
Abstract
Current in vitro gut models lack physiological relevance, and various approaches have been taken to improve current cell culture models. For example, mimicking the three-dimensional (3D) tissue structure or fluidic environment has been shown to improve the physiological function of gut cells. Here, we incorporated a collagen scaffold that mimics the human intestinal villi into a microfluidic device, thus providing cells with both 3D tissue structure and fluidic shear. We hypothesized that the combined effect of 3D structure and fluidic shear may provide cells with adequate stimulus to induce further differentiation and improve physiological relevance. The physiological function of our ‘3D gut chip’ was assessed by measuring the absorptive permeability of the gut epithelium and activity of representative enzymes, as well as morphological evaluation. Our results suggest that the combination of fluidic stimulus and 3D structure induces further improvement in gut functions. Our work provides insight into the effect of different tissue environment on gut cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Oral intake of drugs or food is followed by a digestion, absorption, and metabolism. The main sites where absorption and metabolism occur are the intestine and the liver, respectively, which are complex processes with inter-organ signaling (Bieghs and Trautwein 2014; Li and Chiang 2014). Several different versions of in vitro model systems have been developed to mimic the complexity of absorption and metabolism in vivo. The most widely used cell-based model of intestinal absorption is the Caco-2 model system, which uses a human epithelial colorectal adenocarcinoma cell line (Artursson, Palm et al. 2001). Although derived from colorectal cancer, this cell line exhibits traits of small intestinal epithelium, such as polarization, expression of a few transporter and tight junction proteins (Rousset 1986). However, this model is still far from a faithful reproduction of the human gut, because of several shortcomings such as absence of commensal microorganisms (He, Mishchuk et al. 2013), lack of peristaltic movement and flow (Kim, Huh et al. 2012), low expression of mucus proteins (Behrens, Stenberg et al. 2001) and cytochrome P450 enzymes (Galetin and Houston 2006; Kublbeck, Hakkarainen et al. 2016).
Organ-on-a-chip technology has been proposed as a solution to overcome the limitations of current in vitro models, by recapitulating the essential elements of in vivo tissue environment. One of the earliest demonstration was reported by Bhatia et al., who reproduced the oxygen gradient in the liver and observed differential expression of metabolic enzymes in response (Allen and Bhatia 2003). During the last decade, organ-on-a-chip devices aimed at reproducing the functions of different organs have been developed (Sung, Esch et al. 2013). Several notable proof-of-concept studies have demonstrated that recapitulating the key environmental factors contribute to restoring essential functions of human organs.
Several different approaches have been taken to overcome the aforementioned limitations and improve current models for gut absorption and metabolism. One of the earliest approach was to co-culture Caco-2 cells with supporting cells to compensate for missing functions. Co-culturing Caco-2 cells with goblet cells have been widely used to improve mucus production (Beduneau, Tempesta et al. 2014). Caco-2 cells have been co-cultured with immune cells to observe immune response of the gut epithelium (Ramadan and Jing 2016). Co-culture of commensal bacteria has also been attempted to mimic the crosstalk between the gut epithelium and the microbes (Haller, Serrant et al. 2002; He, Mishchuk et al. 2013). Another notable approach is to provide cells with mechanical stimulus, such as fluidic shear stress or mechanical strain. Exposing Caco-2 cells to fluidic shear or periodic strain has been shown to improve gut functions, by inducing expression of metabolic enzymes, mucus proteins, as well as formation of villi-like structures (Kim, Huh et al. 2012; Kim and Ingber 2013). One of the unique characteristics of gut epithelium is its inherent three-dimensional (3D) structure, shown as finger-like protruding structures with a length of up to 1 mm (Tortora and Grabowski 1993). Known as intestinal villi, these structures are believed to aid in increasing the absorptive surface area, and involved in differentiation of gut epithelial cells (Rao and Wang 2010; Kim and Ingber 2013). This has led researchers to hypothesize that culturing gut cells on a scaffold mimicking the structure of 3D villus and crypt may induce improved differentiation of the cells (Wang, Murthy et al. 2010). We previously developed a novel method of fabricating a collagen scaffold mimicking the shape and density of the human intestinal villi, and showed that culturing Caco-2 cells on this scaffold resulted in a tissue structure with striking resemblance to the human intestinal villi (Sung, Yu et al. 2011). Further studies revealed that simply culturing Caco-2 cells on this villi-shaped scaffold induces changes in the physiological functions of the cells, such as mucus production and its resistance to bacterial invasion (Kim, Chi et al. 2014). It was also shown that other gut-related traits of Caco-2 cells are also enhanced, such as drug absorption kinetics (Yu, Peng et al. 2012), expression of alkaline phosphatase and mucus proteins (Costello, Hongpeng et al. 2014a), host-microbial interaction (Costello, Sorna et al. 2014b).
These studies suggest that recapitulation of gut tissue environment can influence the behavior of gut cells. In fact, a similar principle was verified with other cell types, including those originating from the liver (Ware and Khetani 2016), blood vessel (Zheng, Chen et al. 2012; Zhou and Niklason 2012), lung (Huh, Matthews et al. 2010), and bone (Kuttenberger, Polska et al. 2013; Torisawa, Spina et al. 2014). Various environmental factors, including the fluidic shear (Nieskens and Wilmer 2016), 3D architecture (Jackson and Lu 2016), cell-cell interaction (Chen, Srigunapalan et al. 2013), and chemical concentration gradient (Allen and Bhatia 2003) can elicit improved responses from cultured cells. However, as organ-on-a-chip technology has made significant progress, an attempt to combine different environmental factors together and evaluate their combined effect has been limited. An increasing number of combinations of various environmental factors makes it difficult to explore all combinations of the factors. For example, the gut environment is a complex and dynamic combination of various factors, such as various cell types (van der Flier and Clevers 2009), complex tissue architecture (Lodish 2000), presence of microbiota (Quigley 2013), and peristaltic movement (Huizinga and Lammers 2009). While it is probably almost impossible at this point to recapitulate all these factors to perfectly match the in vivo condition, it is probably worthwhile to elucidate the contribution of each factor and find out what are the key players in maintaining the essential functions of the tissue.
In this study, we aimed to reproduce the two major characteristics of the gut tissue, the 3D villi structure, and the fluidic shear in a microfluidic chip. Although both factors are known to affect the gut cell functions (Kim and Ingber 2013; Costello, Hongpeng et al. 2014a; Costello, Sorna et al. 2014b; Chi, Yi et al. 2015), a systematic study of how these two factors contribute to the gut cell functions have not been done yet. We compared the tissue morphology and essential functions of Caco-2 cells in 2D monolayer culture on transwells, 2D monolayer culture on microfluidic chips, and 3D culture on microfluidic chips. Comparison of these three conditions gave insight into how these environmental factors affect the intestinal cell physiology. Our 3D microfluidic gut chip may potentially offer information about how gut tissue environment affect the cells, and work as an improved gut model for drug development and studying gut-related diseases. In particular, our gut chip provides a step towards an integration of the gut and the liver, which is quite an important requirement for reproducing the first-pass metabolism in humans (Choe, Ha et al. 2017).
2 Materials and methods
2.1 Fabrication of microfluidic chip and 3D villi scaffold
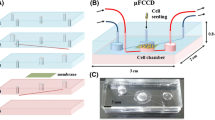
A microfluidic chip consists of three layers of polydimethylsiloxane (PDMS, Corning), a slide glass and a polyester (PET) membrane which was manually separated from transwell-clear inserts (Corning, CLS3450). The schematics of the chip is provided in Fig. 1a, b. The size of all three PDMS layers are 40 mm × 20 mm. The first layer (layer 1, 4 mm thick) provides reservoirs for cell culture media. A biopsy punch (Miltex, 8 mm) was used to punch five holes in the first layer. The second layer (layer 2, 3 mm thick) provides fluidic channels connecting to the apical side of the gut, and has a 6 mm hole at the center and four 1 mm holes at the corners. The third layer (layer 3, 0.3 mm thick) has an 8 mm hole at the center and two 2 mm holes at the corners. The layer 1 were made by simply pouring uncured PDMS into an empty dish to reach the desired thickness and cured at 60 °C for 2 h. The layer 2 was made by pouring PDMS onto a wafer with a SU-8 mold of fluidic channels. The layer 3 was made by spin coating PDMS on the same wafer mold as the layer 2, at 300 RPM for 30 s (Spin-1200D, Midas, Korea). The wafer mold has fluidic channels 120 μm wide and 200 μm tall. The layer 2 and 3 are made from the same wafer mold, but are bonded in different directions, that is, the layer 2 has channels facing up, whereas the layer 3 has channels facing down. A slide glass, all PDMS layers, and a PET membrane was plasma treated (CUTE, Femto Sciences, Korea), and bonded together. The order of bonding was the slide glass, layer 3, PET membrane, layer 2, and layer 1, from the bottom. Figure 1c shows the picture of an assembled chip.

a Schematics of a gut chip b Side-view of a gut chip c Picture of a gut chip showing two sets of reservoirs for the apical (red) and basolateral sides (blue)
The 3D villi structure was fabricated following the previously reported method (Sung, Yu et al. 2011; Kim, Lee et al. 2013; Kim, Chi et al. 2014). To briefly explain, a wafer mold with the inverse structure of the villi was fabricated using photolithography technique. A SU-8 mold with the villi structure was replicated from the wafer mold, and bonded to a gasket to make the alginate inverse mold. Finally, collagen villi scaffold was replicated from the alginate mold, which was then dissolved. The fabricated collagen villi structure was bonded to a porous membrane support fixed inside the PDMS chip, using collagen as a glue.
2.2 Cell culture
Human colon carcinoma cell line, Caco-2 (KCLB # 30037.1, passage number between 30 and 50) was used. When culturing on 24-well transwell insert, 100 μL of cell suspension was seeded at 2 × 105 cells/ml density. A high-glucose DMEM (Gibco, 11,995–065) with 10% FBS (Welgene) and 1% penicillin-streptomycin (Welgene) was changed every two days for 21 days of culture. When culturing cells on chip, cells were seeded at the same density, but were cultured for only 14 days. The chip with cells was placed inside a sterile dish, containing wet filter papers to maintain humidity. The chip was set still for initial 7 days, and was operated with gravity flow device for the next 7 days. The gravity flow device was custom-built mechanical stage with computer control. The tilting angle and frequency of tilting can be controlled. For this study, we used a tilting angle of 10 degrees, velocity of 0.1 degrees/s, and 500 s interval between changing the angle. This gave a flow rate of approximately 100 μL/min through the gut cell culture chamber. This flow rate determined based on our previous studies, where the flow rate higher than this sometimes resulted in cells detaching from the surface, as well as based on the actual flow rate inside the gut (Kim, Lee et al. 2013; Chi, Yi et al. 2015; Choe, Ha et al. 2017). The whole gravity flow device containing the chips was placed inside a cell culture incubator maintained at 37 °C and 5% CO2.
2.3 RT-PCR (reverse transcription polymerase chain reaction)
Total RNA was extracted from cells cultured in either transwell or chip using RNeasy Micro Kit (Qiagen, Germany). cDNA fragments were the synthesized from the extracted RNA using an AccuPower® Cycle Script RT PreMix kit (Bioneer, Korea). Gene expression was determined by RT-PCR using Lightcycler® Nano System (Roche, Germany). Each 100 ng cDNA was mixed with iQ SYBR Green premix (Bio-Rad, Hercules, CA, USA) and specific primers. The PCR primers were as follows: Forward: 5′-CTT CAT CGA GTC ACT GCC TAA T -3′, reverse: 5′- TAA CAA GGG CAC GAG CTA TG-3′ for hP-gp; forward: 5′-AAG AGT TGA CAG TCC CAT GGC ATA C -3′, reverse: 5′-ATC CAC AGG CGA AGT TAA TGG AAG -3′ for hOccludin; forward: 5′-CGG TCC TCT GAG CCT GTA AG-3′, reverse: 5′-GGA TCT ACA TGC GAC GAC AA-3′ for hZo-1; forward: 5′- AGG TGG TGA AGC AGG CGT CGG AGG G-3′, reverse: 5′- CAA AGT GGT CGT TGA GGG-3′ for hGAPDH as housekeeping gene. For data analysis, the raw threshold cycle (CT) value was first normalized to a housekeeping gene for each sample for ΔCT. The normalized Δ CT was then calibrated to the control cell samples, resulting in a ΔΔCT.
2.4 Immunostaining
Cell culture media was removed from the chip and cells were washed with DPBS. Cells were then fixed by treating with 4% (w/v) paraformaldehyde (Sigma, 158,127) for 1 h. Then cells were washed with 0.1% BSA (Gibco, 30,063) three times for five minutes. Cells were permeabilized by treating with 0.3% (w/v) Triton-X 100 (Junsei, 49,415–1601, Japan) for 1 h. Cells were washed with 0.1% BSA for five minutes. To stain actin, Rhodamine phalloidin (Sigma, P1951) was diluted in DPBS at 1/100 ratio before treating cells for 90 min. To stain nucleus, Hoechst 33,342 (Invitrogen) was diluted in DPBS at 1/600 ratio before treating cells for 30 min. After staining process is complete, cells were washed with DPBS for 5 min. Stained cells were examined under confocal laser scanning microscopy (CLSM, Zeiss LSM700, Carl Zeiss).
2.5 Transmission electron microscopy (TEM) and H & E staining
The tissue morphology of Caco-2 cells was examined by transmission electron microscopy (TEM). For cells cultured as 2D monolayer, the porous membrane with cells attached was separated from the chip or transwell, fixed with 4% (w/v) paraformaldehyde for 24 h. In case of cells cultured in 3D forms, the villi scaffold with cells was separated and fixed in 2.5% (v/v) glutaldehyde solution for 24 h. Cells were imaged using Cryo-Transmission Electron Microscope (Cryo Tecnai F20). The tissue slide was placed in an autostainer (Leica) for staining. After staining, the tissue slide was placed in xylene four times for 10 min. Xylene was removed by immersing in absolute ethanol twice for 1 min, and in 95% ethanol twice for 1 min. Then the sample was washed in water, stained with Meyer’s hematoxylin solution (Merck, 1.09249) for 7 min and rinsed with water again. The sample was stained with 1% acidic eosin B solution (Sigma, 861,006) for 3 s, and rinsed with water. For dehydration, the sample was washed in 95% ethanol three times for 10 s. After staining is complete, the sample was mounted and examined.
2.6 Permeability assay
For culture of Caco-2 cells in the chip, cells were seeded into a transwell insert (Costar, 3470, Corning) or collagen villi scaffold at 6 × 104 cells/cm2 density. Media was removed from the transwell insert or gut chip, and washed with Hank’s Balanced Salt Solution (HBSS, Sigma, H6648) for 1 h at 37 °C. In case of using a transwell for influx permeability assay, 200 μL of 3.33 μg/ml Fluorescein (Sigma, 46,955) or 25 μg/ml Rhodamine 123 (Sigma, 83,702) was injected to the apical side, and 1 ml of HBSS was inserted into the basal side. In case of efflux permeability assay, 1 ml of rhodamine 123 solution was injected to the basal side, and 200 μL of HBSS was inserted to the apical side. In case of measuring the influx permeability of in the chip, 325 μL of 3.33 μg/ml Fluorescein or 25 μg/ml Rhodamine 123 was inserted into the cell chamber and medium reservoir (roughly 150 μL in the chamber and 175 μL in the medium reservoir for the apical layer), and HBSS was inserted into the reservoir for the basal layer. For measuring the efflux permeability in the chip, HBSS buffer was inserted into the reservoir for the apical layer, and Rhodamine 123 solution was inserted into the reservoir for the basal side. 100 μL of sample was taken every hour, and fluorescent intensity was measured using a fluorescence spectrophotometer (Cary Eclipse Fluorescence Spectrophotometer, Agilent Technologies) at 494 nm excitation and 512 nm emission wavelength for Fluorescein and 511 nm excitation and 533 nm emission wavelength for Rhodamine 123. After the measurement, the sample was placed back into the transwell or the chip. Fluorescent intensity was converted to concentration using a standard curve of known concentrations, and the permeability was calculated using following equation, where dQ refers to the molecular flux, C0 refers to the initial concentration in the donor compartment, and A denotes the surface area.
2.7 Measurement of enzyme activity
Aminopeptidase and cytochrome P450 (CYP) 3A4 activities of Caco-2 cells were measured. In case of a transwell, enzyme activity was measured directly in the transwell, and in case of chip culture, the membrane or collagen villi scaffold was separated from the chip and transferred to a 96-well plate before measurement. For aminopeptidase activity measurement, the enzyme’s substrate, L-alanine-4-nitroanilide hydrochloride (A4N; Sigma, A9325), was added to cell culture media at 1.5 mM concentration. Cell culture media was removed and 100 μL media containing the substrate was added to the well, and incubated for 2 h at 37 °C. 100 μL sample was taken, and transferred to Costar assay plate (96-well, no lid, Corning 3631) and absorbance was measured at 405 nm using Microplate Spectrophotometer (Multiskan™, GO, Thermo Scientific™). The concentration of product, 4-nitroaniline (pNA) was calculated from the absorbance data using a pre-generated standard curve using known concentrations.
The activity of CYP450 3A4 was measured in a similar manner. We used VIVID BOMCC substrate (Life technologies, P2975) as a substrate for CYP450 3A4 enzyme. The VIVID BOMCC substrate was diluted to 10 μM concentration and incubated with cells for 30 min at 25 °C. 100 μL sample was taken and transferred to Costar assay plate, and fluorescence spectrophotometer was used to measure fluorescent intensity at 460 nm excitation and 495 emission wavelength. A standard curve was drawn using known concentrations of BOMCC product. To obtain a specific enzyme activity per cell, the cell number was counted using Cell Counting Kit-8 (Dojindo, CK04). After the enzymatic reaction, media was removed, 100 μL of media containing CCK-8 in 1:10 ratio was added to each well. After reaction for 90 min at 37 °C, 100 μL of sample was transferred to a Costar assay plate, 96-well, and absorbance was measured at 450 nm wavelength, using Microplate Spectrophotometer (Multiskan™, GO, Thermo Scientific™). The statistical significance of data was examined using the student-t test.
2.8 Statistical analysis
All of quantitative data represents means of at least more than three samples (triplicates), and the error bars in the graph represents standard deviations. The statistical significance of all data was evaluated using students-t method, and in the figures, * denotes P < 0.05, ** denotes P < 0.01, and *** denotes P < 0.001.
3 Results
3.1 Cell morphology
To evaluate the formation of gut epithelium by Caco-2 cells, actin and nucleus of cells were stained with Phalloidin and DAPI, respectively. Figure 2 shows the confocal microscopy images of Caco-2 cells in the gut chip after staining. Fourteen days after Caco-2 cells were seeded, cells were evenly spread over the surface of the membrane or collagen scaffold. Figures 2a, c show images of cells cultured on a porous membrane, as 2D monolayer in the chip. They show that Caco-2 cells were able to cover the membrane to confluency. Figures 2d, f show images of cells cultured on villi scaffold in the chip. It can be verified that the Caco-2 cells has spread well and formed uniform monolayer across the surface of the collagen villi. The height of the original villi before cell seeding is estimated to be roughly 300 μm (data not shown). After two weeks of cell culture on the villi scaffold, villi were observed to become shorter, to about 100 ~ 150 μm. This is thought to be the result of cell spreading and contraction. However, the overall shape of the villi was still conserved, as can be seen from the confocal images in Fig. 2 and H&E stained image in Fig. 3. Judging from the images, the aspect ratio of the villi, that is, the ratio of the height to the diameter was approximately 1.5, whereas the original villi before cell culture had the aspect ratio between 4 and 5 (Sung, Yu et al. 2011).

Confocal microscopy images of Caco-2 cells cultured in the chip as 2D monolayer (a) nucleus stained with DAPI (blue) (b) actin stained with Rhodamine Phalloidin (red) c Merged image showing both nucleus and actin. Confocal microscopy images of Caco-2 cells cultured in the chip on 3D villi scaffold (d) nucleus stained with DAPI (blue) (e) actin stained with Rhodamine Phalloidin (red) f Merged image showing both nucleus and actin. Scale bar 50 μm

H & E stained image of Caco-2 cells cultured (a) as 2D monolayer in the chip and (b) on 3D villi scaffold in the chip. TEM image of Caco-2 cells cultured (c) as 2D monolayer in the chip and (d) on 3D villi scaffold in the chip
The tissue morphology was examined more closely with H & E staining. Figure 3a shows the H & E stained cells cultured as 2D monolayer in the chip, and Fig. 3b shows the H & E stained cells cultured as 3D on villi scaffold in the chip. Both figures show formation of an epithelial barrier across the membrane or villi surface. A difference in the cell morphology was noted, however, cells on 3D villi scaffold showed more prismatic morphology, resembling that of the human intestinal epithelium better than the cells in 2D monolayer (Pusch, Votteler et al. 2011). In contrast, cells in 2D monolayer showed morphology that is more flat and attached more closely to the membrane. A transmission electron microscope (TEM) images of the cells in the chip show formation of microvilli on cell surface in both 2D and 3D culture of Caco-2 cells (Fig. 3c, d). However, the shapes of microvilli were better conserved in case of 2D culture in a chip, whereas in case of 3D villi in the chip, microvilli were damaged in some cases. We speculate that this was caused by the villi scaffold embedded inside the chip, since the presence of the villi would have caused the fluidic shear to act more strongly on the cells than the case of 2D culture inside the chip.
3.2 Enzyme activity assay
Although majority of metabolic activity is exerted by the liver, a significant portion of metabolism occurs in the gut also. In particular, high activity of cytochrome P450 3A4 was detected in the human gut epithelium (Zhang, Dunbar et al. 1999). However, Caco-2 cells are known to express a very low level of P450 3A4 enzyme, which is thought to be one of the major drawbacks of the cell line as a gut model (Pang 2003). The specific activity of P450 3A4 in Caco-2 cells were compared between the 2D well, 2D chip, and 3D villi chip condition (Fig. 4a). The specific activity in the 2D well, 2D chip, and 3D villi chip condition was found to be 1.66 × 10−6, 3.894 × 10−6, and 1.18 × 10−5 mol/1010 cells/h, respectively. P450 3A4 activity was enhanced about two-fold when Caco-2 cells were cultured in a fluidic condition, and was enhanced even further, about additional three-fold higher when the cells were cultured in 3D in fluidic condition. In overall, Caco-2 cells in 3D, fluidic condition showed P450 3A4 activity that is approximately 7 times higher than the same cells cultured in static, 2D condition. This result shows that the presence of mechanical stimulus, that is, culturing cells in a chip, together with morphological and biochemical stimulus, that is, culturing cells on villi-shaped scaffold made of collagen, both enhances the metabolic function of Caco-2 cells.

a Cytochrome P450 3A4 activity of Caco-2 cells in three different culture conditions b Aminopeptidase activity of Caco-2 cells in three different culture conditions
Aminopeptidase activity of Caco-2 cells in different conditions were also compared (Fig. 4b). Aminopeptidase is a apical brush border enzyme highly expressed in the small intestine, and is frequently used as a marker of differentiation of gut cells (Piana, Gull et al. 2007). The aminopeptidase activity of Caco-2 cells was 0.26, 1.23, and 0.92 fmol/min/cell for 2D well, 2D chip, and 3D villi chip condition, respectively. Similar to the case of P450 activity, aminopeptidase activity was enhanced about five-fold when Caco-2 cells were cultured in fluidic condition. However, culturing cells on the 3D villi scaffold did not induce enhancement of aminopeptidase activity, because the enzyme activity measured in the 3D villi chip condition was slightly lower than the 2D chip condition. In contrast to the case of P450 3A4 activity, aminopeptidase activity of Caco-2 cells was enhanced in fluidic condition better than the 3D culture condition.
3.3 Measurement of permeability
The absorptive permeability of model molecules was measured. In particular, model molecules that are absorbed via passive paracellular pathway, Fluorescein and Rhodamine 123, were tested (Leonard, Collnot et al. 2010). In addition to the passive paracellular absorption in influx direction, Rhodamine 123 is known to be transported via carrier-mediated route in case of efflux transport (Yumoto, Murakami et al. 1999; Yumoto, Murakami et al. 2001; Troutman and Thakker 2003). Figure 5a shows the absorption permeability of Fluorescein in influx direction across Caco-2 cell layer in three different conditions, 2D well, 2D chip, and 3D chip. Fluorescein was absorbed about 5-fold more slowly when Caco-2 cells were cultured in monolayer in the chip, compared to a well culture condition. In contrast, culturing cells in 3D form resulted in an increase in the permeability, even when they were cultured in the chip. This suggests that the mechanical stimulus in the chip caused reduction in the absorptive permeability, whereas 3D culture induced more efficient absorption. The trend was different for Rhodamine 123, however, which showed increased absorption permeability in influx direction in both cases, 2D chip and 3D chip (Fig. 5b). In case of efflux transport of Rhodamine 123, permeability decreased about two-fold when Caco-2 cells were cultured in the chip (2D chip), and was further reduced about two-fold when the cells were cultured in 3D in the chip (Fig. 5c). The presence of fluidic stimulus in the chip and 3D culture caused a decrease in the efflux transport of Rhodamine 123.

Permeability measurement across Caco-2 gut epithelium in three different culture conditions. a Influx absorption permeability of Fluorescein, b influx absorption permeability of Rhodamine 123 c efflux permeability of Rhodamine 123
4 Discussion
Gut epithelial cells are known to proliferate from the crypt region, and go through further differentiation while migrating towards the top of the villi (Wang, Murthy et al. 2010; Wang, Ahmad et al. 2014). This implies that there exist spatial heterogeneity in the gut epithelium in terms of cells’ physiology (Leonard, Collnot et al. 2010). It has been shown that the pattern of bacterial invasion in the gut epithelium also shows spatial variation (Costello, Sorna et al. 2014b). These studies suggest that the three-dimensional topology of the gut tissue may influence the physiology of cells. In fact, it has been shown that culturing Caco-2 cells on a villi-shaped scaffold resulted in reduction of absorptive permeability of model drugs, compared to the same cells cultured in 2D monolayer (Yu, Peng et al. 2012). In this study, we hypothesized that the combination of fluidic shear and 3D tissue architecture may induce further physiological changes in Caco-2 cells. In fact, previous studies also reported that exposing cells to mechanical strain and fluidic shear resulted in formation of villi-like structure and increased expression of differentiation markers (Kim, Huh et al. 2012; Kim and Ingber 2013).
When Caco-2 cells were cultured on the 3D villi scaffold in the chip, significant reduction in the villi height was observed. This was also observed in case of static cell culture condition, that is, Caco-2 cells cultured 3D villi scaffold in wells (Sung, Yu et al. 2011). This was probably because the gut epithelial cells caused considerable amount of contraction (Hu, Shi et al. 2010), as well as degradation by secretion of protease proteins (Salmela, Pender et al. 2004). The presence of fluidic shear also could have contributed to the deformation of 3D villi scaffold. However, even after two weeks of culture in the chip, the villi shape was relatively well maintained, with height of approximately 100 μm, which was still considerably greater than that reported in other studies (Kim, Huh et al. 2012; Kim and Ingber 2013).
Caco-2 cells in both 2D monolayer culture and 3D culture in the chip showed reasonably good morphology. Several minor differences were noted, for example, cells in 3D culture in the chip showed more prismatic morphology compared to monolayer culture in the chip (Fig. 3). It is still unclear if this the result of 3D culture or fluidic culture condition. It has been reported that culturing gut epithelial cells in a perfusion culture condition resulted in more prismatic morphology with increased polarization (Pusch, Votteler et al. 2011). In case of 3D culture in the chip, the presence of villi scaffold could have caused the cells to be exposed to a greater amount of shear compared to 2D culture in the chip, because cells are located nearer to the fluidic channels. The presence of 3D villi topology itself could cause heterogenic fluidic pattern near the surface, resulting in higher fluidic shear in some regions. Although adequate amount of fluidic shear can be beneficial to Caco-2 cells (Chi, Yi et al. 2015), excessively high shear stress probably has detrimental effect on the cells. Further in-depth study of fluidic pattern near the villi scaffold in the chip may help optimize the fluidic shear stress on cells. The formation of proper, intact intestinal barrier should be confirmed by staining with tight junctions proteins such as ZO-1 and occludin, and we are planning for this experiment in our subsequent paper.
After ingestion of a drug, a significant portion of metabolism also occurs in the gut (Lin, Chiba et al. 1999). Among various metabolic enzymes, we studied cytochrome P450 (CYP) 3A4 because it is known to be a major active form in the gut, and Caco-2 cells are known for its very low expression level (Zhang, Dunbar et al. 1999; Pang 2003). Our result suggests that chip culture condition and 3D culture condition both contribute to enhanced activity of P450 3A4. A recently published paper suggest that mechanical stimuli, including fluidic shear and mechanical strain, induce increased activity of P450 enzyme in gut cells (Kim, Huh et al. 2012; Kim and Ingber 2013). A similar study using different cell types, for example hepatic cells, also shows a similar evidence that mechanical stimulus induces positive effect on cell’s metabolic activity (Prot, Aninat et al. 2011). Our study shows a result consistent with these studies, but further suggests that combination of mechanical stimulus and 3D culture environment induces additional improvement. Relative contribution of the 3D and fluidic shear on the enhancement of P450 enzyme activity is also an interesting point. In a separate study, we observed about two-fold increase in the enzyme activity when Caco-2 cells were cultured on the 3D villi, but in a static environment (unpublished result). From this, we can conclude that the enhancement of the P450 activity results from combination of 3D and fluidic environment, with roughly equal contribution from the both factors.
In case of aminopeptidase activity, the fluidic culture condition improved the enzyme activity, but no further positive effect of 3D culture was observed. Other studies also confirm that a mechanical stimulus can induce enhancement of aminopeptidase activity (Kim, Huh et al. 2012; Chi, Yi et al. 2015). One possible reason for the lack of observed improvement by the 3D culture condition may be the fact that microvilli was more damaged in case of 3D culture in the chip than 2D culture in the chip. Aminopeptidase is a brush border enzyme known to be expressed mainly in the microvilli of the gut epithelium (Tobey, Heizer et al. 1985).
In our study, we cultured Caco-2 cells on chip for 14 days before conducting experiment. This was mainly based on our observation that 14-day on-chip cultures were sufficient to induce differentiation of Caco-2 cells. This is consistent with several previous literatures, where Caco-2 cells were cultured for 14 days because culturing longer results in degradation of the scaffold (Sung, Yu et al. 2011; Yu, Peng et al. 2012). More interestingly, exposing Caco-2 cells to the fluidic environment shortened the time required for complete differentiation. Several groups reported differentiation of Caoc-2 cells after on-chip culture for considerably shorter time, for examples 3 to 5 days (Kim, Huh et al. 2012; Kim and Ingber 2013; Chi, Yi et al. 2015).
Influx and efflux permeabilities of two model molecules, Fluorescein and Rhodamine 123, were examined. Fluorescein was chosen as model for influx transport via passive, paracellular route (Leonard, Creed et al. 2000). Rhodamine 123 was chosen as a model molecule for influx transport via passive, paracellular route and efflux transport via carrier-mediated route (Yumoto, Murakami et al. 1999; Yumoto, Murakami et al. 2001; Troutman and Thakker 2003). Results show that for Fluorescein, influx permeability decreases in chip culture condition, but increases in 3D culture condition (Fig. 5a). Exposing Caco-2 cells to fluidic shear has been associated with stronger formation of tight junctions and increase in trans epithelial electrical resistance (TEER) (Chi, Yi et al. 2015). This is likely to have resulted in decreased absorptive permeability of Fluorescein in this study. This is also consistent with expression of tight junction proteins, Occludin and ZO-1, both of which was increased in the chip culture condition (See Supplementary Information). Culturing cells in 3D in the chip, however, resulted in higher permeability, at a similar level to cells in monolayer in the transwell condition. This is probably due to the increased absorptive surface area of the villi scaffold. A similar observation was made by a different research group, who reported reduced TEER values and increased paracellular transport when Caco-2 cells were cultured on a villi scaffold in a transwell condition (Yu, Peng et al. 2012). Measuring TEER values in our device would reveal more information about the integrity of the gut epithelium. However, commercially available TEER measurement devices are designed to fit into conventional transwell culture plates, but not into our microfluidic device. Measuring TEER in our device would require re-design of the electrodes or embedding the electrodes inside the chip (van der Helm, Odijk et al. 2016).
Rhodamine 123 is transported via paracellular route, but also is transported via carrier-mediated pathway. The influx permeability of Rhodamine 123 showed that both chip and 3D culture conditions resulted in higher influx permeability. We speculate that this is because of the presence of efflux transport. Since a significant amount of efflux transport exist, this could compensate the influx transport. This explains the low influx permeability in 2D well condition, since efflux transport is highest in this condition. Consistent with this hypothesis, when comparing Fig. 5b, c, an inverse relationship is observed between influx and efflux transport. However, this is not consistent with increased expression of p-gp in chip and 3D culture condition (Supplementary Information). This may be partly explained by interference caused by CYP450 3A4 enzyme. It has been noted that most of substrates for p-gp are also substrates for CYP450 3A4, resulting in potential drug-drug interaction issues (Wacher, Silverman et al. 1998; Yumoto, Murakami et al. 2001). In our study, CYP450 3A4 activity was considerably enhanced in the chip and 3D culture conditions. Therefore, it is more likely to have interacted with p-gp substrate, Rhodamine 123 during the absorption experiment. In summary, culture conditions, whether 3D culture or perfusion culture conditions, exerted a significant influence on the transport of drugs across the gut epithelium.
In our study, we used Caco-2 cells to construct an in vitro gut model. Although Caco-2 cells are one of the most widely used cell line for representing the human gut epithelium, there are several important limitations. These include lack of metabolizing enzymes, mucus layer, and unrealistic absorption profiles for hydrophilic drugs (Artursson, Palm et al. 2001). Using a more physiologically relevance cell sources, such as primary cells or stem cells, is likely to improve our gut model (Noel, Baetz et al. 2017). Another potentially important issue is the use of PDMS as a material for the gut chip, mostly due to the known issue of nonspecific binding of hydrophobic molecules onto PDMS surface (Berthier, Young et al. 2012). In our study, the model molecules we used for gut absorption are relatively hydrophilic and not likely to cause any significant problem. However, for future studies using actual drugs in the gut chip, measures to minimize the absorption of molecules need to be taken (Choe, Ha et al. 2017), or the use of PDMS should be determined more carefully. Another fact that needs some consideration is that our gut chip provides bidirectional flow, that is, the direction of the flow alternates periodically. Although we have not noticed any difference in the behavior of Caco-2 cells, it is possible that the direction of the flow has some kind of effect on the cell functions. Further in-depth study may provide important information on the effect of flow on gut cell physiology.
5 Conclusion
In this study, we used our gut microfluidic chip and 3D villi scaffold to culture Caco-2 cells in different culture environments. We examined the separate and combined effect of 3D culture and perfusion culture condition on cell’s physiological functions, related to differentiation, drug absorption, and metabolism. Cells were able to proliferate well and form intestinal barriers in both perfusion and 3D conditions. Interestingly, exposing cells to perfusion culture in 3D culture condition improved their metabolic activity considerably. Absorption of model drugs were also affected, probably due to combination of changes in absorptive surface, expression of tight junction and transporter proteins. Our gut microfluidic chip, integrated with a 3D villi scaffold, may potentially work as a novel platform with improved physiological relevance to the native human gut.
6 Acknowledgments
This work was supported by Ministry of Trade, Industry and Energy (MOTIE), Republic of Korea (10,050,154, Establishment of Infrastructure for industrialization of Korean Useful Microbes, R0004073), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2016R1C1B1009747), and KFRI (Korea Food Research Institute, grant no: E0121705), and Hongik University Research Fund.
References
J.W. Allen, S.N. Bhatia, Biotechnol. Bioeng. 82(3), 253–262 (2003)
P. Artursson, K. Palm, K. Luthman, Adv. Drug Deliv. Rev. 46(1–3), 27–43 (2001)
A. Beduneau, C. Tempesta, S. Fimbel, Y. Pellequer, V. Jannin, F. Demarne, A. Lamprecht, Eur. J. Pharm. Biopharm. 87(2), 290–298 (2014)
I. Behrens, P. Stenberg, P. Artursson, T. Kissel, Pharm. Res. 18(8), 1138–1145 (2001)
E. Berthier, E.W. Young, D. Beebe, Lab Chip 12(7), 1224–1237 (2012)
V. Bieghs, C. Trautwein, Hepatobiliary Surg Nutr. 3(6), 377–385 (2014)
M.B. Chen, S. Srigunapalan, A.R. Wheeler, C.A. Simmons, Lab Chip 13(13), 2591–2598 (2013)
M. Chi, B. Yi, S. Oh, D.J. Park, J.H. Sung, S. Park, Biomed. Microdevices 17(3), 9966 (2015)
A. Choe, S.K. Ha, I. Choi, N. Choi, J.H. Sung, Biomed. Microdevices 19(1), 4 (2017)
C.M. Costello, J. Hongpeng, S. Shaffiey, J. Yu, N.K. Jain, D. Hackam, J.C. March, Biotechnol. Bioeng. 111(6), 1222–1232 (2014a)
C.M. Costello, R.M. Sorna, Y.L. Goh, I. Cengic, N.K. Jain, J.C. March, Mol. Pharm. 11(7), 2030–2039 (2014b)
L.G. van der Flier, H. Clevers, Annu. Rev. Physiol. 71, 241–260 (2009)
A. Galetin, J.B. Houston, J. Pharmacol. Exp. Ther. 318(3), 1220–1229 (2006)
D. Haller, P. Serrant, G. Peruisseau, C. Bode, W.P. Hammes, E. Schiffrin, S. Blum, Microbiol. Immunol. 46, 195–205 (2002)
X. He, D.O. Mishchuk, J. Shah, B.C. Weimer, C.M. Slupsky, Sci Rep. 33, 3416 (2013)
M.W. van der Helm, M. Odijk, J.P. Frimat, A.D. van der Meer, J.C. Eijkel, A. van den Berg, L.I. Segerink, Biosens. Bioelectron. 85, 924–929 (2016)
K. Hu, H. Shi, J. Zhu, D. Deng, G. Zhou, W. Zhang, Y. Cao, W. Liu, Biomed. Microdevices 12(4), 627–635 (2010)
D. Huh, B.D. Matthews, A. Mammoto, M. Montoya-Zavala, H.Y. Hsin, D.E. Ingber, Science 328 (2010)
J.D. Huizinga, W.J. Lammers, Am. J. Physiol. Gastrointest. Liver Physiol. 296(1), G1–G8 (2009)
E.L. Jackson, H. Lu, Integr Biol (Camb) 8(6), 672–683 (2016)
H.J. Kim, D.E. Ingber, Integr Biol (Camb) 5(9), 1130–1140 (2013)
H.J. Kim, D. Huh, G. Hamilton, D.E. Ingber, Lab Chip 12(12), 2156–2164 (2012)
S.H. Kim, J.W. Lee, I. Choi, Y.C. Kim, J.B. Lee, J.H. Sung, J. Nanosci. Nanotechnol. 13(11), 7220–7228 (2013)
S.H. Kim, M. Chi, B. Yi, S.H. Kim, S. Oh, Y. Kim, S. Park, J.H. Sung, Integr Biol (Camb) 6(6), 1222–1232 (2014)
J. Kublbeck, J.J. Hakkarainen, A. Petsalo, K.S. Vellonen, A. Tolonen, P. Reponen, M.M. Forsberg, P. Honkakoski, J. Pharm. Sci. 105(2), 941–949 (2016)
J. Kuttenberger, E. Polska, B.M. Schaefer, Clin Oral Investig. 17(6), 1547–1555 (2013)
M. Leonard, E. Creed, D. Brayden, A.W. Baird, Pharm. Res. 17(10), 1181–1188 (2000)
F. Leonard, E.M. Collnot, C.M. Lehr, Mol. Pharm. 7(6), 2103–2119 (2010)
T. Li, J.Y. Chiang, Pharmacol. Rev. 66(4), 948–983 (2014)
J.H. Lin, M. Chiba, T.A. Baillie, Pharmacol. Rev. 51(2), 135–158 (1999)
H.F. Lodish, Molecular cell biology (W.H. Freeman, New York, 2000)
T.T. Nieskens, M.J. Wilmer, Eur. J. Pharmacol. 790, 46–56 (2016)
G. Noel, N.W. Baetz, J.F. Staab, M. Donowitz, O. Kovbasnjuk, M.F. Pasetti, N.C. Zachos, Sci Rep. 7, 45270 (2017)
K.S. Pang, Drug Metab. Dispos. 31(12), 1507–1519 (2003)
C. Piana, I. Gull, S. Gerbes, R. Gerdes, C. Mills, J. Samitier, M. Wirth, F. Gabor, Differentiation 75, 308–317 (2007)
J.M. Prot, C. Aninat, L. Griscom, F. Razan, C. Brochot, C.G. Guillouzo, C. Legallais, A. Corlu, E. Leclerc, Biotechnol. Bioeng. 108(7), 1704–1715 (2011)
J. Pusch, M. Votteler, S. Gohler, J. Engl, M. Hampel, H. Walles, K. Schenke-Layland, Biomaterials 32(30), 7469–7478 (2011)
E.M. Quigley, Gastroenterol Hepatol (NY) 9(9), 560–569 (2013)
Q. Ramadan, L. Jing, Biomed. Microdevices 18(1), 11 (2016)
J. N. Rao, J. Y. Wang (2010). Regulation of Gastrointestinal Mucosal Growth. San Rafael (CA)
M. Rousset, Biochimie 68, 1035–1040 (1986)
M.T. Salmela, S.L. Pender, M.L. Karjalainen-Lindsberg, P. Puolakkainen, T.T. Macdonald, U. Saarialho-Kere, Scand. J. Gastroenterol. 39(11), 1095–1104 (2004)
J.H. Sung, J. Yu, D. Luo, M.L. Shuler, J.C. March, Lab Chip 11(3), 389–392 (2011)
J.H. Sung, M.B. Esch, J.M. Prot, C.J. Long, A. Smith, J.J. Hickman, M.L. Shuler, Lab Chip 13(7), 1201–1212 (2013)
N. Tobey, W. Heizer, R. Yeh, T.I. Huang, C. Hoffner, Gastroenterology 88(4), 913–926 (1985)
Y.S. Torisawa, C.S. Spina, T. Mammoto, A. Mammoto, J.C. Weaver, T. Tat, J.J. Collins, D.E. Ingber, Nat. Methods 11(6), 663–669 (2014)
G.J. Tortora, S.R. Grabowski, Principles of anatomy and physiology (HarperCollins College, New York, 1993)
M.D. Troutman, D.R. Thakker, Pharm. Res. 20(8), 1200–1209 (2003)
V.J. Wacher, J.A. Silverman, Y. Zhang, L.Z. Benet, J. Pharm. Sci. 87(11), 1322–1330 (1998)
L. Wang, S.K. Murthy, G.A. Barabino, R.L. Carrier, Biomaterials 31(29), 7586–7598 (2010)
Y. Wang, A.A. Ahmad, C.E. Sims, S.T. Magness, N.L. Allbritton, Lab Chip 14(9), 1622–1631 (2014)
B.R. Ware, S.R. Khetani, Trends Biotechnol. 8(30), 19343–19352 (2016)
J. Yu, S. Peng, D. Luo, J.C. March, Biotechnol. Bioeng. 109(9), 2173–2178 (2012)
R. Yumoto, T. Murakami, Y. Nakamoto, R. Hasegawa, J. Nagai, M. Takano, J. Pharmacol. Exp. Ther. 289(1), 149–155 (1999)
R. Yumoto, T. Murakami, M. Sanemasa, R. Nasu, J. Nagai, M. Takano, Drug Metab. Dispos. 29(2), 145–151 (2001)
Q.Y. Zhang, D. Dunbar, A. Ostrowska, S. Zeisloft, J. Yang, L.S. Kaminsky, Drug Metab. Dispos. 27(7), 804–809 (1999)
Y. Zheng, J. Chen, M. Craven, N.W. Choi, S. Totorica, A. Diaz-Santana, P. Kermani, B. Hempstead, C. Fischbach-Teschl, J.A. Lopez, A.D. Stroock, Proc. Natl. Acad. Sci. U. S. A. 109(24), 9342–9347 (2012)
J. Zhou, L.E. Niklason, Integr Biol (Camb) 4(12), 1487–1497 (2012)
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOCX 131 kb)
Rights and permissions
About this article

Cite this article
Shim, KY., Lee, D., Han, J. et al. Microfluidic gut-on-a-chip with three-dimensional villi structure. Biomed Microdevices 19, 37 (2017). https://doi.org/10.1007/s10544-017-0179-y
Published:
DOI: https://doi.org/10.1007/s10544-017-0179-y