study. Pediatrics. 2006;117:2231–43.
Salhab WA, Wyckoff MH, Laptook AR, Perlman JM. Initial hypoglycemia and neonatal brain injury in term infants with severe fetal acidemia. Pediatrics. 2004;114:361–6.
De Leon DD, Stanley CA. Mechanism of disease: advances in diagnosis and treatment of hyperinsulinism. Nat Clin Pract Endocrinol Metab. 2007;3:57–8.
Meissner T, Brune W, Mayatepek E. Persistent hyperinsulinaemic hypoglycaemia of infancy: therapy, clinical outcome and mutational analysis. Eur J Pediatr. 1997;156:754–7.
Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–9.
Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart A, et al. Early insulin therapy in very low birth weight infants. N Engl J Med. 2008;359:1873–84.
Cruikshank DP, Pitkin RM, Varner MW, et al. Calcium metabolism in diabetic mother, fetus, and newborn infants. Am J Obstet Gynecol. 1983;135:1010–6.
Banerjee S, Mimouni FB, Mehta R, et al. Lower whole blood ionized magnesium concentration in hypocalcemic infants of gestational diabetic mothers. Magnes Res. 2003;16:127–30.
Mimouni F, Loughead J, Miodovnik M, et al. Early neonatal predictors of neonatal hypocalcemia in infants of diabetic mothers: an epidemiologic study. Am J Perinatol. 1990;7:203–6.
Demarini S, Minouni F, Tsang RC, et al. Impact of metabolic control of diabetes during pregnancy on neonatal hypocalcemia: a randomized study. Obstet Gynecol. 1994;83:918–22.
Widness JA. Fetal risks and neonatal complications of diabetes mellitus and metabolic and endocrine disorders. In: Brody SA, Ueland K, editors. Endocrine disorders in pregnancy. Norwalk (CT): Appleton-Lange; 1989. p. 273–97.
Widness JA, Susa JB, Garcia JF, et al. Increased erythropoiesis and elevated erythropoietin in infants born to diabetic mothers and hyperinsulinemic rhesus fetuses. J Clin Invest. 1981;67:637–42.
Warshaw JB, Hay Jr WW. Infant of the diabetic mother. In: McMillan J, DeAngelis C, Feigin R, Warshaw JB, editors. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2006. p. 423–7.
Cetin H, Yalaz M, Akisu M, Kultursay N. Polycythaemia in Infants of Diabetic Mothers: β-Hydroxybutyrate Stimulates Erythropoietic Activity. J Int Med Res. 2011;39:815–21.
Manco-Johnson MJ, Carver T, Jacobson LJ, et al. Hyperglycemia-induced hyperinsulinemia decreases maternal and fetal plasma protein C concentration during ovine gestation. Pediatr Res. 1994;36:293–9.
Gewob IH, O’Brien J. Surfactant secretion by type II pneumocytes in inhibited by high glucose concentrations. Exp Lung Res. 1997;23:245–55.
Gewolb IH. Effect of high glucose on fetal lung maturation at different times in gestation. Exp Lung Res. 1996;22:201–11.
Gewolb IH, Torday JS. High glucose inhibits maturation of the fetal lung in vitro. Morphometric analysis of lamellar bodies and fibroblast lipid inclusions. Lab Invest. 1995;73:59–63.
Rayani HH, Gewolb IH, Floras J. Glucose decreases steady state mRNA content of hydrophobic surfactant proteins B and C in fetal rat lung explants. Exp Lung Res. 1999;25:69–79.
Sugahar K, Lyama K, Sano K, Morioka T. Differential expressions of surfactant protein SP-A, SP-B, and SP-C mRNAs in rats with streptozotocin-induced diabetes demonstrated by in situ hybridization. Am J Respir Cell Mol Biol. 1994;11:397–404.
Ikeda H, Shiojima I, Oka T, et al. Increased Akt-mTOR signaling in lung epithelium is associated with respiratory distress syndrome in mice. Mol Cell Biol. 2011;3:1054–65.
• Lisowski LA, Verheijen PM, Copel JA, et al.: Congenital heart disease in pregnancies complicated by maternal diabaetes mellitus. An international clinical collaboration, literature review, and meta-analysis. Herz 2010, 35:19–26. This article reviews the current experience with the high incidence of congenital heart disease in diabetic pregnancies.
Rolo LC, Marcondes Machado Nardozza L, Araujo Junior E, et al. Reference curve of the fetal ventricular septum area by the STIC method: preliminary study. Arg Bras Cardiol. 2011;96:386–92.
Mongiovi M, Fesslova V, Fazio G, et al. Diagnosis and prognosis of fetal cardiomyopathies: a review. Curr Pharm Des. 2010;16:2929–34.
Verhaeghe J, van Bree R, Van Herck E. Oxidant balance markers at birth in relation to glycemic and acid–base parameters. Metabolism. 2011;60:71–7.
Kaiser N, Sasson S, Feener EP, et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes. 1993;42:80–9.
Clark RJ, McDonough PM, Swanson E, et al. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278:44230–7.
Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–8.
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25.
Ralser M, Wamelink MM, Kowald A, et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol. 2007;6:10–28.
Du X, Matsumura T, Edelstein D, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–57.
Gabbay KH, Merola LO, Field RA. Sorbitol pathway: presence in nerve and cord with substrate accumulation in diabetes. Science. 1966;151:209–10.
McLellan AC, Thornalley PJ, Benn J, Sonksen PH. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin Sci (Lond). 1994;87:21–9.
Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–66.
Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circ Res. 2003;93:1159–69.
Ahmed MU, Brinkmann FE, Degenhardt TP, et al. N-epsilon-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J. 1997;324:565–70.
Ikeda K, Higashi T, Sano H, et al. (epsilon)-(carboxymethyl)lysine protein adduct is a major immunological epitope in proteins modified with advanced glycation end products of the Maillard reaction. Biochemistry. 1996;35:8075–83.
Casson F, Clarke CA, Howard CV, et al. Outcomes of pregnancy in insulin dependent diabetic women: results of a five year population study. Br Med J. 1997;315:275–8.
Jenson DM, Damm P, Moelsted-Pederson L, et al. Outcomes in type 1 diabetic pregnancies: a nationwide, population-based study. Diabetes Care. 2004;27:2819–23.
Doblado M, Moley KH. Glucose metabolism in pregnancy and embryogenesis. Curr Opin Endocrinol Diabetes Obes. 2007;14:488–93.
Kalhan SC, Parimi PS, Lindsay CA. Pregnancy complicated by diabetes mellitus. In: Fanaroff AA, Martin RJ, editors. Neonatal-Perinatal Medicine: diseases of the Fetus and Infant. 7th ed. Philadelphia: Mosby; 2002. p. 1357–62.
Volpe JJ. Neonatal seizures. In: Volpe JJ, editor. Neurology of the newborn. 4th ed. Philadelphia: WB Saunders; 2001. p. 178–216.
Bromiker R, Rachamim A, Hammerman C, et al. Immature sucking patterns in infants of mothers with diabetes. J Pediatr. 2006;149(5):640–3.
Barnes-Powell LL. Infants of diabetic mothers: The effects of hyperglycemia on the fetus and neonate. Neonatal Netw. 2007;26:283–90.
Petry CD, Wobken JD, McKay H, et al. Placental transferrin receptor in diabetic pregnancies with increased fetal iron demand. Am J Physiol. 1994;267:E507–14.
Eidelman AI, Samueloff A. The pathophysiology of the fetus of the diabetic mother. Semin Perinatol. 2002;26:232–6.
Lozoff B, Jimenez E, Hagen J, et al. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics. 2000;105:E51.
Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93.
deUngria M, Rao R, Wobken JD, et al. Perinatal iron deficiency decreases cytochrome c oxidase (cytox) activity in selected regions of neonatal rat brain. Pediatr Res. 2003;133:1174–9.
Beard J. Neonatal iron deficiency results in irreversible changes in dopamine function in rats. J Nutr. 2003;133:1174–9.
Rao R, deUngria M, Sullivan D, et al. Perinatal brain iron deficiency increases the vulnerability of rat hippocampus to hypoxic ischemic insult. J Nutr. 1999;129:199–206.
deReginer RA, Nelson CA, Thomas KM, et al. Neurophysiologic evaluation of auditory recognition memory in healthy newborn infants and infants of diabetic mothers. J Pediatr. 2000;127:777–84.
Ellis H, Kumar R, Kostyrka B. Neonatal small left colon syndrome in the offspring of diabetic mothers-an analysis of 105 children. J Pediatr Surg. 2009;44:2343–246.
Dabelea D. The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care. 2007;30 Suppl 2:S169–74.
Baptiste-Roberts K, Nicholson WK, Brancati FL. Gestational diabetes and subsequent growth patterns of offspring: The National Collaborative Perinatal Project. Matern Child Health J 2011, [Epub ahead of print].
Thorn SR, Regnault TRH, Brown LD, et al. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces hepatic and skeletal muscle mRNA translation initiation and nutrient sensing. Endocrinology. 2009;150:3021–30.
Thorn SR, Rozance PJ, Brown LD, Hay Jr WW. The Intrauterine Growth Restriction (IUGR) phenotype: fetal adaptations and potential implications for later life insulin resistance and diabetes. Semin Reprod Med. 2011;29:225–36.
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–7.
•• Dabelea D, Crume T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes 2011, 60:1849–1855. Diabetes begets diabetes, by epigenetic as well as genetic processes. This article reviews the environmental input to the epigenetic processes, which are becoming more well recognized as causes of later diabetes in the offspring and particularly GDM in female offspring.
Burguet A. Long-term outcome in children of mothers with gestational diabetes. Diabetes Metab. 2010;36:682–94.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hay, W.W. Care of the Infant of the Diabetic Mother. Curr Diab Rep 12, 4–15 (2012). https://doi.org/10.1007/s11892-011-0243-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11892-011-0243-6