
Abstract
Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) is a fatty acid oxidation disorder with especially high mortality and uncertain long-term outcome. The aim of the study was to analyze the influence of diagnostic approach on survival in 59 affected children. Referral to a metabolic center was replaced over time by urine/blood testing in centralized metabolic laboratory (selective screening) and by pilot tandem mass spectrometry newborn screening (NBS). Molecular analysis revealed the prevalent mutation in the HADHA gene in all 58 examined cases. Twenty patients died. The number of detections and number of deaths were respectively 9 and 4 (44%) in the patients recognized by differential diagnosis, 28 and 9 (32%) - by selective screening, and 11 and 1 (9%) - by NBS. In 80% of cases the death occurred before or within 3 weeks from the identification. Urgent and active metabolic service remarkably influenced the surviving. The current age of 39 survivors is 0.5 to 23 yrs (mean 7.2 yrs). The disease frequency estimated on the patients number was 1: 115 450, whereas in the pilot NBS - 1: 109 750 (658 492 neonates tested). Interestingly, the phenylalanine level in asymptomatic neonates frequently exceeded the cut-off values. Conclusions: 1) Urgent metabolic intervention decreases mortality of LCHAD-deficient patients, but the prognosis is still uncertain. 2) Emergent metabolic reporting and service are crucial also for the survival of neonates detected by NBS. 3) The nationwide selective screening appeared efficient in LCHADD detection in the country. 4) Transient mild hyperphenylalaninaemia may occur in LCHAD-deficient newborns.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (OMIM# 609016) is a very rare fatty acid beta-oxidation (FAO) disorder (Roe and Coates 1995). Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD, EC 1.1.1.211) is one of the three enzymes contained in the mitochondrial trifunctional protein (MTP), which catalyzes the third step of the long-chain fatty acid beta-oxidation. The two remaining MTP enzymes, long-chain enoyl-CoA hydratase (LCEH) and long-chain ketoacyl-CoA thiolase (LCKAT), catalyze the last steps of the beta-oxidation spiral and they are located in the N-terminus of the α subunit (HADHA gene) and in the β subunit (HADHB gene), respectively. The domain with LCHAD activity is localized in the C-terminus of the α subunit of MTP and is encoded by the HADHA gene (2p23). Depending on the disturbance of particular enzyme activity that is affected, three categories of MTP deficiency are distinguished: isolated LCHAD deficiency (the most frequent), complete MTP deficiency (the less common) and isolated LCKAT deficiency (recently identified) (Das et al. 2006).
Isolated LCHAD deficiency (LCHADD) is associated with the common HADHA gene mutation, c.1528 G > C [p.E474Q], that is located in the catalytic site of the LCHAD domain. In the majority of LCHAD deficient patients the prevalent substitution is detected at least at one allele. The frequency of this mutation is high and ranges from 71% (Ibdah et al. 1999) to 87% (Ijlst et al. 1996).
Already in early infancy the affected patients usually show rapid clinical progression with liver failure, coma triggered by fasting or catabolic status and frequently sudden death. Detection of the disease requires efficient differential diagnosis based on the history (also family history) and clinical picture, which should suggest the need for the further diagnostics. Assessment of acylcarnitine profile by tandem mass spectrometry (TMS) is a method of choice. In the literature the reported patients presented with particularly high mortality and poor long-term outcome (Sewell et al. 1994; Tyni et al. 1997a; den Boer et al. 2002; Olpin et al. 2005). Since the application of TMS into expanded newborn screening (NBS) the detection of LCHADD has increased. According to the American College of Medical Genetics Report 2006, in the recommended uniform panel for newborn screening programs, 3-hydroxypalmitoylcarnitine (C16-OH) and/or 3-hydroxyoleoylcarnitine (C18:1-OH) are primary biomarkers for both disorders: LCHAD and MTP deficiencies (Sweetman et al. 2006). It has not been proven yet that early detection of the defect significantly improves the prognosis.
To date several cases with diagnosis of LCHADD have been described in Poland (Pronicka et al. 1998; Sykut-Cegielska 2006). The aim of the study was to analyze the influence of the diagnostic approach mode on the detection rate and the risk of death in the group of LCHAD deficient patients identified during the period of 1992-2009.
Patients
A cohort of 59 patients (30 boys and 29 girls) coming from 55 families with LCHADD was enrolled in this study. Two affected patients from two pairs of twins (one pair from in vitro fertilization) were included. The diagnosis was established at the Children’s Memorial Health Institute (CMHI) and in the Institute of Mother and Child (IMC), both located in Warsaw, with contribution of molecular study in Aarhus University Hospital, Skejby.
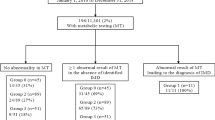
The study group included 44 patients diagnosed by metabolic testing after the disease symptoms appeared (group A), and 15 affected neonates identified presymptomatically (group B).
In the symptomatic group, there were four different detection modes specified in the study as follows (Table 1 and Table 2):
A1. The patients transferred from the whole country for a metabolic work-up at the metabolic center (CMHI) serving specialized diagnostics (differential diagnosis group, 9 patients).
A2. Urine and dry blood spot samples submitted to CMHI with a short clinical description for a metabolic testing. A newly detected patient is immediately and continuously followed-up by a CMHI pediatrician experienced in metabolic medicine, who recommends further metabolic management (urgent metabolic service group, 17 patients).
A3. Acylcarnitine profile determination by TMS method available from the IMC on request since 2001. The results reported by the laboratory to the referring hospital, without involvement of a metabolic expert (laboratory testing group, 11 patients).
A4. Post mortem identification of LCHADD by histological suspicion of FAO defects at autopsy, followed by detection of mutations in the HADHA gene (post mortem group, 4 patients).
A5. Diagnoses established abroad, with unavailable data on detection mode (3 patients).
Presymptomatic group consists of the following three subgroups:
B1. Siblings from the families at risk diagnosed by TMS at neonatal period (family at risk group, 4 patients).
B2. Neonates detected by TMS pilot screening (NBS group, 6 patients)
B3. Additional LCHAD-deficient neonates detected by chance. All routine PKU samples with borderline or higher than cut-off Phe value were obligatory verified by TMS in the central NBS laboratory (IMC) (Phe group, 5 patients).
Incidence of the disease was assessed taking into account the number of detected unrelated patients and the number of births in the relevant time. This incidence was roughly evaluated for symptomatic patients and those, diagnosed by the pilot TMS newborn screening, which covered 20% of population; from four voivodships from the eastern part of the country, while the highest carrier frequency has been noted in another northern part of Poland (Piekutowska-Abramczuk et al. 2008).
The study was approved by the Bioethics Commission of the Children’s Memorial Health Institute. The clinical characteristics of 41 patients from this cohort was included in an earlier multi-center report summarizing 55 cases from 7 European metabolic centers (Spiekerkoetter and Sykut-Cegielska 2007).
Methods
Organic acid analysis in urine by GC-MS method
The analysis was performed using a method based on one described by Chalmers and Lawson (1975) using GC-MS equipment (Fisons Instruments) and a non-polar capillary column type OV1. Trimethylsilyl- and methoxy-derivatives were analyzed. The analytical procedure is under the quality control of the ERNDIM (European Research Network for Inherited Disorders of Metabolism) qualitative organic acids in urine scheme. Increased 3-hydroxydicarboxylic C6-C10 aciduria (3-OHDCA, 3-hydroxyderivatives of adipic, suberic, sebacic acids) with or without low ketonuria (3-hydroxybutyric acid, acetoacetic acid) was interpreted as a profile indicating suspicion of LCHADD.
Acylcarnitine analysis in dry blood spot on paper by tandem MS method
Blood samples were collected on Schleicher and later on Whatman 903 papers, after 48 h of life (mainly 48 – 120 hrs) in NBS and by random sampling in older patients. Tandem MS was performed in API 2000 (Sciex Applie Biosystems). The analysis procedure is under the quality control of CDC (Centers for Disease Control and Prevention). Laboratory cut-off values for C16-OH acylcarnitines were established at 0.22 micromol/l (for newborns) and 0.33 (after the first month of life), and for C18:1-OH acylcarnitines - at 0.15 and 0.18, respectively. Laboratory cut-off value for phenylalanine in NBS was 150 micromol/l.
Molecular analysis
Total DNA of whole blood leukocytes, dry blood spots, cultured fibroblasts or muscle biopsies was isolated by standard proteinase K digestion and phenol/chloroform extraction.
From 1998 to 2003 the molecular analysis was based on the PCR-RFLP method, described by Ijlst et al. (1996). Since 2003 direct sequencing of PCR products was used. Amplification of the coding sequence of the HADHA gene was always started from exon 15, where the common mutation is localized. When necessary, all remaining exons and intron-exon boundaries of the HADHA and HADHB genes were amplified using specific oligonucleotide primers (sequences available on request). DNA samples of both parents were also examined, if available, to confirm the segregation of identified mutations. The control panel included 50 unrelated healthy subjects from a geographically representative (Polish) population group.
Enzyme activity assays
Enzymatic assays for MTP components were performed abroad by the courtesy of Lyon Hopital Debrousse (C. Vianey-Saban) and in Amsterdam Academic Medical Center (R. Wanders).
Results
The patients’ birth year, gender, age at onset, age at diagnosis, age at death and genotype are summarized in the Table 1. Among 59 patients with detected LCHADD 39 are alive (66%) with ages ranging from 6 months to 23 years and 10 months (mean 7 years and 3 months). Initial clinical symptoms usually appeared in the first year of life, but later in some patients. In the oldest patient (patient 1) clinical onset occurred at the time of diagnosis, at the age of 18 years. Seven patients reached an age above 15 years.
Twenty patients (34%) died at the age of 4 days to 10 years (mean 1 year 10 months, median 6 months). The number of deaths differed remarkably depending on an availability of the diagnostic and therapeutic expertise and the emergency approach, presented by the mode of LCHADD detection (Table 2). In the symptomatic group there were 4 out of 9 deaths ( 44%) among the patients diagnosed at the metabolic ward or out-patient service, and with remarkably delayed appropriate management. Remaining 28 cases were identified by the selective screening (GC-MS and/or TMS). Among them, in the cases when urgent proper metabolic expert supervision was provided, four patients died out of 17 (24%), but in the cases without metabolic expertise included in the laboratory data, five patients died out of 11 (46%).
In the presymptomatic detection group only one out of 11 (9%) died among the cases detected in the NBS pilot study, and none of four siblings detected by metabolic testing of the families at risk (experienced by having or loss of the older affected proband).
Table 3 shows the summary of the deceased patients. The deaths were most frequently unexpected, after a short period of clinical deterioration; as sudden infant death syndrome (patient 46), before admission to the hospital (patient 48), but also during proper monitoring of disease treatment (patient 13). They occurred in local hospitals, academic hospitals, and our referral metabolic center, as well. In 80% of the patients the death occurred before or within 3 weeks from the LCHADD diagnosis, especially in the patients with limited approach to the active metabolic expertise, but regardless of the detection procedure. In seven patients data about death cause were unavailable. Death cause as sudden cardiac arrest was specified in six cases, as acute severe liver failure in four cases and as multiorgan insufficiency in three cases.
The results of laboratory methods applied for the LCHADD detection are summarized in Table 4. False negative results were obtained in some patients at the initial testing by TMS and by GC-MS, as well. In as many as 15 out of 51 analyses the free carnitine concentration was very low; below 7 μmol/l, indicating initially a primary carnitine deficiency.
A mildly increased or borderline phenylalanine level was observed in five LCHAD-deficient neonates. They presented with abnormal results of the routine NBS and were identified by chance, when re-investigated by TMS method (Table 2, group B3).
Molecular analysis was performed in all LCHAD-deficient patients, except one (inaccessible biological material). The presence of the common c.1528 G > C mutation was demonstrated in 103 out of 113 alleles tested (overall allele frequency: 91%). Forty-five homozygotes and thirteen heterozygotes were identified in the studied group (Table 1). DNA analysis of the further HADHA gene fragments revealed five novel mutations (one in-frame deletion, three missense mutations, and one splice-site variant) in seven patients from six families. None of them was present in control 100 alleles studied. In three probands only one of two mutant alleles was found, despite analysis comprising all HADHA and HADHB exons. In the remaining three children screening for a second mutation has not been conducted yet. Moreover, eight prenatal diagnoses in seven families were performed (all based on DNA analysis but one – on enzymatic method) with detection of healthy fetuses in all investigations.
On the basis of the number of 29 detected symptomatic and molecularly confirmed LCHAD-deficient patients in 2001-2009 period and the number of total live births in Poland in the same time (3 348 000), the incidence of LCHADD was approximately 1: 115 450. During the same period six patients were detected by the pilot tandem MS study (group B2) among 658 492 examined newborns, so the estimated prevalence of the disease from this evaluation was approximately 1: 109 750.
The comparison of the number of identified patients, who were born since 1986 with the number of expected cases born in subsequent years (estimated on the basis of mean incidence calculated from the pilot TMS study) is shown in Fig. 1. It may be assumed that several dozen LCHAD-deficient infants may have died without diagnosis in this period of time, including older sibs of the patients reported in this study (family history revealed at least 12 sibs’ deaths). On the other hand, since year 2000 when NBS was initiated, the number of patients diagnosed with LCHADD exceeds the expected number.

The number of diagnosed LCHAD deficiency patients, according to their age birth (dark diagram) versus the expected number of affected cases born in subsequent years (light diagram)
Discussion
The LCHADD has been known since 1989, and is reported as a severe, unforeseen disorder, frequently lethal. The patients often die immediately after clinical onset, sometimes too quickly to establish a final diagnosis, not to mention instituting proper therapy. Mortality of up to 50% was reported in some series (Sewell et al. 1994; Tyni et al. 1997b) and usually is about 30% of symptomatic patients (den Boer et al. 2002; Spiekerkoetter et al. 2009). The deaths of LCHAD-deficient patients were described in the majority of case reports (Dionisi-Vici et al. 1990; Duran et al. 1991; Jackson et al. 1992; Isaacs et al. 1996; Van Maldergem et al. 2000; Hintz et al. 2002) including first descriptions (Wanders et al. 1990; Wanders et al. 1992; Roe and Coates 1995) and even in newborns identified presymptomatically by NBS (Wilcken et al. 2003; Wilcken et al. 2009).
This study included the large cohort of 59 LCHAD-deficient patients diagnosed and followed in one metabolic center. The majority of 20 deaths took place before or at the time of diagnosis and the start of proper treatment. In our material, a lower number of deaths was observed when metabolic expert, after information about LCHADD detection from the lab, immediately made a call to a referring doctor, telling about the diagnosis and suggestions for proper management. Laboratory report without such metabolic expert’s contribution was insufficient in our experience, probably due to a lack of knowledge of the risk among referring doctors in the country. Percentage of the deaths in the above situations differed, and were respectively 24% and 46%.
The first hours of the disease rather than days, are critical for the efficacy of the entire detection process (and prognosis) starting from clinical suspicion at the local hospital, through sample taking and sending, metabolic work-up, information transfer, to implementation of systematic metabolic management (treatment and monitoring). In order not to miss any affected patients, 24-hour alert of the local doctors in charge and contact with the metabolism specialists (emergency services) are needed. The availability of metabolic consultation by phone is of great importance. The emergency approach (prompt analysis and reporting) is also necessary in the NBS labs, when an abnormal acylcarnitine profile typical for LCHADD is found. Most of the affected neonates detected by NBS are asymptomatic, but sometimes clinical onset is sudden, leading to early death. In such situations only immediate information transfer about a suspected diagnosis prevents catastrophic consequences. One out of six affected neonates identified by the pilot screening in this study had died before the already known result was reported.
For short-term prognosis the urgent metabolic service at first clinical symptoms (or presymptomatically) is even more important than following specific therapeutic options. In our experience a presence of unconsciousness or liver failure at the start of proper treatment may not influence poor outcome.
Differential clinical diagnosis is difficult and requires marked experience in the field. Unspecific and frequent 3-hydroxydicarboxylic C6-C10 aciduria without ketonuria may be important in initial suspicion, especially in the context of the clinical picture. Acylcarnitine analysis remains the investigation of choice, when LCHADD is suspected but it should be remembered then that the specific biomarkers may be in upper normal reference values, and so may be overlooked. Enzymatic assays in skin fibroblasts should be performed only in c.1528 G > C heterozygotes for a diagnosis confirmation.
In our experience, acylcarnitine analysis for LCHADD should be recommended for those neonates, in whom the phenylalanine level in NBS for PKU, are increased. TMS showed a transient mild increase of phenylalanine level (above cut-off value) in 45% of asymptomatic LCHAD-deficient newborns. At least three similar notes are found in the literature (Hagenfeldt et al. 1990; Sewell et al. 1994; Frazier et al. 2006). This phenomenon needs further considerations.
To date, 29 different mutations in the HADHA gene have been reported; we identified 5 novel molecular variants in Polish patients. Despite direct sequencing of all HADHA and HADHB gene exons, only one causative mutation was detected in three children. Similar findings have been reported earlier (Olpin et al. 2005; Sander et al. 2005) suggesting the existence of rare intronic variants, potentially vital for protein function. The frequency of the common substitution was very high in our study (91% alleles tested) in comparison with other reports (Ijlst et al. 1996; Ibdah et al. 1999), reflecting potential population differences in the mutation spectrum. LCHADD seems extremely rare in Australia and Northern America (Zytkovicz et al. 2001; Wilcken et al. 2003; Frazier et al. 2006; Wilcken et al. 2009) with estimated frequencies of 1:193 430 and 1:314 700 births, respectively. Some data indicate that the condition is more frequent in Europe, especially around the Baltic Sea (Hagenfeldt et al. 1995; Tyni and Pihko 1999; den Boer et al. 2000; Klose et al. 2002). The later study shows that Poland also belongs to the regions of relatively high prevalence of LCHADD. Interestingly, the current results reveal an increased with time number of diagnosed symptomatic patients, in some years even exceeding the incidence estimated in the pilot TMS study. We speculate that this estimation may be lowered by the fact, that the pilot study did not include the Polish region with the highest disease frequency. Further studies are in progress to explain this discrepancy (Piekutowska-Abramczuk et al. 2008).
In summary, our results indicate that the efficient detection of symptomatic patients with LCHAD deficiency may be achieved through a specific, multi-specialty and systematic approach, including fast and directed management. Availability of quick and professional metabolic advice is of great importance. Moreover, the continuous care of all affected families identified in one metabolic center gives a special opportunity to perform reliable assessment of the follow-up. Also Wilcken and coauthors stress the advantages of a centralized metabolic service (Wilcken et al. 2009). Nevertheless, even strict patient’s monitoring may not prevent clinical deterioration. Asymptomatic children identified by NBS also require immediate further diagnostics and vigilant care, because they may develop sudden fatal clinical symptoms. The main diagnostic analysis for LCHAD deficiency is the acylcarnitine profile, but special attention should be paid to samples with low free carnitine concentration and borderline values of diagnostic biomarkers. Transient mild hyperphenylalaninemia detected in NBS requires determination of the acylcarnitine profile in each case in order to exclude LCHADD.
The study results confirm that LCHADD is a relatively frequent condition in Poland and that early symptomatic identification and pilot newborn screening have improved detection of the affected patients and positively influenced risk of mortality.
References
Chalmers RA, Lawson AM (1975) Human metabolic diseases. Chem Brit 11:290–295
Das AM, Illsinger S, Lücke T et al. (2006) Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem 52:530–534
den Boer ME, Ijlst L, Wijburg FA et al. (2000) Heterozygosity for the common LCHAD mutation (1528 G > C) is not a major cause of HELLP syndrome and the prevalence of the mutation in the Dutch population is low. Pediatr Res 48:151–154
den Boer ME, Wanders RJ, Morris AA, IJlst L, Heymans HS, Wijburg FA (2002) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109:99–104
Dionisi-Vici C, Bertini E, Burlina A et al. (1990) Neuromuscular involvement in two unrelated children with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Pediatr Res 28:305
Duran M, Wanders RJA, deJager JP et al. (1991) 3-hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: protective effect of medium-chain trigliceride treatment. Eur J Pediatr 150:190–195
Frazier DM, Millington DS, McCandles SM et al. (2006) The tandem mass spectrometry newborn screening experience in North Carolina: 1997-2007. J Inherit Metab Dis 29:76–85
Hagenfeldt L, Venizelos N, von Dobeln U (1995) Clinical and biochemical presentation of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 18:245–248
Hagenfeldt L, von Dobeln U, Holme E et al. (1990) 3-Hydroxydicarboxilic aciduria – a fatty acid oxidation defect with severe prognosis. J Pediatr 116:387–392
Hintz SR, Matern D, Strauss A et al. (2002) Early neonatal diagnosis of long-chain 3-hydroxyacyl coenzyme a dehydrogenase and mitochondrial trifunctional protein deficiencies. Mol Genet Metab 75:120–127
Ibdah JA, Dasouki MJ, Strauss AW (1999) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: variable expressivity of maternal illness during pregnancy and unusual presentation with infantile cholestasis and hypocalcaemia. J Inherit Metab Dis 22:811–814
Ijlst L, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ (1996) Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest 98:1028–1033
Isaacs JD Jr, Sims HF, Powell CK et al. (1996) Maternal acute fatty liver of pregnancy associated with fetal trifunctional protein deficiency: molecular characterization of a novel maternal mutant allele. Pediatr Res 40:393–398
Jackson S, Singh-Kler R, Bartlett K (1992) Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest 90:1219–1225
Klose DA, Kolker S, Heinrich B et al. (2002) Incidence and short-term outcome of children with symptomatic presentation of organic acid and fatty acid oxidation disorders in Germany. Pediatrics 110:1204–1211
Olpin SE, Clark S, Andresen BS et al. (2005) Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J Inherit Metab Dis 28:533–544
Piekutowska-Abramczuk D, Olsen RKJ, Wierzba J et al. (2008) High frequency of LCHAD deficiency carriers in the northern Poland. Eur J Hum Genet 16(suppl 2):381
Pronicka E, Vianey-Saban Ch, Roe ChR et al. (1998) Clinical, biochemical, and molecular characteristics of five Polish patients with long chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. J Inherit Metab Dis 21(suppl 2):66
Roe CR, Coates PM (1995) Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) Metabolic and molecular basis of inherited disease. McGraw-Hill, New York, pp 1501–1533
Sander J, Sander S, Steuerwald U et al. (2005) Neonatal screening for defects of the mitochondrial trifunctional protein. Mol Genet Metab 85:108–114
Sewell AC, Bender SW, Wirth S, Munterfering H, Ijlist L, Wanders RJA (1994) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a severe fatty acid oxidation disorder. Eur J Pediatr 153:745–750
Spiekerkoetter U, Lindner M, Santer R et al. (2009) Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis 32:488–497
Spiekerkoetter U, Sykut-Cegielska J (2007) Prognosis and treatment of LCHAD deficiency. Workshop of European Metabolic Group, Milupa Foundation, Warsaw 2-3 June, pp. 16-19
Sweetman L, Millington DS, Therrell BL et al. (2006) Naming and counting disorders (conditions) included in newborn screening panels. Pediatrics 117:S308–S314
Sykut-Cegielska J (2006) Mitochondrialne zaburzenia utleniania kwasów tłuszczowych. Rozprawa habilitacyjna, IPCZD Warszawa : 11-163
Tyni T, Palotie A, Viinikka L et al. (1997a) Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528G mutation: Clinical presentation of thirteen patients. J Pediatr 130:67–76
Tyni T, Rapola J, Peatau A, Palotie A, Pihko H (1997b) Pathology of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by G1528C mutation. Pediatr Pathol Lab Med 17:427–447
Tyni T, Pihko H (1999) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr 88:237–245
Van Maldergem L, Tuerlinckx D, Wanders RJ et al. (2000) Long chain 3-hydroxy-CoA dehydrogenase deficiency and early-onset liver cirrhosis in two siblings. Eur J Pediatr 159:108–112
Wanders RJA, Ijlst L, Poggi F et al. (1992) Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid oxidation. Biochem Biophys Res Commun 188:1139–1145
Wanders RJ, Ijlst L, van Gennip A et al. (1990) Long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid oxidation. J Inherit Metab Dis 13:311–314
Wilcken B, Haas M, Joy P et al. (2009) Expanded newborn screening; Outcome in screened and unscreened patients at age of 6 years. Pediatrics 124:e241–e248
Wilcken B, Wiley V, Hammond J, Carpenter K (2003) Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med 348:2304–2312
Zytkovicz TH, Fitzgerald EF, Marsden D et al. (2001) Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from New England Newborn Screening Program. Clin Chem 47:1945–1955
Acknowledgements
The authors are very grateful to Orly Elpeleg, Charles Roe, Priscille Divry, Christine Vianey-Saban, Peter Vreken, Hans Waterham, Ronald Wanders and others who had contributed in developing LCHADD detection in Poland. Many thanks to the SSIEM for creating a friendly European metabolic platform to meet, learn and collaborate. European Metabolic Group contribution is also appreciated. The authors thank Beata Gruszczyńska, Tomasz Kurył, Monika Pohorecka and Ewa Twardo from the CMHI for their participation in the work.
The work was supported by State Committee for Scientific Research grants No PB 0709/P05/99/17, PB0314/S4/94/07 and PB0678/B/P01/2007/33.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ronald J.A. Wanders
Jolanta Sykut-Cegielska, Wanda Gradowska and Dorota Piekutowska-Abramczuk contributed equally
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Sykut-Cegielska, J., Gradowska, W., Piekutowska-Abramczuk, D. et al. Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J Inherit Metab Dis 34, 185–195 (2011). https://doi.org/10.1007/s10545-010-9244-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9244-x