
Abstract
Mitochondrial trifunctional protein (MTP) deficiency caused by HADHA or HADHB gene mutations exhibits substantial molecular, biochemical, and clinical heterogeneity and ranks among the more severe fatty acid oxidation (FAO) disorders, without pharmacological treatment. Since bezafibrate has been shown to potentially correct other FAO disorders in patient cells, we analyzed its effects in 26 MTP-deficient patient fibroblasts representing 16 genotypes. Overall, the patient cell lines exhibited variable, complex, biochemical profiles and pharmacological responses. HADHA-deficient fibroblasts showed markedly reduced alpha subunit protein levels together with decreased beta-subunit abundance, exhibited a −86 to −96 % defect in LCHAD activity, and produced large amounts of C14 and C16 hydroxyacylcarnitines. In control fibroblasts, exposure to bezafibrate (400 μM for 48 h) increased the abundance of HADHA and HADHB mRNAs, immune-detectable alpha and beta subunit proteins, activities of LCHAD and LCKAT, and stimulated FAO capacities, clearly indicating that MTP is pharmacologically up-regulated by bezafibrate in human fibroblasts. In MTP-deficient patient fibroblasts, which were found markedly FAO-deficient, bezafibrate improved FAO capacities in six of 26 (23 %) cases, including three cell lines heterozygous for the common c1528G > C mutation. Altogether, our results strongly suggest that, due to variable effects of HADHA and HADHB mutations on MTP abundance and residual activity, improvement of MTP deficiency in response to bezafibrate was achieved in a subset of responsive genotypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondrial trifunctional protein (MTP) is a large hetero-multimeric enzyme associated with the inner mitochondrial membrane that catalyzes the three final steps of long-chain mitochondrial fatty acid β-oxidation (FAO). MTP is composed of four α- and four β-subunits encoded by the HADHA and HADHB genes, respectively. Both genes are located head-to-head on chromosome 2 (2p23.3) and share a common promoter (Orii et al 1999). The alpha subunit carries the long-chain enoylCoA hydratase (LCEH) and the long-chain 3-hydroxyacylCoA dehydrogenase (LCHAD) activities, whereas the ß-subunit harbors the long-chain 3-ketoacylCoA thiolase (LCKAT) activity (Eaton et al 2000). Disorders of the MTP complex, initially described in the 1990s, are rare diseases with an estimated prevalence of 1:200,000 newborns, classified in three groups (IJlst et al 1996; Fletcher et al 2012). The majority of patients have an isolated LCHAD deficiency with moderate or no reduction in the hydratase and thiolase activities. Patients with decreased levels of all three enzymes activities (general MTP deficiency) are also observed (Boutron et al 2011; Sykut-Cegielska et al 2011). Finally a small proportion of patients exhibit an isolated long-chain 3-ketothiolase deficiency (Das et al 2006).
The molecular basis of these disorders is complex and only partially understood, and measurement of enzyme activities is the only means to distinguish between the various forms of the disorder (Choi et al 2007; Purevsuren et al 2008; Sperk et al 2010; Fletcher et al 2012). All patients with isolated LCHAD deficiency harbor at least one allele with the c. 1528G > C mutation, which affects the alpha subunit of the enzyme on position 474 (p.Glu474Gln), and lies within the LCHAD catalytic site (Spiekerkoetter et al 2004). Other mutations in HADHA or HADHB gene often result in general MTP deficiency (Spiekerkoetter et al 2004). To date, the Human Gene Mutation database reports 59 HADHA and 53 HADHB gene mutations, many of which have not been functionally characterized (Boutron et al 2011; Fletcher et al 2012).
These mutations are associated with a variety of clinical manifestations ranging from severe life-threatening phenotypes in the neonate to a mild adolescent-onset myopathic form with episodes of rhabdomyolysis (Olpin 2013). In addition to recapitulating symptoms of other FAO disorders, LCHAD deficiency is unique because patients often develop long-term peripheral neuropathy and retinopathy, with progressive loss of vision (Fletcher et al 2012; Olpin 2013). Altogether, deficiencies of the MTP complex are associated with a high mortality and morbidity and rank among the more severe FAO disorders (Sykut-Cegielska et al 2011). The common c. 1528G > C mutation, either in the homozygous or heterozygous state, is often associated with severe phenotypes and high mortality rates, but has also been reported in milder phenotypes (Sykut-Cegielska et al 2011). Overall, LCHAD and MTP deficiencies exhibit substantial molecular, biochemical, and clinical heterogeneity and no clear genotype-phenotype correlation (Olpin et al 2005; Boutron et al 2011; Sykut-Cegielska et al 2011). To date, management of the patients relies on nutritional supplementation with medium chain triglycerides, but no pharmacological treatments have been tested, either in pre-clinical or clinical studies (Gillingham et al 2003).
In recent years, we have shown that bezafibrate, a widely prescribed hypolipidemic drug, corrects, in some cases, CPT2 or VLCAD deficiency ex vivo in primary human cells (Djouadi and Bastin 2008). In these experiments, the effects of bezafibrate were mediated by activation of peroxisome proliferator activated receptor (PPAR) nuclear receptors, which triggered a transcriptional stimulation of CPT2 and VLCAD gene expression. Data obtained in mice suggest that expression of HADHA and HADHB genes is controlled by the PPAR signaling pathway (Aoyama et al 1998), and might therefore be stimulated in response to bezafibrate. There are, however, no data on a possible control of MTP by PPAR in human cells. In the present study, we therefore raised the question whether bezafibrate might be beneficial for improvement or correction of MTP deficiency in a panel of patient fibroblasts harboring different mutations of HADHA or HADHB gene.
Materials and methods
Cell culture
Human skin fibroblasts were obtained from cell repositories of different reference centers for the diagnosis of inborn metabolic diseases. All the patients’ fibroblasts originated from individuals in which diagnosis of MTP deficiency had been previously established on the basis of clinical, biochemical, and molecular data. This panel included all the cells available at the time of the study, without any selection, except for a few cell lines that were ruled out because of insufficient growth. The patient genotypes are given in Tables 1 and 2. The present study included fibroblasts from 26 patients, homozygous or heterozygous for the common c.1528G > C HADHA gene mutation, or harboring various other HADHA or HADHB gene mutations. Control fibroblasts were obtained from three healthy individuals. Cells from patients or control individuals were grown in Ham’s F10 media (Invitrogen, Cergy-Pontoise, France) with glutamine, 12 % fetal bovine serum and 100 U/ml penicillin and 0.1 mg/ml streptomycin under standard conditions (5 % CO2, 37 °C). For treatment, cells were incubated in fresh media containing 400 μM bezafibrate (Sigma) or vehicle only (0.01 % DMSO) for 48 h.
Fatty acid oxidation (FAO) measurements
FAO flux was determined by quantifying the production of 3H2O from (9.10-3H) palmitate (Perkin Elmer), in cultured fibroblasts, as previously described (Manning et al 1990). Palmitate bound to fatty acid-free albumin was used at the final concentration of 125 μM (60 Ci/mmol). For each cell line, FAO measurements were performed in triplicate and repeated in two to three independent experiments. The oxidation rates were expressed as nmol of 3H fatty acid oxidized per hour per mg of cell protein (nmol 3H. FA/ h/ mg prot).
Acylcarnitine analysis
The method used has been described previously (Djouadi et al 2005a, b). Briefly, fibroblasts were cultured in complete Ham’s F10 medium containing 200 μM palmitate and 400 μM carnitine for 72 h at 37 °C. The culture medium was collected, extracted, and analyzed for acylcarnitine content by electrospray MS-MS, using an API3000 triple quadrupole mass spectrometer (Sciex, Applied Biosystems, USA) detecting the precursors of an m/z ratio of 85, by reference to added internal deuterated standards. Long-chain acylcarnitines were quantified by reference to standard curves. The values were expressed relative to protein content determined by the Lowry method.
Enzyme activity
All enzyme activities were measured essentially as described previously using UHPLC-analysis to quantify substrate production and calculate enzyme activities (Wanders et al 2010).
Western blot analysis
Mitochondria were isolated from fibroblasts using a mitochondrial/cytosol fractionation kit (Abcam, Cambridge, UK) according to the manufacturer’s protocol. Cultured fibroblasts or isolated mitochondria from fibroblasts were harvested in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 % NP40, 0.25 % sodium deoxycholate, 0.1 % SDS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride) to prepare protein extracts; 25 μg of total protein or 15 μg of mitochondrial protein per lane were resolved by 12 % SDS-PAGE and transferred to Hybond-P PVDF membrane (Amersham Biosciences, Freiburg, Germany). The following antibodies were used: mouse monoclonal anti-HADHA (dilution 1/1000) and anti-HADHB (dilution 1/500) (Abcam, Cambridge, UK) and mouse monoclonal anti-actin (dilution 1/2000) (Chemicon International, Temecula, USA). The primary antibodies were detected using horseradish peroxidase (HRP) conjugated anti-mouse IgG, and the chemiluminescent reagent, ECL (Amersham Biosciences, Freiburg, Germany). Immunoreactive bands corresponding to the proteins were scanned by densitometry with a computerized video densitometer.
Statistical analysis
Data are the means ± SD. Unpaired t-tests were used for the comparisons between control and deficient fibroblasts, and paired t-test were used to analyze the effects of drug versus vehicle. P < 0.05 was considered significant.
Results
The patient fibroblasts were first screened for their response to bezafibrate in tritiated palmitate oxidation assays. Basal oxidation rates in vehicle-treated fibroblasts revealed a marked long-chain fatty acid oxidation (FAO) deficiency in all the patients’ fibroblasts harboring HADHA (Fig. 1a) or HADHB (Fig. 1b) mutations. Indeed, 16 of 26 patient cell lines (patients 2–4, 9–14, 17, 18, 20–24, 26) exhibited basal FAO rates that did not exceed 1 nmol 3HFA/ h/ mg prot, indicating marked FAO deficiencies relative to the control value (4.8 nmol 3HFA/ h/ mg prot). In seven cell lines (patients 1, 7, 8, 15, 16, 19, 25), basal FAO rates were in the range of 1 to 2 nmol 3HFA/ h/ mg prot, and only two patients’ cell lines (patients 5 and 6) exhibited values close to 3 nmol 3HFA/ h/ mg prot in the absence of treatment.

Palmitate oxidation flux in cultured fibroblasts from (a) HADHA- or (b) HADHB-deficient patients, treated with bezafibrate (400 μM, 48 h; dark bars), or with vehicle only (open bars). The results are means ± SD of at least three experiments. Control values are from three different healthy individuals. The genotypes are numbered as in Tables 1 and 2. *p < 0.05; ** p < 0.01; *** p < 0.001 compared with vehicle-treated fibroblasts
Exposure to 400 μM bezafibrate for 48 h resulted in a significant increase (+31 %; p < 0.001) in FAO capacity in control fibroblasts. There was substantial variability in the response to bezafibrate in our panel of patient cells harboring HADHA (Fig. 1a) or HADHB (Fig. 1b) gene mutations. Although many increases in FAO were found statistically significant, only those reaching at least 3 nanomol 3HFA/h/mg prot, i.e., 62 % of the control value, were considered deserving of further investigation. Thus, in HADHA-deficient fibroblasts, only five of 18 cell lines (patients 1, 5, 6, 7, 8) fitted this criterion. Analysis of data also showed that none of the four cell lines homozygous for the common c.1528G > C HADHA gene mutation (patients 9–12) significantly responded to bezafibrate. Interestingly, however, three of the six bezafibrate-responsive fibroblasts (patient 6, 7, 8) harbored this c.1528G > C mutation in the heterozygous state. Marked increases in FAO capacity were observed in two HADHA-deficient cell lines (patient 7, 8) that initially exhibited very low basal FAO values (−75 % compared to control), as well as in three cell lines (patients 1, 5, 6) with milder FAO deficiencies (−63 to −33 % versus control). In this group of HADHA-deficient cells, the amplitude of FAO changes triggered by bezafibrate varied from 1.4- (patient 6) to 3.2-fold (patient 7). Overall, after treatment by bezafibrate, only one cell line (patient 5) was restored to control levels.
In HADHB-deficient fibroblasts (Fig. 1b), all cell lines except patient 19 initially exhibited profound FAO deficiency (−80 to −95 % relative to control) under standard culture conditions. In this group, exposure to bezafibrate induced significant –but generally modest- increases in FAO flux values, which therefore remained quite deficient in most treated cells. By contrast, in patient 19, treatment with bezafibrate triggered a marked FAO stimulation, from 1.8 to 3.9 nanomol 3HFA/h/mg prot (+117 %, p < 0.001). Thus, only patient 19 was further investigated.
Next, we analyzed the effects of bezafibrate on LCHAD activity, an α-subunit function (Fig. 2a), and LCKAT activity, the β-subunit enzyme (Fig. 2b), in four fibroblasts in which FAO flux was significantly induced by the drug (patients 5, 6, 7, 19) and in 2two cells lines (patients 11 and 12, homozygous for c.1528G > C) that did not respond to bezafibrate. These cells were chosen because they reflect the two kinds of response to bezafibrate, and because enough cell material could be obtained to perform these assays. Short-chain hydroxyacyl-CoA dehydrogenase (SCHAD) activity was measured in parallel (Fig. 2c). SCHAD was used as an internal standard to better evaluate the amplitude of defects in LCKAT and LCHAD activities in patient cells.

Measurements of MTP enzyme activities (a) Long-chain hydroxyacylCoA dehydrogenase, LCHAD, (b) Long-chain 3-ketoacylCoA thiolase, LCKAT, and (c) of short-chain hydroxyacylCoA dehydrogenase, SCHAD, in MTP-deficient patients fibroblasts, treated with bezafibrate (400 μM, 48 h; dark bars), or with vehicle only (open bars). The results are mean ± SD. The genotypes are numbered as in Table 1. *p < 0.05; ** p < 0.01; *** p < 0.001 compared with vehicle-treated fibroblasts
The results show (Fig. 2a) that all the HADHA-deficient genotypes considered (patients 5, 6, 7, 11, and 12) were associated with a marked defect in LCHAD enzyme activity in the patient fibroblasts, which ranged from 14 % (patient 5) down to 4 % (patient 12), of control values. The residual levels of LCHAD enzyme activity were increased significantly, from +40 to +68 % in three (patients 5, 6, and 7) of the five cell lines harboring HADHA mutations. Interestingly, bezafibrate induced no changes in LCHAD activity in both cell lines homozygous for c.1528G > C (patients 11 and 12). In HADHB-deficient patient 19 fibroblasts, treatment with bezafibrate resulted in a significant increase (+92 %, p < 0.001) in LCHAD residual enzyme activity. Finally, control fibroblasts exhibited a robust increase in LCHAD enzyme activity, from 82 to 122 nmol/min/mg prot (+46 %, p < 0.001), in response to bezafibrate.
Under basal conditions, LCKAT activity was unchanged (patient 12), or modestly reduced (patient 11) in the two cell lines homozygous for the c.1528G > C mutation, compared to control (Fig. 2b). In patients 6 and 7 (c.1528G > C on one allele), LCKAT activity was close to 50 % of normal value. In contrast, there was marked LCKAT deficiency in patient 5 (10 % of control value) and in patient 19 (4 % of control value) fibroblasts. As observed for LCHAD, LCKAT enzyme activity was markedly up-regulated in control fibroblasts (from 79 to 123 nmol/min/mg prot) after treatment with bezafibrate. Drug-induced increases in LCKAT were also observed in the two cell lines homozygous for the c.1528G > C mutation, which reached normal activity levels after treatment with bezafibrate, and in patient 6 and patient 7 fibroblasts, which reached 75 % of normal values. In patient 5 and patient 19 fibroblasts, bezafibrate induced a small but significant, increase in LCKAT activity, but nevertheless both cell lines remained extremely deficient. Finally, SCHAD enzyme activity levels were normal, or close to normal, in all the patient cells, under basal conditions, indicating that mitochondrial short-chain fatty acid ß-oxidation is unaffected by MTP deficiency, and does not greatly vary among the various fibroblasts cell lines. In control and patient cells as well, SCHAD activity was generally induced (+34 to +64 %) in response to bezafibrate (Fig. 2c).
Accumulation of long-chain hydroxyl-acylcarnitines is a hallmark of HADHA deficiency, which can be observed in vitro by incubating patient cells with long-chain fatty acids. We thus analyzed acylcarnitine production in fibroblasts with three distinct HADHA-deficient genotypes and investigated possible effects of bezafibrate (Fig. 3). We chose two patients in which bezafibrate increased the FAO flux (patients 5 and 8) and one with severe LCHAD mutations, in which bezafibrate had no effect (patient 11). Under standard culture conditions, two hydroxylated acylcarnitine species, namely C14-OH and C16-OH, were present in large excess in patients’ fibroblasts, compared to control, with the highest levels found in patient 11, homozygous for the c.1528G > C mutation. A modest decrease in these acylcarnitine species was observed in bezafibrate-treated patient 11 fibroblasts (−16 and −28 %, respectively). Interestingly, however, exposure to bezafibrate resulted in a marked reduction of C14-OH and C16-OH acylcarnitines levels in patient 5 (−62 and −77 %, respectively) and in patient 8 (−56 and −61 %, respectively).

Analysis of acylcarnitine profiles was performed in cultured control or patients’ fibroblasts incubated with palmitate and carnitine for 72 h, in the presence or absence of 400 μM bezafibrate. The histograms represent the quantification of C14- and C16- hydroxylated (OH)- acylcarnitines. Values are means ± SD. *** p < 0.001 compared with vehicle-treated fibroblasts
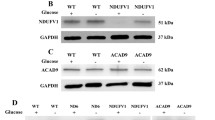
We then analyzed by western blot the abundance of HADHA and HADHB protein levels in a panel of patient cells. A first set of experiments was performed using total protein extracts from control and HADHA-deficient fibroblasts. Unfortunately, and despite repeated tests, the commercially available antibody against the MTP alpha subunit did not allow detection of this protein in whole cell extracts. In contrast, the beta subunit protein could be immuno-detected and quantified in this material, and western blot analysis revealed a marked decrease in beta-subunit protein levels in HADHA-deficient fibroblasts, compared to control (Fig. 4a). Interestingly, bezafibrate treatment led to increases in beta-subunit protein abundance in both control and HADHA-deficient fibroblasts. We were able to grow enough fibroblasts from patients 7 and 8 to isolate mitochondria and, in these preparations HADHA and HADHB could be detected in parallel (Fig. 4b). This revealed extremely low residual alpha-subunit protein levels in both HADHA-deficient cells, and confirmed the relative diminution in beta-subunit abundance in these cells, compared to control fibroblasts (Fig. 4b). Quantification of the signals showed that bezafibrate significantly increased the abundance of alpha (×3) and beta (×1.5) subunit proteins in control fibroblasts, consistent with the up-regulation of the corresponding mRNA, by 1.3- and 1.6-fold for HADHA and HADHB genes, respectively (data not shown). In response to bezafibrate, parallel increases in the abundance of alpha and beta subunit proteins were also observed in mitochondrial fractions from both HADHA-deficient fibroblasts. Table 3 provides a flowchart of the different results obtained in the various HADHA or HADHB-deficient cell lines.

Quantification by western blot analysis of (a) HADHB protein levels in whole protein extracts from control or patients fibroblasts, treated with bezafibrate (400 μM, 48 h; dark bars), or with vehicle only (open bars) and normalized to actin abundance. (b) HADHA and HADHB protein levels in mitochondria-enriched extracts from control or patients fibroblasts, treated with bezafibrate (400 μM, 48 h; dark bars), or with vehicle only (open bars) and normalized to porin abundance. ***p < 0.001 compared with vehicle-treated fibroblasts
Discussion
The most common genetic defect affecting the mitochondrial trifunctional protein is the c1528G > C mutation in the HADHA gene, which results in LCHAD deficiency (Spiekerkoetter et al 2004; Sykut-Cegielska et al 2011). However, numerous other mutations affecting either the HADHA or the HADHB gene have now been described, leading to various forms of MTP deficiency (Boutron et al 2011). Many of these mutations have not been thoroughly characterized at the biochemical level, particularly as measurement of the three enzyme activities in patient cells is relatively complex, and since in vitro systems are difficult to use to analyze the effects of mutations, due to the hetero-multimeric state of the MTP. For these reasons, there is at the moment limited data on biochemical characterization of HADHA and HADHB deficiency in regards to the number of mutations and genotypes described. In order to address the functional consequences of various HADHA and HADHB mutations, we first performed parallel analysis of FAO capacities in 26 deficient cell lines, representing 16 different genotypes. In the absence of treatment with bezafibrate, the large majority of HADHA and HADHB fibroblasts exhibited extremely low (<20 % of control or lower) residual FAO capacities, indicating that most mutations have severe negative consequences on the use of long-chain fatty acids as energy substrates. In particular, the four cell lines homozygous for the c1528G > C mutation, which completely abolishes LCHAD activity, were extremely deficient in this assay system. Accordingly, in our HADHA or HADHB-deficient fibroblasts, the defects in long-chain fatty acid ß-oxidation enzymes isoforms were not compensated by the activity of medium- or short-chain-specific isoforms, as suggested in some previous studies (Olpin et al 2005). We, and others, previously showed that the FAO assay provides a sensitive tool to evaluate the functional consequences of ß-oxidation enzyme deficiency on palmitate oxidation in patient fibroblasts. These marked ß-oxidation flux deficiencies in MTP-deficient fibroblasts are in line with the severe clinical manifestations of HADHA and HADHB defects, for which few treatments exist to date.
Previous studies in patient fibroblasts with CPT2 or VLCAD-deficiencies (Djouadi and Bastin 2008), established that FAO rate provided a sensitive parameter to reveal pharmacological stimulation of mitochondrial ß-oxidation by bezafibrate, or by other molecules (Aires et al 2014). In these studies, kinetics and dose–response experiments showed that maximal stimulation of FAO was reached using 400 μM bezafibrate for 48 h (Djouadi et al 2005a, b). In the present study, we therefore raised the question whether this treatment by bezafibrate could induce a stimulation of palmitate oxidation in our panel of HADHA or HADHB-deficient patient fibroblasts. The results obtained show that bezafibrate significantly improved the FAO deficiency in six cell lines. Specifically, after exposure to bezafibrate, fibroblasts from patients 1, 6, 7, 8, and 19 reached FAO values equivalent to 60-80 % of normal value, and patient 5 was restored to control level. This reflected a stimulation of FAO capacities by bezafibrate varying from 1.3 to almost threefold in these cell lines. Interestingly, such pharmacological increases in FAO were observed in three cell lines (patient 6, 7, 8) heterozygous for the common c1528 G > C mutation.
There were, to our knowledge, no data on possible effects of fibrates or PPAR agonists on the expression of mitochondrial trifunctional protein in human cells. Our western blot studies show that, in control fibroblasts, both HADHA and HADHB protein levels were significantly up-regulated in response to bezafibrate. Additionally, both LCHAD and LCKAT enzyme activities were markedly induced by bezafibrate in control fibroblasts. Altogether, these data therefore indicate that mitochondrial trifunctional protein was up-regulated in bezafibrate-treated control fibroblasts, which accounted for the parallel increases in LCHAD and LCKAT enzyme activities, and for the resulting up-regulation of FAO capacities.
We then addressed the question of whether pharmacological inductions of MTP protein subunits and enzyme activities could also be detected in those HADHA-deficient fibroblasts that were responsive to bezafibrate in palmitate oxidation assays. Quantification of HADHA protein was not possible in whole fibroblasts, but analysis of mitochondria-enriched fractions revealed extremely low levels of α-subunit protein and reduced levels of β-subunit proteins, in the HADHA-deficient fibroblasts. As observed in control fibroblasts, both the alpha and β-subunit proteins appeared up-regulated in response to bezafibrate. However, in line with the protein misfolding effects of mutations, the amounts of mutated alpha subunit in mitochondria of treated patient cells remained much lower than that found in mitochondria from treated control fibroblasts. It was difficult to precisely evaluate the residual HADHA protein levels in patients’ cells and their changes in response to bezafibrate. Indeed, detection of mutant proteins by western blot is often difficult in patient cells with FAO deficiency. This is why we preferentially used the tritiated palmitate oxidation rate as primary endpoint to assess the effects of bezafibrate in MTP-deficient fibroblasts.
In parallel, our results indicated significant, but modest, increases in LCHAD residual enzyme activity in those patient cell lines that responded to bezafibrate in FAO assays. Regarding LCKAT activity, the effects of bezafibrate varied among the different patient cells. In c.1528G > C homozygous genotypes, induction of LCKAT was in the same order of magnitude as in control fibroblasts factors, whereas, in contrast, a complete absence of response was observed in other deficient cells, which remained extremely deficient for this thiolase activity. As mentioned above, MTP has three distinct catalytic functions, and the changes in enzyme activities associated to various genotypes of MTP deficiency are still poorly delineated. In line with this, further studies would be needed to precisely evaluate the effects of bezafibrate on LCHAD and LCKAT in MTP-deficient fibroblasts with various genotypes. Finally, analysis of acylcarnitines in bezafibrate-responsive patient cells showed that accumulation of long-chain hydroxyl-acylcarnitines was markedly reduced after exposure to bezafibrate, but was not restored to control levels. Taken together, these data suggest that, in bezafibrate responsive fibroblasts, drug-induced increases in mutant proteins’ levels and residual enzyme activities did occur, but were often not sufficient to fully restore the FAO flux, and hence to clear the accumulation of acylcarnitine species.
MTP is probably the more complex enzyme in the mitochondrial fatty acid ß-oxidation pathway, encoded by two genes, assembled into a hetero-multimeric structure, and finally embedded in the inner mitochondrial membrane. Very little is known about the factors and signaling pathways that might be involved in regulating MTP expression. The work of Aoyama et al, in mice, established that expression of both HADHA and HADHB genes was strongly inducible in vivo in wild-type animals by treatment with WY14643, a potent PPAR alpha agonist, which led to up-regulation of both MTP subunits proteins. These effects were entirely PPAR-dependent since they were fully abolished in PPAR alpha knockout mice (Aoyama et al 1998). In our study, large scale sequencing of gene transcripts (RNAseq; data not shown) established that HADHA and HADHB gene expression levels were induced by bezafibrate (1.3-fold and 1.6-fold respectively), and we also found that the alpha- and ß-subunit protein levels, as well as the LCHAD and LCKAT activities were robustly induced, in response to bezafibrate, in human fibroblasts. It should be noted that RNAseq studies (Brage S. Andresen, personal communication) and previous data (Djouadi et al 2005a, b), suggest that bezafibrate triggers a coordinate increase in gene expression of several other mitochondrial FAO enzymes. Likewise, this and previous studies indicate that, in control fibroblasts, pharmacological stimulation of long-chain FAO capacities by bezafibrate results from a coordinate up-regulation of multiple enzymes, including CPT2, involved in long-chain fatty acid import, as well as all the enzymes catalyzing the four steps of Lynen helix within the mitochondria, i.e., VLCAD and MTP.
In our study, however, treatment by bezafibrate often failed to improve FAO flux in HADHA- or HADHB-deficient fibroblasts, in contrast with similar experiments performed in CPT2- or VLCAD-deficient fibroblasts. This could be explained by several factors. First, it should be recalled that, in the case of CPT2 or VLCAD deficiency, a response to bezafibrate was specifically observed in fibroblasts from patients with the mild muscular form of the disease, but not in cells from patients with the severe phenotype (Djouadi et al 2003; Gobin-Limballe et al 2007). In both disorders, the positive pharmacological response was clearly ascribed to parallel increases in mutated gene mRNA, protein levels, and residual enzyme activity, resulting from the activation of PPAR nuclear receptors by bezafibrate (Djouadi and Bastin 2008). Consistent with this scheme, no response was observed in patients harboring severe CPT2 or VLCAD gene mutations, which led to a complete defect of the corresponding enzyme, and therefore remained extremely deficient after exposure to bezafibrate. Incidentally, this supports the notion that possible inductions by bezafibrate of other enzymes in the FAO pathway could not compensate for a complete deficiency at the CPT2 or VLCAD step. In the present study, we also found a number of cell lines that remained extremely deficient after treatment with bezafibrate. Accordingly, it can be thought that the HADHA and HADHB gene mutations in our panel of cells were generally severe (frameshift, non sense, splicing, etc.…), and incompatible with the production of mutant protein, hence incompatible with pharmacological stimulation. Furthermore, while a clear-cut distinction between mild and severe phenotypes exists in CPT2 and VLCAD deficiency, there is a continuum of decreasing severity from the severe neonatal MTP phenotype to the mildest MTP phenotype, and possible correlations between phenotype, genotype, and residual FAO capacities are not fully elucidated in this disorder.
Finally, some of the cell lines analyzed in the present study were functionally improved after treatment with bezafibrate. It appears unlikely that this effect was due to a compensatory stimulation of medium- or short-chain specific FAO enzymes, for several reasons. First, if this were the case, then stimulatory effects of bezafibrate would be found in all the HADHA- or HADHB-deficient treated cell lines, since medium-chain or short-chain specific FAO enzymes are normally operative in all the patients cells. Second, when bezafibrate treatment did induce FAO stimulation, it was also found to up-regulate the levels of mutant MTP alpha or MTP beta subunit, and it also tended to decrease the accumulation of long-chain hydroxyacylcarnitines, which are highly specific of MTP deficiency. Altogether, this clearly suggests that, when observed, the beneficial effects of bezafibrate on FAO capacities were primarily related to increases in residual MTP protein and enzyme activities.
Overall, the results obtained from control fibroblasts clearly identify both MTP alpha and beta subunits as possible targets of bezafibrate since, upon cell exposure to the drug, the protein abundance of each subunit, and the LCHAD and LKCAT activities, increased in parallel, and this was accompanied with a marked stimulation of FAO capacity. Nevertheless, the nature of several of the mutations in HADHA and HADHB appeared to result in little or no significant response to bezafibrate, most probably because these mutations abolished, or severely compromised, the production of mutant protein, as well as the expression of residual enzyme activity. As a result, positive effects of bezafibrate on FAO capacities were only observed in 23 % of cases, in our panel of patient cells. Finally, the present study should be considered as exploratory, and has obvious limitations. In particular, considering the genetic heterogeneity of the disease, additional studies will be needed to fully analyze the potential of bezafibrate as a function of genotype, or to really evaluate the proportion of response to bezafibrate among all the different MTP-deficient genotypes. Furthermore, the possible relations between the disease phenotypes and the response to bezafibrate would also need to be investigated.
To conclude, it is worth mentioning that very recent pharmacological therapies developed for Duchenne myopathy (Haas et al 2015) or cystic fibrosis (Massie et al 2014) are based on the restoration of low levels of the mutated protein function, and are effective only in a restricted number of disease genotypes (Perez et al 2012). Thus, although not necessarily applying to a majority of patients, the search of mutation-specific therapies can, in some cases, represent a valuable approach to progress in the treatment of rare genetic disorders.
References
Aires V, Delmas D, Le Bachelier C et al (2014) Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts. Orphanet J Rare Dis 9:79
Aoyama T, Peters JM, Iritani N et al (1998) Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 273:5678–5684
Boutron A, Acquaviva C, Vianey-Saban C et al (2011) Comprehensive cDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a French cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab 103:341–348
Choi JH, Yoon HR, Kim GH, Park SJ, Shin YL, Yoo HW (2007) Identification of novel mutations of the HADHA and HADHB genes in patients with mitochondrial trifunctional protein deficiency. Int J Mol Med 19:81–87
Das AM, Illsinger S, Lucke T et al (2006) Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem 52:530–534
Djouadi F, Bastin J (2008) PPARs as therapeutic targets for correction of inborn mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis 31:217–225
Djouadi F, Bonnefont JP, Thuillier L et al (2003) Correction of fatty acid oxidation in carnitine palmitoyl transferase 2-deficient cultured skin fibroblasts by bezafibrate. Pediatr Res 54:446–451
Djouadi F, Aubey F, Schlemmer D, Bastin J (2005a) Peroxisome proliferator activated receptor {delta} (PPAR{delta}) agonist but Not PPAR{alpha} corrects carnitine palmitoyl transferase 2 deficiency in human muscle cells. J Clin Endocrinol Metab 90:1791–1797
Djouadi F, Aubey F, Schlemmer D et al (2005b) Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet 14:2695–2703
Eaton S, Bursby T, Middleton B et al (2000) The mitochondrial trifunctional protein: centre of a beta-oxidation metabolon? Biochem Soc Trans 28:177–182
Fletcher AL, Pennesi ME, Harding CO, Weleber RG, Gillingham MB (2012) Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol Genet Metab 106:18–24
Gillingham MB, Connor WE, Matern D et al (2003) Optimal dietary therapy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 79:114–123
Gobin-Limballe S, Djouadi F, Aubey F et al (2007) Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet 81:1133–1143
Haas M, Vlcek V, Balabanov P et al (2015) European medicines agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord 25:5–13
IJlst L, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ (1996) Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest 98:1028–1033
Manning NJ, Olpin SE, Pollitt RJ, Webley J (1990) A comparison of [9,10-3H]palmitic and [9,10-3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inherit Metab Dis 13:58–68
Massie J, Castellani C, Grody WW (2014) Carrier screening for cystic fibrosis in the new era of medications that restore CFTR function. Lancet 383:923–925
Olpin SE (2013) Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J Inherit Metab Dis 36:645–658
Olpin SE, Clark S, Andresen BS et al (2005) Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J Inherit Metab Dis 28:533–544
Orii KE, Orii KO, Souri M et al (1999) Genes for the human mitochondrial trifunctional protein alpha- and beta-subunits are divergently transcribed from a common promoter region. J Biol Chem 274:8077–8084
Perez B, Rodriguez-Pombo P, Ugarte M, Desviat LR (2012) Readthrough strategies for therapeutic suppression of nonsense mutations in inherited metabolic disease. Mol Syndromol 3:230–236
Purevsuren J, Fukao T, Hasegawa Y, Fukuda S, Kobayashi H, Yamaguchi S (2008) Study of deep intronic sequence exonization in a Japanese neonate with a mitochondrial trifunctional protein deficiency. Mol Genet Metab 95:46–51
Sperk A, Mueller M, Spiekerkoetter U (2010) Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol Genet Metab 101:205–207
Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW (2004) General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res 55:190–196
Sykut-Cegielska J, Gradowska W, Piekutowska-Abramczuk D et al (2011) Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J Inherit Metab Dis 34:185–195
Wanders RJ, Ruiter JP, IJLst L, Waterham HR, Houten SM (2010) The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis 33:479–494
Acknowledgments
This study was supported by grants of the Association Française contre les Myopathies (AFM).
Compliance with ethics guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Jörn Oliver Sass
Rights and permissions
About this article

Cite this article
Djouadi, F., Habarou, F., Le Bachelier, C. et al. Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: effects of bezafibrate. J Inherit Metab Dis 39, 47–58 (2016). https://doi.org/10.1007/s10545-015-9871-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9871-3