
Abstract
The technical advances in mass spectrometry, particularly the development of (ultra)-high-resolution/mass accuracy measurement capabilities in combination with refinement of soft ionization techniques, have increased the application and success of lipidomics to answer biological questions in relation to lipid metabolism. Together with other omics technologies, lipidomics has become an important tool to practice systems biology as lipids comprise a very significant part of the metabolome and play pleiotropic roles in cellular functions. As an increasing number of disorders are linked to lipid metabolism, lipidomics is used to search for biomarkers, understand disease mechanism and follow the efficacy of therapeutic options. This review provides a first introduction to the major methodological strategies currently used for mass spectrometry-based lipidomics and associated data pre-processing and analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids are highly diverse molecules which are traditionally best known for their role in the formation of biological membranes in cellular systems and as a way to store energy. In the last decade, lipids have outgrown this rather dull image and have taken center stage in apoptosis, cell signaling, inflammation, immunity and, last but not least, inborn errors of metabolism. Lipids are sometimes simply defined as molecules that are insoluble in water and soluble in organic solvents. As this is not true for all lipids, a more generally accepted definition has been described by the International Lipid Classification and Nomenclature Committee of lipids: “hydrophobic or amphipathic small molecules that may originate entirely or in part by carbanion-based condensation of thioester (fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, and polyketides) and/or by carbocation-based condensations of isoprene units (prenol lipids and sterol lipids)”. This designation describes eight main categories that are subdivided based on their chemical properties. This classification also has been used by the LIPID Metabolites And Pathways Strategy (LIPID MAPS) consortium, a multi-institutional effort to identify and quantitate lipid species in mammalian cells (Fahy et al 2009). This classification system has been widely accepted and the LIPID MAPS database contains more than 37,500 unique structures for biologically relevant lipids from mammals, plants, bacteria, fungi, algae, and marine organisms ((Fahy et al 2009), Table 1).
Lipids were traditionally analyzed by thin-layer chromatography, gas chromatography, and mass spectrometry. Technical advances in mass spectrometry have paved the way for the realization of a new type of metabolomics: lipidomics. Lipidomics aims to study the pathways and networks of cellular lipids by characterization and quantitation of all lipids present in a biological system. Especially the development of “soft” ionization techniques as electrospray ionization (ESI) and exact mass resolution, high resolution mass spectrometers have greatly propelled the field of lipidomics. In addition, new bioinformatics tools have been developed to cope with the increasing amounts of raw data and extract relevant information to yield biological insight. The parallel rise of next-generation sequencing and concomitant identification of new inborn errors of metabolism in genes encoding lipid modifying enzymes has brought about the desire to fully characterize the lipidome, further boosting the development and refinement of lipidomic techniques. Lipidomics is being applied to find biomarkers suitable for diagnosis, follow-up and ideally prognosis in order to characterize the course of the patient’s disorder. In addition to linking new monogenetic inborn errors of lipid metabolism there is a growing number of links being uncovered between lipid metabolism and complex genetic traits as obesity, diabetes, atherosclerosis,and cancer (Hyotylainen and Oresic 2014). The corresponding research communities increasingly use lipidomics underscoring the broad application of this technique for research in general.
This paper provides a first introduction to the major methodological strategies currently used for lipidomics, and by no means attempts to provide a complete and in-depth overview. Instead, we frequently refer the reader to excellent reviews for further reading. We focus on lipidomic techniques that employ mass spectrometry and the pre-analytical/analytical phase but will also discuss the data (pre)-processing and analysis, which has become an important part of lipidomics workflow. The latter emphasizes that metabolomics in general is becoming a multidisciplinary field that requires input and knowledge from many specialists to succeed, including physicians, technicians, analytical chemists, bioinformaticians, and clinical biochemists.
Experimental design
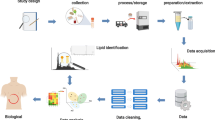
The lipidomics experiment comprises of several steps which are depicted in Fig. 1. After defining the biological question, the experiment is carefully formulated into a protocol which 1. takes into account the statistical considerations, 2. describes the collection of samples and 3. describes sample preparation and data acquisition (the actual measurement). In the post-analytical phase, bioinformatics approaches are used for data visualization, data pre-processing to translate the raw data into a list of detected and quantified peaks, (multivariate) statistical analysis for the comparison of samples, and various types of downstream analysis depending on the specific biological question.

Lipidomics workflow. The different steps in the lipidomics experiment
Like any metabolomics experiment, a lipidomics experiment must be meticulously designed from beginning to end in order to avoid pitfalls and to ensure that the required information can be obtained from the data. Many factors before, during, and after the actual analysis can greatly (and negatively!) influence the outcome of the experiment. Typically, experimental design involves decisions about the number of samples, group size (i.e., technical and biological replicates), and quality control samples (Box et al 2005). A main requirement for experimental design is its ability to account for unwanted effects (experimental bias) such as contamination of the chromatographic column during a study. Randomization and blocking of samples are two vehicles toward this requirement. In addition, choices concerning sample collection (timing, procedure, processing, storage), sample preparation (extraction, derivatization), and analytical method (method/reagents, internal standards, type of instrument) greatly influence the end result and require careful consideration. In the post-analytical bioinformatics phase data pre-processing (peak detection and integration, isotope correction, normalization, compound identification) and (statistical) data analysis are key steps that need to be meticulously prepared and controlled.
Conducting a power analysis on metabolomics data is difficult, since in general, the effect size and variance of metabolites across samples are unknown a priori and depend on differences in metabolite concentrations between experimental groups (e.g., control versus diseased) as well as biological and technical variation of the samples within a group. Sometimes this information can be obtained from previous similar studies or from conducting a small pilot study before investing time and effort into a large scale study. For longitudinal studies or studies with a large number of samples, it is prudent to include a number of Quality Control (QC) samples in the study design, i.e., samples of a consistent composition which are included in the measurements repeatedly and which allow for the correction of run to run effects (Hendriks et al 2011). Depending on the study objective, other experimental design issues may need consideration. For example, to determine fluxes in metabolic pathways (e.g., flux balance analysis (FBA)) one should ensure that the measured metabolites sufficiently constrain the putative fluxes (Orth et al 2010).
Sample preparation/workup
Extraction of lipids from the sample is the first step toward their isolation, enrichment, and concentration but also serves to remove salts and proteins, which interfere with separation techniques such as liquid chromatography and subsequent mass spectrometric analysis. Liquid-liquid extraction is mostly used but solid phase extraction is also employed. Lipid liquid-liquid extraction usually makes use of a two-phase system (organic/aqueous) where lipids partition into the organic phase whereas water soluble molecules remain in the aqueous phase, and proteins are precipitated. Two established methods have been described long ago by Folch et al (Folch et al 1957) and Bligh and Dyer (Bligh and Dyer 1959) and these procedures are still widely used, but alternatives have been developed and compared to these established standards (Reis et al 2013). For example, the use of methyl-tert-butyl ether is gaining in popularity (Matyash et al 2008; Abbott et al 2013) as it has a lower density than water (making it the upper layer in contrast to chloroform-based procedures) which makes it more suitable for high-throughput applications and automation. Given the high level of complexity and physicochemical diversity of lipid molecules it is as yet impossible to extract all lipid species using a single extraction procedure. This makes the measurement of the complete lipidome in a single analysis, which of course is the Holy Grail of lipidomics and metabolomics in general, impossible at this time. The choice for an extraction method therefore depends on the aim of the experiment; either target/optimize for a specific lipid category or try a general method that extracts as many lipids as possible. Yet, researchers have experimented with serial extractions to create fractions that partition the different lipid categories as efficiently as possible (Han et al 2012).
Another important aspect of the sample preparation and for the lipidomics experiment itself is the addition of stable isotope labeled compounds that serve as internal standards. One or more of these internal standards can be added for each class of lipids and be used to perform ratiometric comparisons with the molecular species of that lipid class to allow (semi)quantitative analysis of these analytes. In addition these stable isotope labeled molecules can be used for pulse/chase experiments to perform fluxomics studies (Mueller and Heinzle 2013).
Lastly, care should be taken during sample preparation and storage to protect lipids from being chemically or enzymatically modified as this obviously negatively influences the outcome of the lipidomics experiment. Prior to extraction samples should preferably be stored at −80 °C. Lipids in tissues and cells are relatively protected by natural antioxidant systems and compartmentalization. After sample homogenization, however, cellular content is mixed and unavoidably diluted, which renders lipids more prone to chemical or enzymatic modification. Some lipids are very stable (sterols, bile acids) while others are more prone to chemical oxidation (plasmalogens, lipids containing polyunsaturated fatty acids) or oxidation by light (7-dehydrocholesterol, ergosterol) (Wolf and Quinn 2008). Enzymatic modification can be prevented by working at temperatures close to 0 °C and adding a small percentage of organic solvent to the homogenization buffer. After extraction, lipids are much more prone to chemical oxidation and extracts therefore should preferably be stored in glass vials, solubilized in sufficient organic solvent at −80 °C and care should be taken to eliminate air/oxygen by flushing with inert gasses and storing in full containers.
Data acquisition
Common techniques to separate lipids before MS analysis include high/ultra-high performance liquid chromatography (HPLC/UHPLC) and capillary electrophoresis. Alternatively, direct infusion of the lipid extract into the mass spectrometer (e.g., no prior chromatographic separation), so called “shotgun lipidomics”, is also frequently used.
For convenience, several technical terms in relation to mass spectrometry are listed in box 1. To appreciate the different approaches in lipidomics one must first understand the ionization process and the types of mass spectrometers and techniques that are used to separate/filter ions. Basically, there are three types of mass filters namely quadrupoles (Q), time-of-flight (TOF), and ion traps. By using combinations of these mass filters, different mass spectrometers can be constructed which are called hybrids. Frequently used types of machines include triple quadrupole instruments (QqQ), Quadrupole time-of-flight (QTOF), and ion traps.
Box 1 Mass spectrometry lexicon
Amu | Stands for atomic mass unit, which is the same as Dalton (Da). One (unified) atomic mass unit is defined as one twelfth of the mass of an unbound neutral atom of 12C in its nuclear and electronic ground state and is equivalent to 1 g/mol |
Acquisition rate | Amount of scans that can be performed per second for a certain mass range |
Accurate mass | The measured exact mass |
Data-dependent scanning | Automated real-time selection of ions for MSn analysis. |
Exact mass | The calculated mass of an ion based on the sum of the (monoisotopic) masses of each atom in the molecule |
Ion suppression/enhancement | The negative (or positive) effect on the ionization of the molecules of interest by the presence of other endogenous or exogenous molecules. |
Linear dynamic range | This is the range over which ion signal is linear with the analyte concentration |
m/z | Mass (m) to charge (z) ratio, i.e., the mass of the ionized molecule divided by its charge |
Mass range | The range of m/z that can be covered by a certain mass spectrometer |
Mass accuracy | The difference between the exact mass and the measured mass divided by the exact mass (e.g., │(exact mass – accurate mass) │/exact mass). In other words, how close is the measured mass to the exact mass. Mass accuracy is usually measured in parts per million (ppm) or amu/Da |
MSn | Mass spectrometry where after fragmentation, fragments are either analyzed of fragmented again n-times. Tandem mass spectrometry as performed by QqQ machines is termed MS2 |
Resolution | Resolution is defined as the m/Δm, where m is the mass of the peak and Δm is the peak width at half height and is a measure of the ability to distinguish two peaks of slightly different mass-to-charge ratios, in a mass spectrum. The higher the value the better the resolution. Also sometimes indicated as “full width at half maximum” = FWHM |
Sensitivity | Amount of moles (usually femtomol/attomol range) that can be detected by the instrument |
Triple quadrupole instruments (low resolution)
Triple quadrupole instruments (QqQ) are commonly used in the targeted quantification of metabolites in biological samples. The two quadrupoles (Q) are separated by a collision cell (q) which is used to fragment ions originating from the first quadrupole, making it possible to perform different types of scans (Han et al 2012). Typically, a combination of constant neutral loss scans, precursor ion scans and product ion spectra is used to detect metabolites in biological matrices (Liu 2012). The limited resolution of the quadrupole (up to 7500, FWHM) is not sufficient for metabolite identification based on the measured mass in contrast to high resolution instruments as will be dealt with next. At higher acquisition rates and when scanning a wide mass range, sensitivity is considerably reduced, making QqQ instruments less suitable for biomarker discovery. QqQ instruments are, however, the best option for sensitive targeted quantitative analysis (especially when combined with UHPLC) of a limited amount of metabolites using their tandem MS capabilities in multiple reaction monitoring (MRM).
Time-of-flight (TOF), ion traps, and hybrids (high resolution)
TOF mass spectrometers combine high-resolution with mass accuracy, which increases the possibility of determining elemental compositions of molecules and also provides high specificity of detection. By combining TOF’s with quadrupoles, hybrid machines have been developed allowing novel scan modes together with high resolution capabilities. Machines as QqTOFs are, just like QqQ instruments, capable of selecting and fragmenting ions that can be subsequently separated by the TOF and detected at high resolution. The resolution (up to 70,000, FMWH) is sufficient for metabolite identification and TOF instruments can cope with a high acquisition rate which combines well with fast chromatographical systems such as UHPLC. Resolution is an important parameter of mass spectrometry and is explained in more detail in Fig. 2.

Mass resolution. One sample measured using three different resolutions; 10,000 (10K), 50,000 (50K) and 100,000 (100K) on a Thermo Q-Exactive. Increasing resolution enables the detection of more ions at higher mass accuracy which makes identification of the corresponding compounds more dependable
Ion traps, Orbitraps in particular, have developed into mass filters with very high resolution (up to 450,000 at m/z 200) making it possible to determine the exact mass of the compounds but also to resolve isobaric species and reveal isotope fine structures. Another advantage of some ion trap machines is that these instruments have MSn capability, meaning that ion fragments can be further fragmented and characterized, which is particularly useful for structural elucidation of molecules. When compared to TOF instruments, ion traps have a relatively slow acquisition rate, especially at high resolution and resolution declines at higher m/z values. Ion traps are therefore less suitable for fast chromatography. By interfacing Orbitraps with linear ion traps or quadrupole mass filters many of these limitations have partially been overcome.
An interesting and useful feature that is especially useful when using hybrid machines is the capability to perform so called data-dependent scanning. This technique uses specific criteria to select one or more ions of interest for subsequent fragmentation, meaning that a product ion scan is performed for these selected ions providing more structural information of the compound. Scanning in the data-dependent mode starts with a survey scan to identify ions and their abundances in the sample. This survey scan is then followed by the acquisition of a fragment spectrum from the automatically selected precursors. In this way, product ion scans are obtained (or in some ion traps even MSn spectra) that can aid in the identification of the precursor ions. Although this type of scanning is biased toward the more abundant ions it provides valuable structural information of the ions (Bhattacharya 2013).
Ionization techniques (ESI, APCI, and MALDI)
Before any molecule can be analyzed by the mass spectrometer it first has to be ionized so that it can be manipulated using electric fields and mass filters and detected by the detector. For lipidomics, three ionization techniques are most used, 1. Electrospray ionization (ESI), 2. Atmospheric pressure chemical ionization (APCI), and 3. Matrix-assisted laser desorption/ionization (MALDI). There are many other ways to ionize analytes (Li et al 2014) but these will not be discussed here. The three ionization techniques mentioned above are all so called “soft ionization” techniques as very little fragmentation occurs during the ionization process and mainly monocharged molecular ions, which is ideally suited for metabolomics purposes.
ESI is most widely used for thermally labile and mostly non-volatile molecules and therefore can be applied to almost all lipid categories. The sample is nebulized through a highly charged capillary using heated nitrogen gas, producing a fine aerosol. This results in evaporation of the solvent and ionization of the molecules after which the ions enter the mass spectrometer.
With APCI, the sample is nebulized and heated so that both solvent and analytes are in the gas phase followed by a corona discharge which ionizes the solvent molecules that subsequently also ionize the analyte molecules. APCI generally yields monocharged ions and is mainly used with small thermally stable nonpolar molecules (<1500 Da). For lipidomics, APCI is mostly applied for neutral lipids including triglycerides, sterols, and fatty acid esters (Byrdwell 2001; Li et al 2014).
For MALDI, the sample is mixed with a matrix that readily forms crystals that aids the ionization process. The fluid mixture of sample and matrix is spotted on a MALDI plate and allowed to dry. To ionize the analyte molecules, a laser is fired at the matrix crystals in the dried-droplet spot, which absorbs the laser energy resulting in desorption and ionization. The ionized matrix molecule then transfers its charge to the analyte, thus ionizing the analyte.
Direct infusion MS, chromatography- and MALDI-based MS analysis
For the data acquisition, essentially two strategies can be chosen, MS preceded by a chromatographic separation (usually LC-MS) or direct infusion, which is also called “shotgun MS”. Both approaches have their advantages and limitations.
In shotgun lipidomics, the lipid extract is introduced directly into the MS and acquisition can proceed for prolonged periods of time to acquire multiple spectra with good signal to noise ratios. Especially when combined with nanomate devices, reproducible direct infusion into the mass spectrometer can be accomplished. As there is no prior separation, analysis time is relatively fast and when using suitable internal standards, the shotgun approach is surprisingly reproducible, even in complex matrices (Jung et al 2011). Multiple analyses are done on multiplex extractions of the same sample using different types of scans on QqQ machines (neutral loss scan, precursor ion scan, and product ion scans) and this information combined is so called multi-dimensional MS (MDMS) which allows identification and quantification of individual lipid species (Han et al 2012). As sensitivity, mass resolution and accuracy, acquisition rate and dynamic range of mass spectrometers have all improved rapidly, shotgun MS is becoming a considerably attractive method of choice for lipidomics. Despite these advances, however, shotgun lipidomics is hampered by ion-suppression/enhancement effects, the inability to distinguish certain isobars/isomers, unreliable quantification of low abundant lipids and difficulty in identifying unknown lipids (Hyotylainen and Oresic 2014). ESI-QTOF instruments are frequently used for shotgun lipidomics as they combine the ability to perform fragmentation with accurate mass measurement. Like hybrid instruments containing ion traps, however, these instruments are limited in their capabilities to perform tandem MS scans typically used in QqQ instruments. As they capture all precursors and their fragments in parallel and in a single scan, the analyses of the data pose a great challenge to relate the fragments to their precursors (Bhattacharya 2013).
LC-MS, which is the most frequently used hyphenated mass spectrometric technique, has the advantage that separation of the different lipid classes/species lowers the complexity of the sample, reduces ion suppression/enhancement effects, and therefore allows a more specific identification of lipid species. Another advantage is that isobaric/isomeric lipids sometimes can be separated and quantified, which is not possible using shotgun MS. For example, bismonoacylglycerolphosphates and phosphatidylglycerols are isobaric/isomeric molecules that cannot be distinguished using MS and have to be separated by chromatography to be detected and quantified separately. Another advantage of chromatographical separation is that some lipids can undergo in source fragmentation that “synthesizes” other lipids. For instance, phosphatidylserine can lose its headgroup and form phosphatidic acid which can be recognized when phosphatidylserine-derived phosphatidic acid is separated from endogenous phosphatidic acid by liquid chromatography prior to MS detection (Knittelfelder et al 2014). When using LC-MS, however, the acquisition rate needs to be sufficient to fully designate an eluting peak with enough sensitivity to allow reproducible quantification and identification. This can be problematic when using ion traps as the technical setup of these instruments is such that at higher resolutions acquisition rates are significantly reduced. In addition, although ion suppression/enhancement is considerably less when using LC, the different elution time of internal standards and analytes calculated using those internal standard can introduce variation because of variable amounts of ion suppression during the chromatographical run. The LC step also necessitates extra data pre-processing efforts as retention time correction and correct peak grouping needs to be implemented in the bioinformatics pipeline.
MALDI-based MS analysis in lipidomics is mostly used to directly detect lipids from surfaces including tissue sections and TLC plates, but spotted lipid extracts can also be analyzed (Berry et al 2011; Ellis et al 2013). The relatively simple sample preparation and the fact that considerable amounts of impurities are tolerated make MALDI-based MS useful for the analysis of a large number of samples. The MALDI ionization, however, renders it difficult to hyphenate with other techniques, UHPLC in particular, and metabolite quantification is not a strong suit of MALDI-based MS (Fuchs et al 2010). Despite these disadvantages, MALDI-based MS is being used more and more in lipidomics because of the capability to scan tissue slices and provide a spatial distribution map of lipids within a sample, a technique called mass spectrometry imaging or MSI (Berry et al 2011). To be able to visualize the location of different lipids in tissue slices, and even at the cellular level, makes MSI a valuable addition to the techniques to characterize the lipidome.
Bioinformatics data pre-processing
Metabolomics experiments generate large amounts of data, which can be processed by various bioinformatics methods to detect and quantify metabolite peaks, assign compound names, and perform further downstream analysis such as statistical, biological pathway, or metabolic flux analysis. Pre-processing is the first part of this bioinformatics workflow and comprises the application of methods needed to generate a peak table for each sample which contains all detected peaks (positions) and their relative concentrations (peak intensities and/or areas). Subsequently, to facilitate comparative analysis, matching peaks representing the same metabolite from different samples are grouped together in a peak group list (Boccard et al 2010). Sometimes, chromatogram alignment is required to adequately group matching peaks (see below). In essence, the challenge of data pre-processing is to collect as many true metabolite signals from the data as possible, while at the same time minimizing the number of detected artefact peaks (e.g., noise, spikes). Highly abundant metabolites usually yield strong, unmistakable signals, but as the metabolite concentrations reach the detection limit, it becomes harder to automatically identify and quantify the peaks. In practice time-consuming curation and improvement of the peak table by mass spectrometry experts still remains necessary. Generally, a few rounds of pre-processing and curation are required to establish a final peak table.
Although each pre-processing step can be implemented in several ways they generally comprise the following steps (Boccard et al 2010):
-
Peak detection. For LC-MS data, this first step involves the detection of each metabolite peak from the raw data for each individual sample. This results in a list of peaks characterized by its mass (m/z) and retention time position. Subsequent peak quantification is achieved by determining the peak area through peak integration. A detection limit (signal-to-noise level) for the intensity is set a priori and conditions can be placed upon the shape of a peak in order to eliminate noise peaks. For direct infusion data, which lacks chromatographic separation, the intensity of each peak is determined as a (weighted) average over the collection time;
-
Peak quantification/normalization. The response of different lipid classes can be altered by matrix effects and suppression and therefore multiple internal standards are routinely added to correct for these influences. The measured intensities can then be normalized using these internal standards in order to obtain semi-quantitative data that can be further analyzed statistically;
-
Peak grouping. To facilitate the statistical comparison of metabolite peaks across groups of samples one needs to group those peaks that, in terms of peak position, are most similar to each other. Generally, peak matching procedures search for peaks across samples within a group of (biologically) related samples that are within a pre-specified m/z and retention time distance of each other and, consequently, are assumed to represent the same metabolite. Such grouping needs to account for fluctuations in the chromatographic system that leads to non-linear deviations of the retention time of individual peaks. Consequently, (non-linear) time alignment approaches are generally applied to correct time shifts between samples;
-
Imputation of missing peaks. Due to shortcomings in experimental and/or bioinformatics procedures or due to sample outliers, one or few samples in a group of related samples may sometimes miss one or more metabolite peaks. To avoid missing intensities in downstream (statistical) analysis, the intensities of missing peaks can be added to the peak table by assuming that they are located at the same position as the identified peaks in the related samples and, subsequently, integrate the measured intensity within these areas. At this point, a peak group in the list is referred to as a feature, which is defined by its m/z and RT value, and intensity. Further analysis of the dataset is required to determine which features correspond to identified metabolites or noise and which remain unidentified;
-
Visualization. The complexity of metabolomics data dictates the use of visualization methods to explore raw and processed data for quality control, validation of results, and to pinpoint parts of the data or results that requires further attention. For every feature, simple graphs of the contributing peaks (overlays of extracted ion chromatograms) or box plots of the intensities can give rapid insight into the shape of the peaks and the relative differences between groups of samples.
-
Isotope correction. Isotopes are molecules with the same chemical composition, but with one or more heavier atoms (for example 13C instead of 12C) and a correspondingly higher mass. At low resolution, peaks from isotope molecules of one lipid overlap with peaks of a lipid with two more hydrogen atoms (e.g., one less double bond). The intensity of the isotope peak needs to be subtracted from the intensity of the second lipid, before all lipids can be properly quantified. Since the isotope patterns follow the natural abundances of atoms, they can be easily calculated;
-
Compound identification. It is not sufficient to have a peak list that only contains a long list of features without knowing to which metabolites these correspond, since this would prevent any sort of biological interpretation. Therefore, it is essential to assign metabolite (compound) names to these features, which is currently one of the main bottlenecks in metabolomics and for which experimental (e.g., compound fragmentation) and bioinformatics strategies are available. The identification of metabolites is usually based on queries to internal or external public databases of all known lipid species. Mass spectroscopic resolution and the quality of the chromatographic separation of different classes of lipids are key factors for the accuracy of the identification. At low resolution, there is a risk of ambiguous assignments because the peaks for lipids with small mass differences in their mass have a tendency to overlap, and deconvolution can be difficult or even impossible, which makes it impossible to identify these peaks. At high resolution, the m/z value of a feature is much more accurate and the identification of the corresponding lipid molecule is much more reliable. In both cases, the full scan only gives information on the combined side chains of the lipid; further MS fragmentation is necessary for determining the composition of the individual side chains (e.g., PE(20:4/18:4)). The LIPID MAPS consortium has developed an online database of over 37,500 lipid structures, which can be queried (Fahy et al 2009). LipidBlast is a database of in silico generated MS/MS spectra of over 200,000 lipids which can be cross-referenced with experimental data (Kind et al 2013). There are several web-based and commercial services or packages that can search MS files to identify lipids but frequently are MS vendor-specific and generally do not provide the source code of the application to allow user modifications.
There are various commercial and open-source software packages that address pre-processing of LC-MS data (Oresic 2011; Theodoridis et al 2012). The commercial software is efficiently integrated with the machine of the vendor, has advanced graphical interfaces, and is usually well documented. The open-source packages have the advantage that the source code is available and are generally produced through a community effort. Open-source solutions provide more flexibility compared to commercial software, include contributions from multiple academic groups, often provide state-of-the-art methods, and the software can be tailored or extended to the specific needs of the researcher. For direct infusion (shotgun lipidomics) data, the number of software packages that are suitable for untargeted, batch-mode studies is still rather limited (Herzog et al 2012).
Just as there is no standard in procedures and platforms for collecting lipidomics data, there is no uniform data pre-processing strategy. Most labs have their own requirements, and rely on tailor-made pipelines that consist of a combination of the methods described above. Pre-processing is a complicated step in the analysis of lipidomics data and choices of parameter settings greatly influence the outcome (peak table) and therefore potentially the biological interpretation. A lot of fine-tuning is achieved by the regular interaction of technicians or clinical biochemist with bioinformaticians in order to make a bioinformatics pipeline reliable.
Statistical data analysis
Lipidomics experiments yield a wealth of data; hundreds to thousands of features can be observed in a single experiment, corresponding to tens to hundreds of identified lipid species, along with a large number of unidentified features. A large range of statistical approaches is available to analyze the data depending on the biological question. For the comparison of two or more groups one may resort to univariate methods such as analysis of variance (ANOVA). For other types of analysis one may need more advanced methods such as partial-least-squares discriminant analysis (PLS-DA) for biomarker discover (Want and Masson 2011). Principal component analysis (PCA) or cluster analysis for a first exploration of the data provides information about the (separation of) groups of samples and/or metabolites. For an overview see (Hendriks et al 2011).
As part of the statistical data analysis one must deal with the fact that in most studies one will have far fewer features in the peak table than samples measured. One consequence is that the application of univariate statistical methods may result in too many false positive findings due to the many hypothesis tests that are performed. Indeed, many of these findings won’t be confirmed during labor intensive experimental validation and, consequently, one has to account for this multiplicity issue. A common approach is to correct the p-value for the number of statistical tests by, for example, a Bonferroni correction or control the expected proportion of incorrectly rejected null hypotheses through a less conservative false discovery rate (FDR) procedure (Boccard et al 2010). Nevertheless, univariate tests have many advantages, such as applicability to all types of variables and outcomes and simplicity in interpretation (Chadeau-Hyam et al 2013). A disadvantage of univariate tests is that they disregard relations between features which may be important for the biological interpretation, or which may improve statistical inference through their inclusion in the statistical test. Multivariate approaches such as PCA and PLS-DA consider the correlation structure of the data and reduce the dimensionality of the data by constructing so-called latent variables, which are combinations of the original variables (e.g., metabolites). This also facilitates the visualization of the data in two or three dimensions.
There is no pre-defined statistical test that gives the right answer for every lipidomics experiment. The most appropriate test to choose depends on the biological question (differences between groups, e.g., control versus diseased, or the discovery of biomarkers) and the scope, e.g., whether a holistic or detailed analysis is required (Saccenti et al 2014). Even the stage in the data analysis in which the statistical test is applied can be important, whether the test is applied for exploratory data analysis or for the determination of a definitive treatment effect.
Pathway analysis
To facilitate the biological interpretation of lipidomics data a range of approaches can be pursued. Perhaps the most straightforward approach is to integrate and visualize the identified metabolites in the context of metabolic pathways obtained from public databases (Barupal et al 2012; Karnovsky et al 2012; Sreenivasaiah et al 2012). A statistical approach toward pathway analysis could involve metabolite set enrichment analysis (MSEA) (Xia and Wishart 2011) which is based gene set enrichment analysis (GSEA) commonly used in the analysis of gene expression datasets. MSEA can be used to investigate the enrichment of predefined groups of related metabolites instead of individual metabolites. This allows to identify subtle but coordinated changes among related metabolites. MSEA starts with a list of metabolites that have been extracted from the experimental data through statistical approaches such as ANOVA, cluster analysis, PCA, or PLS-DA. Subsequently, one aims to determine whether metabolites from a predefined metabolite set are over-represented in the experimental list of metabolites, that is more than expected by chance. Commonly, the hypergeometric test is used to test for over-representation. The metabolite sets can be constructed according to any criteria such as co-occurrence in the same metabolic pathway, association with disease, or functional class. More advanced systems biology approaches are also feasible. For example, the construction of correlation networks from metabolic profiles to identify metabolites that are related and co-regulated across the conditions that were measured (Kotze et al 2013). These methods do not require any prior knowledge such as pathway stoichiometry.
For the investigation of metabolic adaptations at the molecular level one may not only look at metabolite concentrations by also at the alteration of fluxes in the network. Metabolic fluxes are an ultimate representation of the cellular phenotype since they are shaped through cellular regulatory processes at many different levels such as enzyme activity, substrate affinity, presence of inhibitors and activators, and concentration of the metabolites (Nielsen 2003). The concentrations of the metabolites are a result of the fluxes and this plays an important role in feedback regulation. Unfortunately, intracellular fluxes cannot be measured directly but have to be estimated from concentration measurements. One approach is to use constraint-based methods such as flux balance analysis (FBA) for steady-state systems or dynamic FBA (DFBA) for dynamic systems (Mahadevan et al 2002). These methods, however, do require information about the reaction stoichiometry in addition to the specification of constraints that bound the magnitude of the fluxes and an objective function that defines the phenotype (e.g., maximum growth rate or energetic efficiency) in the form of a biological objective such as biomass production. The objective function quantifies the relative contribution of each reaction to the phenotype.
Finally, one may investigate pathways through the use of detailed mathematical (kinetic) models such as those based on differential equations. Such models predict and quantify responses of a metabolic system and can be used to simulate different scenarios such as gene knock outs (Peskov et al 2012). Generally, these models require a detailed description of the underlying metabolic pathway and kinetic constants. The latter may (to some extent) be estimated from metabolite concentration measurements.
Conclusion
The technical advances in mass spectrometry, particularly the development of (ultra)-high-resolution/mass accuracy measurement capabilities in combination with refinement of soft ionization techniques, have increased the application and success of lipidomics to answer biological questions in relation to lipid metabolism. Together with other omics technologies, lipidomics has become an important tool to practice systems biology as lipids comprise a very significant part of the metabolome and play pleiotropic roles in cellular functions. As an increasing number of disorders are linked to lipid metabolism, lipidomics is used to search for biomarkers, understand disease mechanism, and follow the efficacy of therapeutic options. Still, with a plethora of different techniques, each with their own strengths and weaknesses, it is clear that no single platform is sufficient to fully characterize the lipidome completely. Combining different lipidomics techniques, such as mass spectrometry imaging, shotgun/LC lipidomics, global profiling using LC-high resolution MS, and quantitative targeted lipid analyses with LC-QqQ MS and GC-MS has the potential to yield the most complete lipidomics data sets (Brugger 2014; Hyotylainen and Oresic 2014). Unfortunately, only a few laboratories have the technical, financial, and analytical capabilities to achieve this. Many examples in literature, however, have shown that even a single platform can yield insightful results, which is encouraging more researchers to setup lipidomics platforms for both research and diagnostics.
There is a great need for standardization and consolidation in the field of lipidomics, in terms of sample preparation, data collection methodology, and data management and analysis. The exchange of data and results between different laboratories through publicly accessible repositories (such as Metabolights (Salek et al 2013)), will allow the community to build a knowledge base to map the entire lipidome (Theodoridis et al 2012). The pre-processing of metabolomics data remains a major bottleneck and continues to require the time-consuming step of manual validation. Clearly, more bioinformatics research is required to develop improved algorithms that eventually allow to pre-process data with no or only minimal human intervention. However, this will require the large-scale availability of public metabolomics data. Another bottleneck is the assignment of compound names to identified peaks in the raw data. A solution to this problem should be a joint effort of experimental and bioinformatics approaches. A final challenge is the further development of statistical, bioinformatics, and systems biology methods to enable biological interpretation. In particular, efforts toward the integration and interpretation of metabolomics data with genome data (e.g., SNPs), gene expression data, and proteomics data are expected to become more common and will enable new insights in living systems and the identification of biomarkers.
It is important to realize that in each phase of the metabolomics experiment different specialists play a dedicated role. When researching human disorders, physicians ensure that patient selection is accurate and together with clinical biochemists see to it that no pre-analytical trivialities occur. Technicians and analytical chemists/clinical biochemists need to develop a reproducible and robust method and bioinformaticians are responsible to assist in the statistical design of the experiment and provide reliable and validated data pre-processing and analysis methods to facilitate correct biological interpretation of the results. This requires the presence of dual-thinkers on the lipidomics team and emphasizes that research and development is based on constructive cooperation between different disciplines to obtain the best results.
Abbreviations
- ANOVA:
-
Analysis of variance
- APCI:
-
Atmospheric pressure chemical ionization
- (D)FBA (Dynamic):
-
flux-balance analysis
- ESI:
-
Electrospray ionization
- FDR:
-
False discovery rate
- FWHM:
-
Full width at half maximum
- GSEA:
-
Gene set enrichment analysis
- LC:
-
Liquid chromatography
- MALDI:
-
Matrix-assisted laser desorption/ionization
- MDMS:
-
Multi-dimensional MS
- MSEA:
-
Metabolite set enrichment analysis
- MRM:
-
Multiple reaction monitoring
- PCA:
-
Principal component analysis
- PLS-DA:
-
Partial-least-squares discriminant analysis
- (U)HPLC:
-
(Ultra)-high performance chromatography
- Q:
-
Quadrupole
- QC:
-
Quality control
- QqQ:
-
Triple quadrupole instrument
- TOF:
-
Time-of-flight
References
Abbott SK, Jenner AM, Mitchell TW, Brown SH, Halliday GM, Garner B (2013) An improved high-throughput lipid extraction method for the analysis of human brain lipids. Lipids 48:307–318
Barupal DK, Haldiya PK, Wohlgemuth G et al (2012) MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinforma 13:99
Berry KA, Hankin JA, Barkley RM, Spraggins JM, Caprioli RM, Murphy RC (2011) MALDI imaging of lipid biochemistry in tissues by mass spectrometry. Chem Rev 111:6491–6512
Bhattacharya SK (2013) Recent advances in shotgun lipidomics and their implication for vision research and ophthalmology. Curr Eye Res 38:417–427
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Boccard J, Veuthey JL, Rudaz S (2010) Knowledge discovery in metabolomics: an overview of MS data handling. J Sep Sci 33:290–304
Box GEP, Hunter JS, Hunter WG (2005) Statistics for experimenters: design, innovation, and discovery.: Wiley, New York
Brugger B (2014) Lipidomics: analysis of the lipid composition of cells and subcellular organelles by electrospray ionization mass spectrometry. Annu Rev Biochem 83:79–98
Byrdwell WC (2001) Atmospheric pressure chemical ionization mass spectrometry for analysis of lipids. Lipids 36:327–346
Chadeau-Hyam M, Campanella G, Jombart T et al (2013) Deciphering the complex: methodological overview of statistical models to derive OMICS-based biomarkers. Environ Mol Mutagen 54:542–557
Ellis SR, Brown SH, In Het Panhuis M, Blanksby SJ, Mitchell TW (2013) Surface analysis of lipids by mass spectrometry: more than just imaging. Prog Lipid Res 52:329–353
Fahy E, Subramaniam S, Murphy RC et al (2009) Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res 50(Suppl):S9–14
Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509
Fuchs B, Suss R, Schiller J (2010) An update of MALDI-TOF mass spectrometry in lipid research. Prog Lipid Res 49:450–475
Han X, Yang K, Gross RW (2012) Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev 31:134–178
Hendriks MMWB, Eeuwijk FA, Jellema RH et al (2011) Data-processing strategies for metabolomics studies. TrAC Trends Anal Chem 30:1685–1698
Herzog R, Schuhmann K, Schwudke D et al (2012) LipidXplorer: a software for consensual cross-platform lipidomics. PLoS ONE 7:e29851
Hyotylainen T, Oresic M (2014) Systems biology strategies to study lipidomes in health and disease. Prog Lipid Res 55C:43–60
Jung HR, Sylvanne T, Koistinen KM, Tarasov K, Kauhanen D, Ekroos K (2011) High throughput quantitative molecular lipidomics. Biochim Biophys Acta 1811:925–934
Karnovsky A, Weymouth T, Hull T et al (2012) Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 28:373–380
Kind T, Liu KH, Lee do Y, DeFelice B, Meissen JK, Fiehn O (2013) LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat Methods 10:755–758
Knittelfelder OL, Weberhofer BP, Eichmann TO, Kohlwein SD, Rechberger GN (2014) A versatile ultra-high performance LC-MS method for lipid profiling. J Chromatogr B: Analyt Technol Biomed Life Sci 951–952:119–128
Kotze HL, Armitage EG, Sharkey KJ et al (2013) A novel untargeted metabolomics correlation-based network analysis incorporating human metabolic reconstructions. BMC Syst Biol 7:107
Li M, Yang L, Bai Y, Liu H (2014) Analytical methods in lipidomics and their applications. Anal Chem 86:161–175
Liu ZY (2012) An introduction to hybrid ion trap/time-of-flight mass spectrometry coupled with liquid chromatography applied to drug metabolism studies. J Mass Spectrom 47:1627–1642
Mahadevan R, Edwards JS, Doyle FJ 3rd (2002) Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J 83:1331–1340
Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D (2008) Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res 49:1137–1146
Mueller D, Heinzle E (2013) Stable isotope-assisted metabolomics to detect metabolic flux changes in mammalian cell cultures. Curr Opin Biotechnol 24:54–59
Nielsen J (2003) It is all about metabolic fluxes. J Bacteriol 185:7031–7035
Oresic M (2011) Informatics and computational strategies for the study of lipids. Biochim Biophys Acta 1811:991–999
Orth JD, Thiele I, Palsson BO (2010) What is flux balance analysis? Nat Biotechnol 28:245–248
Peskov K, Mogilevskaya E, Demin O (2012) Kinetic modelling of central carbon metabolism in Escherichia coli. FEBS J 279:3374–3385
Reis A, Rudnitskaya A, Blackburn GJ, Mohd Fauzi N, Pitt AR, Spickett CM (2013) A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J Lipid Res 54:1812–1824
Saccenti E, Hoefsloot HJ, Smilde A, Westerhuis J, Hendriks MWB (2014) Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 10:361–374
Salek RM, Haug K, Steinbeck C (2013) Dissemination of metabolomics results: role of MetaboLights and COSMOS. Gigascience 2:8
Sreenivasaiah PK, Rani S, Cayetano J, Arul N, Kim do H (2012) IPAVS: integrated pathway resources, analysis and visualization system. Nucleic Acids Res 40:D803–808
Theodoridis GA, Gika HG, Want EJ, Wilson ID (2012) Liquid chromatography-mass spectrometry based global metabolite profiling: a review. Anal Chim Acta 711:7–16
Want E, Masson P (2011) Processing and analysis of GC/LC-MS-based metabolomics data. Methods Mol Biol 708:277–298
Wolf C, Quinn PJ (2008) Lipidomics: practical aspects and applications. Prog Lipid Res 47:15–36
Xia J, Wishart DS (2011) Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc 6:743–760
Acknowledgments
We thank Willem Kulik for critical reading of the manuscript and Henk van Lenthe for technical assistance.
Compliance with ethics guidelines
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Matthias Baumgartner
Rights and permissions
About this article

Cite this article
Vaz, F.M., Pras-Raves, M., Bootsma, A.H. et al. Principles and practice of lipidomics. J Inherit Metab Dis 38, 41–52 (2015). https://doi.org/10.1007/s10545-014-9792-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-014-9792-6