
Abstract
The development of an Aptamer based biosensor for the selective detection of human epidermal growth factor receptor 2 (HER2) with high sensitivity and specificity was achieved. A screen-printed carbon electrode was used in the scope of this work. The HER2 Aptamer was immobilized via electrostatic adsorption on the surface of a screen-printed electrode, which was modified with Au Nanoparticles (~ 20 nm diameter) to support the Aptamer immobilization. The Aptasensor was extensively investigated using Cyclic voltammetry, Differential pulse voltammetry, Electrochemical impedance spectroscopy, Fourier transform infrared spectroscopy and Atomic force microscopy. The Aptasensor exhibits a fast response with a binding time of only 5 min and shows a log-linear response over a wide concentration range of 0.001—100 ng/mL. Moreover, it has high sensitivity and enhanced detection limit reaching 52.85 μA/ng/mL, and 0.001 ng/mL, respectively, with a relative standard deviation < 5%. The Aptasensor selectivity was studied by using different interfering substances, and the results demonstrate that the Aptasensor is efficient for the detection of HER2 with approximately 8% extent of the interference.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Breast cancer is considered as one of the main cancer types leading to death universally. This is because it is responsible for about 23% of the cancer death cases in women worldwide (Mahfoud et al. 2014). The critical point to achieve successful management of these diseases is to be detected in its early stages. Current diagnostic techniques, to some extends, are limited, as they have low sensitivity, time-consuming, and high cost. Therefore, until today there is an urgent need for high-performance diagnostic techniques to overcome the limitations of current analysis methods.
In breast cancer, one of the most essential prognoses and diagnosis biomarker is the Human epidermal growth factor receptor 2 (HER2), which is a protein with clinical serum cuts-off at 15 ng/mL (Ravalli et al. 2016); the normal range in healthy women is in the range of 2—15 ng/ml. Hence, HER2 has a vital role in cancer detection systems, HER2 can be found in 20—30% of the breast cancer confirmed cases (Laocharoensuk 2016; Mittal et al. 2017; Boureau et al. 1988; Gullick 2001; Riccio et al. 2009; Mishra and Verma 2010; Hasanzadeh et al. 2017). Some researchers reported that circulating HER2 levels are valuable for diagnosis and therapeutic purpose (Köstler et al. 2004; Ludovini et al. 2008; Carney et al. 2003). The monitoring of HER2 level is also useful in other cancer types such as oral, ovarian, gastric, and lung cancers. Immunohistochemistry and fluorescent in situ hybridization are FDA approved assays for clinical testing, but both methods have limitations in accuracy and reliability. In addition, their procedure is complicated, time-consuming, and requires skilled-qualified staff to execute the appropriate multi-step procedures. Therefore, the development of a new robust, simple, accurate, and fast time response diagnostic technique is needed (Tse et al. 2005; Furrer et al. 2015). Besides, it may lead to revolutions in the cancer biomedical devices industry.
Different types of biosensors were developed in the last decades for HER2 detection. From recognition point of view, antibodies are the most popular choice, here are some recent studies on the development of HER2 immunosensor (Salahandish et al. 2018; Arkan et al. 2015; Tallapragada et al. 2017; Emami et al. 2014; Ahmad et al. 2019; Hartati et al. 2020; Freitas et al. 2019). In comparison with antibody, Aptamer is smaller in size (one-tenth of antibodies), higher specific binding efficiency, and has higher thermostability. Besides, it can be modified with minimal variation when different batches are produced (Sun and Zu 2015). Furthermore, using Aptamer in the biosensor design is showing great potential for developing innovative biosensor. Several HER2 Aptasensors were recently developed. Gold nanoparticles modified with 2,5-bis(2-thienyl)-1H-pyrrole-1-(p-benzoic acid), were used to develop a sandwich based voltammetric sensor for HER2 detection by using graphene carbon electrode (Zhu et al. 2013). In (Ou et al. 2019), a sandwich electrochemical Aptasensor was developed for HER2 detection, the obtained liner range was from 0.1 to 100.0 ng/mL, and the limit of detection was 0.08 ng/mL. Electrodes modified with Au Nanoparticles were used in the development of a HER2 Aptasensor; showing a linear range between 10−5—102 ng/ml, and a limit of detection reaching 5 ng/ml (Chun et al. 2013). Qureshi et al., fabricated an interdigitated Au microelectrode to develop a label-free capacitance-based Aptasensor. In their work, the biosensor reached a linear in the range of 0.2—2 ng/ml, and a LOD of 2 ng/ml (Qureshi et al. 2015). Others reported a 1 pM LOD, and 1 pM to 100 nM linear range using an interdigitated capacitive Aptasensor modified with Au nanoparticles (Arya et al. 2018).
Screen-printed electrodes (SPEs) gained worldwide attraction due to their low cost, and high reproducibility. Therefore, SPEs are used worldwide in the biosensor development for different studies (Beitollahi et al. 2020; Smart et al. 2020; Antuña-Jiménez et al. 2020; Mohanraj et al. 2020); especially in breast cancer biomarkers detection (Tallapragada et al. 2017; Nawaz et al. 2016; Patris et al. 2014; Freitas et al. 2020a, b; Bezerra et al. 2019; Ou et al. 2019). In (Bezerra et al. 2019), Aptasensor was developed using a screen-printed electrode modified with poly-L-lysine, showing a detection limit of 3.0 ng/mL over concentrations range of HER2 protein from 10 to 60 ng/mL. An in vitro assay was developed for HER2 detection by Javed H. Niazi et al., they investigated the affinity separation of carbon nanotubes enveloped with anti-HER2 ssDNA Aptamers (Niazi et al. 2015), showing approximately 1:1 affinity dissociation ratio between H2 Aptamer and HER2 protein.
In this research, screen-printed carbon electrodes modified with Au nanoparticle were used to develop an Aptamer based biosensor for HER2 recognition. The efficiency of each step in the development process, was investigated by using different characterization techniques, such as Atomic force microscopy (AFM), Fourier transform infrared spectroscopy (FTIR), Cyclic voltammetry (CV), Differential pulse voltammetry (DPV), and Electrochemical impedance spectroscopy (EIS). Differential pulse voltammetry and Electrochemical impedance spectroscopy were used to study the Aptasensor response. The developed Aptasensor shows a log-linear response over a wide concentration range from 0.001 to 100 ng/mL and high sensitivity of ~ 52.85 μA/decade, with a very low detection limit reaching 0.001 ng/mL. These findings can lead to enhanced biomedical devices used for cancer diagnosis.
2 Experimental setup
2.1 Materials
DNA Aptamer specific for HER2 with thiol terminal (5'-SH-(CH2)6-GGG CCG TCG AAC ACG AGC ATG GTG CGT GGA CCT AGG ATG ACC TGA GTA CTG TCC-3') was received from Integrated DNA Technologies (USA) and was stored at − 20 °C until used. Human epidermal growth factor2 (R & D Systems) was prepared with different concentrations in PBS, pH 7.4. Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) was received from Alfa Aesar (USA). Ethylenediaminetetraacetic acid (EDTA) buffer was purchased from Integrated DNA Technologies (USA). 6-Mercapto-1-hexanol (MCH) was received from Merck (USA). Dulbecco's phosphate-buffered saline was procured from Merck (UK). Potassium hex cyanoferrate(II) trihydrate, 98.5%, (K4[Fe(CN6]0.3H2O) was acquired from ACROS Organics (Belgium). Potassium Hexacyanoferrate(III) K3[Fe(CN)6] from Fisher Chemical (UK). Potassium chloride (KCl), 99.5%, from Gainland Chemical Co. All aqueous solutions were prepared using nuclease-free water.
2.2 Measurement and apparatus
The screen-printed carbon electrodes (3.4 × 1.0 × 0.05 cm, length x width x height) were acquired from DropSens (Spain). These SCPEs consist of three electrodes: A Carbon working electrode (WE) with 4 mm diameter, modified with AuNPs (~ 20 nm diameter). A carbon counter electrode (CE), and a reference electrode (RE), are made of a silver composite as (Ag/AgCl). Before starting the experiments, the SPEs were rinsed thoroughly with deionized water and were blown dry with nitrogen gas.
The Aptasensor was characterized and studied with different techniques: Fourier-transform infrared spectroscopy from Perkin-Elmer Spectrum, the AFM images were taken by an AIST-NT SmartSPM™ 1000 scanning probe microscope (SPM, Novato, CA). Also, the EIS, CV, and DPV measurements were performed in an Ivium Compact Potentiostat from Ivium Technologies. The CV measurements were recorded in the potential window of—0.5 to 0.8 V at a scan rate from 10—90 mV/s. The reactant solution was prepared in a 10 mM PBS (pH 7.4), by using a 2 ml of PBS solution after mixing it with a 5 mM [Fe(CN)6]4−, and a 5 mM of [Fe(CN)6]3− as a redox probe couple. The DPV measurements were carried out in the potential region from—0.5 to 0.8 V at a scan rate of 3 mV/s with the excitation of 85 mV amplitude, and a 4 mV step potential. The frequency range used for the EIS measurements were 100 kHz – 0.01 Hz with a 0.01 V signal amplitude.
2.3 Immobilizing capture of the anti-HER2 Aptamer on the SPEs
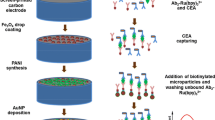
Aptamer resuspension and dilution were done using TE buffer (10 mM Tris, and 0.1 mM EDTA pH 7.4). The immobilization of the thiolated anti-HER2-Aptamer on the modified electrode was performed according to the following procedure: Aptamers stock solution heated to 95 °C gradually, then cooled down to the room temperature. A 25 µM Aptamer solution was added to a 10 mM TCEP, and then, the sample was incubated for 2 h at room temperature in a dark chamber. After the reduction process, the Aptamer solution was diluted to 1 µM supplemented with 5 mM of NaCl and 5 mM of MgCl2. The immobilization of Aptamers was carried out by casting the Aptamers solution (8 µL) onto the SPE/AuNPs surface. The samples were then incubated for 2 h at room temperature in a dark chamber. This step was followed by using PBS for washing purposes. To remove the physically adsorbed Aptamer and to passivate the electrode surface, an 8 μL of 1 mM MCH was positioned subsequently on the SPE/AuNPs in the dark chamber for 2 min. A schematic diagram showing the preparation steps mentioned before is shown in Fig. 1 below.

Schematic diagram for the preparation steps of the developed Aptasensor
2.4 Electrochemical detection of HER2
For HER2 detection, 100 µg/ml of the stock solution was prepared in PBS (pH 7.4). The working concentration of HER2 was prepared by appropriate dilutions of the HER2 stock solution in PBS at pH 7.4 containing a 5 mM [Fe(CN)6]3−/4−. The detection of the HER2 protein was carried out by casting various concentrations of HER2 solution (8 µL) onto the SPE/AuNPs/Aptamer/MCH surface at room temperature, and for five minutes. After this, washing with PBS to remove any unbounded HER2 was performed. The binding reaction needs 10 min at 25 °C. The DPV cathodic peak responses of the SPEs/AuNPs/Aptamer/MCH electrodes were studied at different HER2 concentrations (100, 10, 1, 0.1, 0.01, 0.001, and 0.0001 ng/ml).
The selectivity of the Aptasensor was quantitatively evaluated by incubating the SPE/AuNPs/Aptamer/MCH in [Fe(CN)6]3−/4−/PBS solutions containing non-complementary DNA oligonucle-otide (200 ng/ml), non-complementary RNA oligonucle-otide (100 ng/ml), Midkine protein (150 ng/ml), lgG (1 × 104 ng/ml) and the mixture of these interference substances.
3 Results and discussion
The SPEs/AuNPs, SPEs/AuNPs/Aptamer, and SPEs/AuNPs/Aptamer/MCH electrodes were characterized with CV, DPV, and EIS measurements by using a 50 mM PBS, pH 7, that contains a 5 mM [Fe(CN)6]3−/4, as shown in Fig. 2A, B, C. At first, the CV measurements for each modification were recorded between − 0.5 V—0.8 V with a scan rate from 10—90 mVs−1. The results are showing an increase in the cathodic peak currents and anodic peak currents as increasing the scan rate, also, the value of peak potential (ΔEp); which is the difference between the anodic peak potential (Epa), and the cathodic peak potential (Epc), increases at higher scan rates as shown in Fig. 2D. From comparing these results, it is noted that the electrode has notable current peak changes due to the modification, which alters the kinetics of electron transfer. After Aptamer immobilization, the surface resistance was increased, and the kinetics of electron transfer was decreased, causing current reduction, and peak potential increment. While after the modification with MCH, the nonspecifically adsorbed parts of the Aptamer were displaced, because of the repletion between the negative charge of the MCH terminus and the Aptamer backbones. Simultaneously an increment in the current peaks was noted. (Mehennaoui et al. 2019; Khan et al. 2016; Aghaei et al. 2017).

Cyclic voltammetry results of the (A) SPEs/AuNPs, (B) SPEs/AuNPs/Aptamer and (C) SPEs/AuNPs/Aptamer/MCH electrodes. (D) The relation between the square root of the scan rate and ΔEp
Relative variations of the current were checked using the CV measurements at a 70 mVs−1 scan rate as shown in Fig. 3A. Compared with the control SPEs/AuNPs electrode (the black curve), a noticeable decrease in the peak current of the Aptamer probes modified electrode (the red curve) of about 20 µA was noticed. The increment in the difference of the redox peak potential is related to the repulsive interaction of the [Fe(CN)6]3−/4− anions with the negatively charged backbones of the Aptamer. After blocking the SPEs/AuNPs/Aptamer with MCH, the interfacial electron transfer of the probes was impeded. Therefore, the peak currents increased (~ 6 µA), and the difference of the redox peak potential decreased as seen in the blue curve.

(A) Cyclic voltammograms performed at a scan rate of 70 mv/s, (B) the relation between the square root of scan rate with the current peak, and (C) DPV curves gathered at a 3 mV/s scan rate for: are SPE/AuNPs, SPE/AuNPs/Aptamer, SPE/AuNPs/Aptamer/MCH
As shown in Fig. 3B, the correlation between the peak current (\({\mathrm{I}}_{\mathrm{p}}\)) and the square root of the scan rate \({(\mathrm{V}}^{1/2})\) is linear After Aptamer immobilization, the slop was drastically decreased, which is an indication of the strong binding between the Aptamer and the modified electrode surface. The slope, which is \(\frac{\partial {\mathrm{I}}_{\mathrm{p}}}{\partial {\mathrm{V}}^{1/2}}\) is proportional to the effective surface area (\({\mathrm{Area}}_{\mathrm{effective}})\), which can be calculated from Randles–Ševčík equation (Shi et al. 2011):
where \({\mathrm{Area}}_{\mathrm{effective}}\) is the effective surface area (\({cm}^{2})\),\(D\) is the diffusion coefficient (\(6.7\times {10}^{-6} {cm}^{2}{s}^{-1})\), C is the molar concentration of the electroactive species, which is ferricyanide (5 mM), V is the scan rate (\({mV s}^{-1}\)), and n is the number of transferred electrons for the redox reaction. The n value is equal to one according to the following half-reaction, which takes place at the electrode surface:
The \({\mathrm{Area}}_{\mathrm{effective}}\) is 0.3516 \({cm}^{2}\), which is 2.8 times the geometric surface area. Gold nanoparticles increase the effective surface area and increase the conductivity, which provides stable immobilization and improves the electrons transfer at the electrode–electrolyte interface (Mittal et al. 2017).
Differential pulse voltammetry measurements are shown in Fig. 3C. These measurements were studied for further investigations of the sensing interface construction. For the pristine SPEs/AuNPs electrode, the change of the cathodic peak current (Ip) was recorded to be 108 μA (the black curve). After the Aptamer immobilization on the SPEs/AuNPs electrode, the Ip decreased to 77.6 μA (the red curve). After further modification with MCH, Ip increased to 88.5 μA (the blue curve).
In the EIS measurements, the electron-transfer resistance (Rct) was analyzed after each modification step. It is needed to monitor the change in this parameter as it gives an indication of the change of the redox probe kinetics at the electrode interface. The Rct value of the pristine SPEs/AuNPs was as small as 193.357 Ω (blue curve Fig. 4A). The resistance increased to 618.76 Ω, after Aptamer immobilization (black curve Fig. 4A) (Chun et al. 2013). Which can be attributed to the electrostatic repulsion of the negatively charged Aptamer with the solution-phase [Fe(CN)6]3−/4− at the redox probe. Finally, after MCH blocking, the Rct decreased to 433.852 Ω (red curve in Fig. 4A). The Randle's circuit used to model the measured impedance spectra is shown in Fig. 4B. In this circuit, the electrode solution interface is represented by the parallel combination of the charge, Rct, the solution resistance (Rs), and the double-layer capacitor (Cdl). The diffusion of the redox couple from the electrode and into it is represented by Warburg impedance (Zw), which arises at low frequencies. For each obtained EIS spectra, the Rct can be noted from the diameter of the observed well-defined semicircle. The results of the EIS measurements and analysis also indicate that the Aptasensor was prepared efficiently. In addition, the EIS results obtained during the Aptasensor construction were consistent with both the CV and the DPV measurements, respectively. These results confirm that the SPEs/AuNPs/Aptamer/MCH was successfully assembled.

(A) EIS measurements for SPE/AuNPs, SPE/AuNPs/Aptamer, SPE/AuNPs/Aptamer/MCH, and (B) The equivalent Randle's electrical circuit of the measured electrodes
In order to study the topography and roughness during the Aptasensor development, AFM images were obtained. The AFM results of the SPE/AuNPs, SPEs/AuNPs/Aptamer, and SPEs/AuNPs/Aptamer/MCH surface are shown in Fig. 5. For the SPE/AuNPs electrode, the roughness was Ra = 50.54 nm as shown in Fig. 5A. After the immobilization of the Aptamer probes, the Ra value significantly decreased to 17.39 nm, as shown in Fig. 5B. The change in roughness here explains the modification of the electrode surface via Aptamer probes, which completely cover the gold modified carbon electrode. On the other hand, after binding of the Aptamer probes with the target protein (HER2), the average roughness increased to 40.53 nm, as shown in Fig. 5C. These results confirm the formation of layers and quantification of the volume of the Aptamer and the target on the surface of the electrode.

AFM images of (A) SPE/AuNPs, (B) SPE/AuNPs/Aptamer, (C) SPE/AuNPs/Aptamer/HER2
Fourier-transform infrared spectroscopy results were recorded in the spectral range between 900 – 3800 cm−1, as shown in Fig. 6. These spectra were obtained for the three cases under study, where they show how the spectra change from one modification to the other. In the case of SPE/AuNPs/Aptamer (Fig. 6B), Some bands were found are corresponded to the characteristic Aptamer peaks. The vibrations of C-H may be assigned to the bands at about ~ 2908 and 2980 cm−1, respectively, which is arising from the − CH2- groups in the sugar-phosphate backbone. The bands about 1057 cm−1 are assigned to the vibration of P-O-C groups, the vibration of the thiol (S–H) assigned to the band about 2645 cm−1. After further modification with MCH (Fig. 6c), all the bands did not show significant changes, which means that all the absorption peaks characterized by the MCH groups are masked by the Aptamer absorption peaks. These results agrees with other reported results elsewhere (Mehennaoui et al. 2019; Tak et al. 2017; Wang et al. 2016).

FTIR spectra of (a) Bare SPE/AuNPs, (b) SPE/AuNPs/Aptamer, (c) SPE/AuNPs/Aptamer/MCH measured in the spectral range between 900 – 3800 cm−1
The performance of the Aptasensor to detect HER2 was studied by using DPV measurements, as shown in Fig. 7A. The produced DPV cathodic peak currents response gradually decreased as the concentration of HER2 increased. The Aptasensor SPEs/AuNPs/ Aptamer/MCH electrodes were studied with different concentrations as: 100, 10, 1, 0.1, 0.01, 0.001, and 0.0001 ng/ml. As can be seen, the inherent current peak was recorded at Epa = 0.088 V. When the aptaelectrode interacted with a 100 ng/ml concentration of HER2, the interaction exhibited the lowest peak current intensity (38.6 µA) in comparison with the other concentration. It is concluded that at this concentration, the sensor surface contains the highest ratio of the Aptamer-HER2 complex.

(A) DPVs of the Aptasensor SPEs/AuNPs/Aptamer-MCH electrodes studied with different concentrations (100, 10, 1, 0.1, 0.01, 0.001, 0.0001 ng/ml) of HER2 diluted in PBS. (B) The linear variation in the obtained DPV current with log [HER2 target concentration]
The regression equation of the DPV experimental data is y = A + B(x), where y is the biosensor current response (µA), x is the logarithm of HER2 concentration, and A and B are the biosensor constants. Figure 7B shows the linear fitting of the data points which gives y = -7.3343 + 52.85(x) with 0.992 correlation co-efficient, n = 4, and RSD < 5%. The detection limit was found to be 0.001 ng/ml, as it was calculated using S/N = 3 criteria. One of the attractive features of the developed Aptasensor is the fast measuring time, the Aptasensor exhibits fast response with a binding time of only 5 min. In addition, the Aptasensor showed a stable EIS response for 72 h with RSD around 4% After this period, the stability was considerably decreased with RSD around 30%. Table 1 shows recent researched HER2 Aptasensors, in comparison with other HER2 Aptasensors, this work demonstrated excellent performance showing a very low limit of detection, and wide linear range.
To investigate the specificity of the biosensor non-complementary DNA oligonucle-otide, non-complementary RNA oligonucle-otide, Midkine protein, lgG, and a mixture of these mentioned above with HER2 were analyzed under the same conditions using DPV. As illustrated in Fig. 8, the current intensity changes (ΔI) were very poor with the interfering substances, and the Aptasensor showed negligible cross binding to the interfering substances (not greater than 10% of that observed for HER2). It was found that when mixing certain concentration of HER2 (i.e. 10 ng/ml) with an extremely high concentration of interferences mixture, the extent of the interference was found to be around 8%. These results demonstrate that the developed Aptasensor is highly successful in the selective HER2 detection process.

The selectivity of the Aptasensor for HER2 (10 ng/ml), detected against interferences, including Midkine (150 ng/ml), lgG 1 × 104 ng/ml, RNA 100 ng/ml, and DNA 200 ng/ml in PBS
4 Conclusion
In this work, Aptamer based biosensor for HER2 detection was developed, using Au nanoparticles modified screen-printed carbon electrode. Different techniques (CV, DPV, EIS, FTIR, and AFM) were used to characterize the development process. The Au nanoparticle modified Aptasensor showed a log-linear response over a wide concentration range of 0.001—100 ng/mL, a very low detection limit of about 0.001 ng/ml, and a fast binding time of only 5 min. In addition, the aptasensor showed a stable response for three days with RSD around 4%. Moreover, results of the selectivity study demonstrate that the developed Au nanoparticle modified Aptasensor is highly effective in HER2 selective detection with negligible cross binding to the interfering substances (not greater than 10% of that observed for HER2). The incorporation of gold nanoparticles with the screen-printed technology provides support features showing promising results in the aptasensor development.
References
S.A.A. Ahmad, M.S. Zaini, M.A. Kamarudin, An electrochemical sandwich immunosensor for the detection of HER2 using antibody-conjugated PbS quantum dot as a label. J Pharm Biomed Anal 174, 608–617 (2019)
F. Aghaei, S.M. Seifati, N. Nasirizadeh, Development of a DNA biosensor for the detection of phenylketonuria based on a screen-printed gold electrode and hematoxylin. Anal Methods 9, 966–973 (2017)
D. Antuña-Jiménez, M.B. González-García, D. Hernández-Santos, P. Fanjul-Bolado, Screen-Printed Electrodes Modified with Metal Nanoparticles for Small Molecule Sensing. Biosensors 10, 9 (2020)
E. Arkan, R. Saber, Z. Karimi, M. Shamsipur, A novel antibody–antigen based impedimetric immunosensor for low level detection of HER2 in serum samples of breast cancer patients via modification of a gold nanoparticles decorated multiwall carbon nanotube-ionic liquid electrode. Anal Chim Acta 874, 66–74 (2015)
S.K. Arya, P. Zhurauski, P. Jolly et al., Capacitive aptasensor based on interdigitated electrode for breast cancer detection in undiluted human serum. Biosens Bioelectron 102, 106–112 (2018)
H. Beitollahi, S.Z. Mohammadi, M. Safaei, S. Tajik, Applications of electrochemical sensors and biosensors based on modified screen-printed electrodes: a review. Anal Methods 12, 1547–1560 (2020)
F. Boureau, A. Leizorovicz , F. Caulin, [The placebo effect in bone metastatic pain]. Press médicale (Paris, Fr 1983) 17, 1063–1066 (1988). https://doi.org/10.1038/nature01423.Published
G. Bezerra, C. Córdula, D. Campos et al., Electrochemical aptasensor for the detection of HER2 in human serum to assist in the diagnosis of early stage breast cancer. Anal Bioanal Chem 411, 6667–6676 (2019)
W.P. Carney, R. Neumann, A. Lipton et al., Potential clinical utility of serum HER-2/neu oncoprotein concentrations in patients with breast cancer. Clin Chem 49, 1579–1598 (2003)
L. Chun, S.-E. Kim, M. Cho et al., Electrochemical detection of HER2 using single stranded DNA aptamer modified gold nanoparticles electrode. Sensors Actuators B Chem 186, 446–450 (2013)
M. Emami, M. Shamsipur, R. Saber, R. Irajirad, An electrochemical immunosensor for detection of a breast cancer biomarker based on antiHER2–iron oxide nanoparticle bioconjugates. Analyst 139, 2858–2866 (2014)
D. Furrer, F. Sanschagrin, S. Jacob, C. Diorio, Advantages and disadvantages of technologies for HER2 testing in breast cancer specimens. Am J Clin Pathol 144, 686–703 (2015)
M. Freitas, H.P.A. Nouws, C. Delerue-Matos, Electrochemical sensing platforms for HER2-ECD breast cancer biomarker detection. Electroanalysis 31, 121–128 (2019)
M. Freitas, M.M.P.S. Neves, H.P.A. Nouws, C. Delerue-Matos, Quantum dots as nanolabels for breast cancer biomarker HER2-ECD analysis in human serum. Talanta 208, 120430 (2020a)
M. Freitas, H.P.A. Nouws, E. Keating et al., Immunomagnetic bead-based bioassay for the voltammetric analysis of the breast cancer biomarker HER2-ECD and tumour cells using quantum dots as detection labels. Microchim Acta 187, 1–10 (2020b)
W.J. Gullick, Alternative strategies for targeting the epidermal growth factor system in cancer. Breast Cancer Res 3, 390–394 (2001). https://doi.org/10.1186/bcr328
M. Hasanzadeh, N. Shadjou, M. de la Guardia, Early stage screening of breast cancer using electrochemical biomarker detection. TrAC Trends Anal Chem 91, 67–76 (2017)
Y.W. Hartati, L.K. Letelay, S. Gaffar, et al., Cerium oxide-monoclonal antibody bioconjugate for electrochemical immunosensing of HER2 as a breast cancer biomarker. Sens Bio-Sensing Res 27, 100316 (2020)
W.J. Köstler, B. Schwab, C.F. Singer et al., Monitoring of serum Her-2/neu predicts response and progression-free survival to trastuzumab-based treatment in patients with metastatic breast cancer. Clin Cancer Res 10, 1618–1624 (2004)
R. Khan, M. Pal, A.V. Kuzikov et al., Impedimetric immunosensor for detection of cardiovascular disorder risk biomarker. Mater Sci Eng C 68, 52–58 (2016)
R. Laocharoensuk, Development of Electrochemical Immunosensors towards Point-of-care Cancer Diagnostics: Clinically Relevant Studies. Electroanalysis 28, 1716–1729 (2016)
V. Ludovini, S. Gori, M. Colozza et al., Evaluation of serum HER2 extracellular domain in early breast cancer patients: correlation with clinicopathological parameters and survival. Ann Oncol 19, 883–890 (2008)
O.K. Mahfoud, T.Y. Rakovich, A. Prina-Mello et al., Detection of ErbB2: nanotechnological solutions for clinical diagnostics. RSC Adv 4, 3422–3442 (2014)
S. Mehennaoui, S. Poorahong, G.C. Jimenez, M. Siaj, Selection of high affinity aptamer-ligand for dexamethasone and its electrochemical biosensor. Sci Rep 9, 6600 (2019). https://doi.org/10.1038/s41598-019-42671-3
S. Mittal, H. Kaur, N. Gautam, A.K. Mantha, Biosensors for breast cancer diagnosis: A review of bioreceptors, biotransducers and signal amplification strategies. Biosens Bioelectron 88, 217–231 (2017)
A. Mishra, M. Verma, Cancer biomarkers: are we ready for the prime time? Cancers (basel) 2, 190–208 (2010)
J. Mohanraj, D. Durgalakshmi, A. Rakkesh, Current Trends in Disposable Graphene-Based Printed Electrode for Electrochemical Biosensors. J Electrochem Soc 167, 67523 (2020)
M.A.H. Nawaz, S. Rauf, G. Catanante et al., One step assembly of thin films of carbon nanotubes on screen printed interface for electrochemical aptasensing of breast cancer biomarker. Sensors 16, 1651 (2016)
J.H. Niazi, S.K. Verma, S. Niazi, A. Qureshi, In vitro HER2 protein-induced affinity dissociation of carbon nanotube-wrapped anti-HER2 aptamers for HER2 protein detection. Analyst 140, 243–249 (2015)
D. Ou, D. Sun, X. Lin et al., A dual-aptamer-based biosensor for specific detection of breast cancer biomarker HER2 via flower-like nanozymes and DNA nanostructures. J Mater Chem B 7, 3661–3669 (2019)
S. Patris, P. De Pauw, M. Vandeput et al., Nanoimmunoassay onto a screen printed electrode for HER2 breast cancer biomarker determination. Talanta 130, 164–170 (2014)
A. Qureshi, Y. Gurbuz, J.H. Niazi, Label-free capacitance based aptasensor platform for the detection of HER2/ErbB2 cancer biomarker in serum. Sensors Actuators B Chem 220, 1145–1151 (2015)
A. Ravalli, D. Voccia, I. Palchetti, G. Marrazza, Electrochemical, electrochemiluminescence, and photoelectrochemical aptamer-based nanostructured sensors for biomarker analysis. Biosensors 6, 39 (2016)
G. Riccio, G. Esposito, E. Leoncini et al. Cardiotoxic effects, or lack thereof, of anti-ErbB2 immunoagents. FASEB J 23(9), 3171–3178 (2009). https://doi.org/10.1096/fj.09-131383
P.F. Rostamabadi, E. Heydari-Bafrooei, Impedimetric aptasensing of the breast cancer biomarker HER2 using a glassy carbon electrode modified with gold nanoparticles in a composite consisting of electrochemically reduced graphene oxide and single-walled carbon nanotubes. Microchim Acta 186, 495 (2019)
R. Salahandish, A. Ghaffarinejad, S.M. Naghib et al., Nano-biosensor for highly sensitive detection of HER2 positive breast cancer. Biosens Bioelectron 117, 104–111 (2018)
J. Shi, J.C. Claussen, E.S. McLamore, et al., A comparative study of enzyme immobilization strategies for multi-walled carbon nanotube glucose biosensors. Nanotechnology 22, 355502 (2011)
A. Smart, A. Crew, R. Pemberton, et al., Screen-Printed Carbon Based Biosensors and their Applications in Agri-Food Safety. TrAC Trends Anal Chem 115898 (2020)
H. Sun, Y. Zu, Aptamers and their applications in nanomedicine. Small 11, 2352–2364 (2015)
A. Tabasi, A. Noorbakhsh, E. Sharifi, Reduced graphene oxide-chitosan-aptamer interface as new platform for ultrasensitive detection of human epidermal growth factor receptor 2. Biosens Bioelectron 95, 117–123 (2017)
S.D. Tallapragada, K. Layek, R. Mukherjee et al., Development of screen-printed electrode based immunosensor for the detection of HER2 antigen in human serum samples. Bioelectrochemistry 118, 25–30 (2017)
M. Tak, V. Gupta, M. Tomar, An electrochemical DNA biosensor based on Ni doped ZnO thin film for meningitis detection. J Electroanal Chem 792, 8–14 (2017). https://doi.org/10.1016/j.jelechem.2017.03.032
C. Tse, D. Brault, J. Gligorov et al., Evaluation of the quantitative analytical methods real-time PCR for HER-2 gene quantification and ELISA of serum HER-2 protein and comparison with fluorescence in situ hybridization and immunohistochemistry for determining HER-2 status in breast cancer pa. Clin Chem 51, 1093–1101 (2005)
Q. Wang, F. Gao, J. Ni et al., Facile construction of a highly sensitive DNA biosensor by in-situ assembly of electro-active tags on hairpin-structured probe fragment. Sci Rep 6, 1–10 (2016). https://doi.org/10.1038/srep22441
Y. Zhu, P. Chandra, Y.B. Shim, Ultrasensitive and selective electrochemical diagnosis of breast cancer based on a hydrazine-Au nanoparticle-aptamer bioconjugate. Anal Chem 85, 1058–1064 (2013). https://doi.org/10.1021/ac302923k
Acknowledgements
This research was fully supported by the Royal Hashemite Courte under the Royal initiative for innovative projects in Nanotechnology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Harahsheh, T., Makableh, Y.F., Rawashdeh, I. et al. Enhanced aptasensor performance for targeted HER2 breast cancer detection by using screen-printed electrodes modified with Au nanoparticles. Biomed Microdevices 23, 46 (2021). https://doi.org/10.1007/s10544-021-00586-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10544-021-00586-9