
Abstract
Cadmium (Cd) is an important environmental pollutant that poses a threat to human health and represents a critical component of air pollutants, food sources, and cigarette smoke. Cd is a known carcinogen and has toxic effects on the environment and various organs in humans. Heavy metals within an organism are difficult to biodegrade, and those that enter the respiratory tract are difficult to remove. Autophagy is a key mechanism for counteracting extracellular (microorganisms and foreign bodies) or intracellular (damaged organelles and proteins that cannot be degraded by the proteasome) stress and represents a self-protective mechanism for eukaryotes against heavy metal toxicity. Autophagy maintains cellular homeostasis by isolating and gathering information about foreign chemicals associated with other molecular events. However, autophagy may trigger cell death under certain pathological conditions, including cancer. Autophagy dysfunction is one of the main mechanisms underlying Cd-induced cytotoxicity. In this review, the toxic effects of Cd-induced autophagy on different human organ systems were evaluated, with a focus on hepatotoxicity, nephrotoxicity, respiratory toxicity, and neurotoxicity. This review also highlighted the classical molecular pathways of Cd-induced autophagy, including the ROS-dependent signaling pathways, endoplasmic reticulum (ER) stress pathway, Mammalian target of rapamycin (mTOR) pathway, Beclin-1 and Bcl-2 family, and recently identified molecules associated with Cd. Moreover, research directions for Cd toxicity regarding autophagic function were proposed. This review presents the latest theories to comprehensively reveal autophagy behavior in response to Cd toxicity and proposes novel potential autophagy-targeted prevention and treatment strategies for Cd toxicity and Cd-associated diseases in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Source, species and transportation of cadmium
Cadmium (Cd) is one of the most toxic metals in the environment, and it is mainly produced through two major pathways: natural release and anthropogenic factors. Naturally produced Cd mainly occurs through volcanic eruptions, crustal movement, and vegetation combustion. Cd usually exists with other minerals in nature and forms inorganic salts, such as cadmium oxide (CdO), cadmium sulfide (CdS), cadmium sulfate (CdSO4), cadmium carbonate (CdCO3), and cadmium chloride (CdCl2). In the atmosphere, Cd exists in aerosol form as CdO dust, CdCl2, CdSO4and CdS. The chemical properties of Cd are dominated by inorganic compounds with an oxidation state of + 2 (Nordberg et al. 2018). The rapid development during the human industrial revolution led to an increase in the combustion of fossil fuels and non-ferrous metal mining.Cd has been widely used in steel plating, in plastics as a stabilizer, in nickel-cadmium batteries as an electrode material, and in the production of alloys and semiconductors as a useful element. These factors have dramatically altered the biochemical life cycle of Cd in the environment, making it more mobile in ecosystems. Cd is transported through the atmosphere, which increases depositional contamination in urban, rural, and remote areas between countries, including Antarctica and the Arctic (Chiarelli et al. 2019).
With the recent rapid development of nanotechnology, an increasing number of metallic nanoparticles have been synthesized and have been adopted in multidisciplinary fields (Ghasempour et al. 2023; Sadeghzadeh et al. 2023). Although a biological function for Cd in humans has not been identified, other forms of Cd, such as CdO/CdCO3nanocomposites and CdS nanoparticles, have been developed as anticancer agents for drug delivery, biosensors, and bioimaging applications (Ghasempour et al. 2023; Lefojane et al. 2021). Modifying the chemical and physical characteristics of Cd can significantly change its toxicity and harmful effects, as described by Peana et al. (Peana et al. 2021).
Effects of cadmium on human health
Cd is widespread in industrially contaminated soil, water, and air, and the human body is primarily exposed to Cd through Cd-contaminated air, tobacco smoke, water, and food (Chandler et al. 2019; Ganguly et al. 2018; Satarug et al. 2003). Cd enters the body mainly through the respiratory and digestive tracts, where it is absorbed by the lungs and intestines. Cd is present in organic and inorganic (Cd2+) forms in human biological fluids, including blood. After acute Cd exposure, organic and inorganic Cd can be measured in the blood, and the concentration of Cd2+decreases after 2–5 h (Abbasabadi and Shirkhanloo 2020). Cd can form a complex with certain organic compounds, such as metallothionein (MT), albumin, other plasma proteins, or peptides (e.g., glutathione), and accumulates mainly in the liver and kidneys (Abbasabadi and Shirkhanloo 2020; Satarug 2018; Tinkov et al. 2018). In blood, Cd binds to molecular targets on erythrocytes as well as proteins, peptides, and amino acids; it may also be transported by anion exchangers, thereby facilitating its uptake into erythrocytes in the form of [Cd (OH)(HCO3)2]− and [Cd (OH)(HCO3)Cl]−complexes (Lou et al. 1991). Histidine can also stimulate Cd uptake into human erythrocytes (Horn and Thoma 1996). As mentioned above, MT is an important metal-binding protein enriched in cysteine residues. Heavy metals, such as Cd, can bind to the thiol (-SH) group present in cysteine residues of the protein (Klaassen et al. 1999; Sabolic et al. 2010). Current findings on Cd target molecules have revealed that different MT isoforms I-IV are more common biomolecular targets of Cd in mammalian organs, as reviewed in detail by Hill and Gailer (Hill and Gaile 2021). The authors also summarized other possible biomolecules and mechanisms of Cd targeting in plasma and erythrocytes and provided pathways for its uptake by target organs via blood circulation in the human body.
Cd is present in the environment at extremely low levels; however, it is difficult to eliminate after absorption into the body. Cd is one of the most toxic substances known to accumulate in the body and has a biological half-life of more than 25 years (Nordberg and Nordber 2022). Once Cd is taken up by renal proximal tubule (PT) cells, it induces MT, after which, it is chelated, inactivated, and stored in the cells (Sabolic et al. 2010). Cd can damage various organ systems, including the respiratory, skeletal, reproductive, immune, and nervous systems, as well as the liver and kidneys. Available scientific evidence shows that mammals lack an effective mechanism to eliminate Cd toxicity, with most Cd ions (not bound to MT) persisting indefinitely in PT cells after filtration by the kidneys (Nordberg and Nordber 2022; Satarug 2018). In a mouse model, chronic Cd exposure resulted in its accumulation in most organs and tissues, with the kidneys being the most severely affected (Tai et al. 2022).
Epidemiological studies have shown that Cd toxicity is associated with various diseases. A positive correlation between Cd exposure and diabetes mellitus and prediabetes risk was reported in a meta-analysis by Filippini et al. (Filippini et al. 2022), who observed that the highest and lowest Cd exposure were associated with type 2 diabetes, with relative risks (RRs) of 1.24 (95% CI: 0.96–1.59), 1.21 (95% CI: 1.00–1.45) and 1.47(95% CI:1.01–2.13)for the blood, urinary, and toenail matrices. An increased risk of prediabetes after Cd exposure was also observed based on urine and blood matrices. Furthermore, a negative correlation was reported between serum Cd levels and lung function in patients with chronic obstructive pulmonary disease (COPD) (Jiang et al. 2022). High blood Cd levels may also be associated with stroke and hypertension in the Korean population aged < 60 years (Jeong et al. 2020). Owing to the long half-life of Cd in the human body, certain organs are more vulnerable to Cd-induced carcinogenesis. Cd has been classified as a type I human carcinogen by the International Agency for Research on Cancer (IARC) (Hartwig 2013). Low-dose and long-term exposure to Cd can increase the risk of developing various cancers, including lung, liver, prostate (Waalkes 2000), oral (Satir 2022), endometrial (McElroy et al. 2017), and renal cancers (Song et al. 2015). Cd enhances the risk of lung diseases and promotes the malignant transformation and development of lung cancer, particularly in occupational populations (Humans 2012 Waalkes 2000) exposed to Cd directly through the respiratory route. Accordingly, blood Cd levels may serve as a potential marker to monitor the incidence of early lung cancer in former smokers (Lener et al. 2021). Therefore, Cd is not only closely related to toxicity in various organs and systems but also contributes to the occurrence and development of various diseases, including cancer, in humans.
Previous reviews on Cd toxicity have mainly focused on the toxicity of environmental Cd exposure to plants, crops, soil, and marine organisms, while as far as we know, few have provided assessments of the mechanism underlying Cd toxicity in the human body in terms of defense responses (Thevenod and Lee 2013). Therefore, in this review, we discuss the toxicity of Cd to specific tissues and organs after its entry into the human body and review the representative signaling pathways and molecular mechanisms involved in autophagy-Cd toxicity. The aim of this review is to explore the role of autophagy in response to Cd exposure in organisms and identify methods of better preventing and controlling Cd-induced cell and tissue damage or even cell death by monitoring specific biomarkers in the process of autophagy. In this review, we first summarize the general information about autophagy and its regulation mechanism. We then discuss the toxic effects of Cd through the autophagic pathway in the target organs of organisms, with a focus on hepatotoxicity, nephrotoxicity, respiratory toxicity, and other systemic toxicities, including neurological and intestinal toxicities. Moreover, the characteristics of exposure to Cd in vitro and in vivo with different physicochemical conditions, such as different morphologies and concentrations, are also addressed. We then analyze the molecular defense mechanisms that induce autophagic responses that lead to cell survival, adaptive response, or death after Cd exposure.
Autophagy
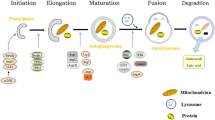
Autophagy is the process in which autophagy-related genes (ATGs) direct cells to wrap misfolded proteins or damaged organelles in the cytoplasm to form phagocytic vesicles and fuse with lysosomes to form autophagic lysosomes that degrade the substances inside the vesicles. Autophagy is an intracellular degradation mechanism that regulates cellular metabolism and maintains intracellular homeostasis (Galluzzi et al. 2017), and it is involved in pathophysiological processes, such as cell development, senescence, starvation, hypoxia, inflammation, oxidative stress, programmed death, and immune responses (Liu and Levine 2015). Autophagy is mainly classified into three categories: macroautophagy, microautophagy, and chaperon-mediated autophagy (Mizushima et al. 2001). Among them, macroautophagy is the most extensively studied and is a highly conserved eukaryotic cellular process (Yamamoto et al. 2023). In this review, we highlight the function and signaling pathways of macroautophagy, hereafter referred to as autophagy.
Autophagy process is controlled tightly by several functional modules including the unc 51-like kinase (ULK) complex (I), the class III phosphatidylinositide 3-kinase (PI3K) complex (II), the ATG2/WD repeat domain phosphoinositide-interacting protein (WIPI) complex and the ATG9 cycling system (III), the ATG12-conjugation system (IV), and the LC3-conjugation system (V) (Keller et al. 2023). Among them, a series of ATGs are involved in the pre-initiation and initiation processes of autophagy, and these processes are the most critical steps for the occurrence of autophagy in the species of Homo sapiens and Saccharomyces cerevisiae (Li. et al. 2020). Moreover, ULK1, Beclin 1 and LC3 (microtubule-associated light chain 3), GABARAP in H. sapiens are equivalent to Atg1, Atg6/Vps30, and Atg8 respectively, in S. cerevisiae(Zhen, et al. 2023). LC3B/MAP1LC3B, one of the Atg8 mammalian orthologs, is well-studied, owing to its role in key steps in the autophagy process. For example, receptors for selective autophagy, such as P62/SQSTM1, NBR1, and TOLLIP, function as connectors between LC3/GABARAP proteins and ubiquitinated substrates in human cells (Zellner. et al. 2021). LC3B is also involved in processes such as autophagosome biogenesis (Weidberg 2010), autophagosome transport, and membrane fusion process. LC3 can be cleaved to LC3-I by ATG4B, which is then lipidated to LC3-II by conjugation with phosphatidylethanolamine, participating in autophagosome formation. LC3-I is regenerated from LC3-II via ATG4B-mediated cleavage of phosphatidylethanolamine for further membrane biogenesisc (Nguyen et al. 2014). Therefore, LC3 and its subfamily (LC3A, LC3B, and LC3C) are considered hallmarks reflecting canonical autophagy (Nieto-Torres et al. 2023). The initiation process of autophagy is complex. During the initiation of the autophagosome, phagophores, the pre-structures for forming autophagosomes, are formed near the ER, elongate, bend, and finally form double-membraned structures, via membrane fission, called autophagosomes (Yamamoto et al. 2023). Therefore, the ER is an important organelle for autophagosome initiation, which is the key process in controlling autophagic flux. In addition to the plasma membrane, recent studies have revealed that phagophores are also derived from lipid droplets, endosomes, contact sites between ER-mitochondria, and the Golgi endosomal membranes (Zhen et al. 2023). Most of the ATG family members participate in the biogenesis of phagophores, whereas the ATG8 (homolog of LC3) family members SNAP29 and VAMP8, along with Syntaxin17, are critical for autophagosome-lysosome membrane fusion in mammalian cells (Li, et al. 2020). The mammalian target of rapamycin (mTOR) is an also crucial signaling pathway for initiating and controlling autophagy and cellular growth under pathological conditions in humans (Panwar et al. 2023; Keller et al. 2023). Autophagosome formation can be negatively regulated by mTOR signaling through AMP-activated protein kinase (AMPK)-Ulk2 interactions; notably, AMPK is potentially involved in all stages of autophagy (Wang et al. 2022c).
Functionally, autophagy primarily serves as a cellular protective mechanism against exogenous stimuli and is considered a pro-survival mechanism (Tsapras and Nezis 2017). However, excessive autophagy or its inhibition usually results in a decreased ability to metabolically degrade foreign substances and an imbalance in cellular homeostasis, potentially resulting in cell death. Notably, various types of cell death are associated with autophagy dysfunction. Apoptosis and autophagy are considered antagonistic processes (Cong et al. 2022); the initiation of autophagy inhibits apoptosis, whereas apoptosis occurs when autophagy induction fails (Kouroumalis et al. 2023). However, excessive autophagy may also induce apoptosis. As indicated in Keller's study (Keller et al. 2023), the Atg12-Atg5-Atg16L1 complex promotes autophagy by converting LC3-I to LC3-II to form autophagosomes. In Atg5-deficient mammalian cells, Atg12 and Atg16L1 lose the ability to interact with Atg12 to induce autophagy (Mizushima et al. 2001). Moreover, unbound Atg5 is cleaved by calpain to Atg5tN, which can mediate cytochrome C release via Bcl-2 proteins in the mitochondria, ultimately leading to apoptosis (Lepine et al. 2011; Cong et al. 2022). In addition, Beclin-1, P53, and Caspase, as well as the PI3K/AKT/mTOR pathway play essential roles in regulating the interactions between autophagic and apoptotic processes (Cong et al. 2022; Tsapras and Nezis 2017).
Ferroptosis is a recently discovered form of cell death characterized by iron overload, lipid peroxidation, and ROS accumulation (Latunde-Dada 2017). Ferroptosis is closely related to autophagy (Chen et al. 2023; Hou et al. 2016), which is classified into selective and non-selective autophagy. As a type of selective autophagy, ferritinophagy is an autophagic degradation process of ferritin, the iron storage protein, which is essential for regulating cellular iron content. FTL1 (ferritin light polypeptide 1) and FTH1 (ferritin heavy polypeptide 1) belong to the ferritin family and function as intracellular iron storage proteins. Ferritin is a critical regulator for maintaining cellular iron levels (Liu et al. 2020c). Increased autophagy degrades ferritin, which increases iron levels and leads to oxidative damage. Hou et al. (Hou et al. 2016) found that knockout ofATG5 and ATG7 genes can inhibit erastin (a ferroptosis inducer)-induced cell death and decrease intracellular Fe2+ and malondialdehyde levels, which is a typical manifestation of ferroptosis. Under ATG5-deficient conditions, FTH1 was remarkably increased in PANC1 (human cancer cell line) and MEF cells (mouse embryonic fibroblasts) irrespective of erastin treatment, indicating that ferritin degradation requires ATG5-mediated autophagy. NCOA4, the cargo receptor, was identified in 2014. It is highly enriched in autophagosomes and can degrade autophagy-dependent ferritin (Mancias et al. 2014). Overexpression of NCOA4 promotes ferroptosis via ferritin degradation (Hou et al. 2016). NCOA4-mediated ferritinophagy provides further evidence of the crosstalk between autophagy and ferroptosis. The discovery of multi-autophagy-dependent ferroptosis builds upon our understanding of iron-based lipid peroxidation and aberrant autophagic degradation pathways that regulate cell death (Chen et al. 2023). Defensive response mechanisms such as autophagy that balance and eliminate internal and external stimuli and adverse stress outcomes caused by excessive or inadequate autophagy, such as cellular damage, inflammation, metabolic imbalance, and even cellular death, are critical issues that require further investigation.
Cadmium and autophagy
Most living cells undergo low levels of basal autophagy to maintain cellular homeostasis under physiological conditions. When cells are subjected to stresses, such as oxidative stress, starvation, and hypoxia, autophagy adapts to changes in the internal environment by either enhancing or weakening autophagy (Phadwal et al. 2020). These dynamic changes in autophagy under different conditions can lead to different cellular outcomes. Considerable evidence indicates that autophagy is closely related to the pathogenesis of human diseases, such as cancer, cardiovascular disease, neurodegenerative disease, lung disease, kidney cancer, bone cancer, and metabolic diseases (Klionsky et al. 2021).
Mammalian cells can initiate selective autophagy to eliminate cellular damage caused by environmental pollutants, particularly industrial products, thereby maintaining cellular homeostasis and protecting cells from endogenous stress (Martinez-Garcia and Marino 2020; Rahman et al. 2023). Stress is a major factor in the induction of autophagy, and ionized Cd can produce excessive reactive oxygen species by inducing cellular oxidative stress. Environmental pollutants, particularly industrial compounds, modulate autophagic flux by either increasing it as a protective response, blocking it, or switching its protective role toward a pro-cell death mechanism.(Martinez-Garcia and Marino 2020). Under certain circumstances, exposure to low Cd concentrations induces apoptosis and autophagy; this depends on the duration of Cd exposure and autophagic signaling, which may differ in various cell types (Tuffour et al. 2023). Initially, low-dose Cd exposure may induce autophagy as a cytoprotective mechanism; however, prolonged exposure may disrupt the autophagic process and lead to apoptosis.
The relationship between cytotoxicity and the cellular stress response is one of the most important drivers of the acute exposure effects of Cd; however, this relationship remains uncertain (Man et al. 2023). Therefore, exploring the interaction between Cd and autophagy in organisms has great biological significance for understanding the molecular action in response to metal toxicity. Recent studies have shown that enhanced autophagy improves stress response and reduces genetic damage. Rahman et al. (Rahman et al. 2023) in 2023 provided a detailed review of the mechanisms of autophagy as a target of environmental pollutants and assessed current research on autophagy pathway responses and protective effects when the body is exposed to certain environmental pollutants, particularly heavy metals (including Cd) and particulate matter.
Toxic effects of Cd-induced cellular autophagy on human health
Although the mechanisms of cadmium-induced cytotoxicity remain unclear, many studies have indicated that in addition to oxidative damage and apoptosis, autophagy dysfunction is one of the main mechanisms leading to Cd cytotoxicit (Genchi et al. 2020). Numerous studies have reported that Cd-induced autophagy produces different biological effects in different organs and cells. The biological effects, pathway alterations, and pathological outcomes induced by Cd in different cells depend on the time of Cd entry into the body, duration of accumulation, dose, and route of exposure (Guo et al. 2022). In this section, we summarize the latest research on the toxic action of Cd on its main targets (kidney, liver, lung, and others) associated with autophagy and discuss the possible role of autophagy in Cd organ-based toxicity.
Nephrotoxicity
The kidney is an important target organ of Cd, and renal proximal tubule (PT) epithelial cells are the main targets of Cd nephrotoxicity (Thevenod 2003). MTs are critical modulators of the toxicokinetics and biochemistry of essential and non-essential metals (Nordberg et al. 2018). Cd complexes bound to MTs, other plasma proteins, peptides and small organic molecules circulate through the bloodstream to the kidneys, where they are reabsorbed by PT cells via various uptake mechanisms and trigger PT cells to induce MTs (Thevenod and Wolff 2016). MT enrichment in the kidneys and liver may be partially responsible for the higher Cd concentrations in these two organs (Genchi et al. 2020; Guo et al. 2022). Approximately 30–40% of the Cd entering the body is deposited in the kidneys (Niture et al. 2021). It has been suggested that when the accumulation of Cd exceeds the ability of PT cells to synthesize MT, unbound Cd causes cellular oxidative stress, alteration of autophagic fluxes, and acute loss of cells, which exceeds the regeneration of PT cells and ultimately leads to cell death and luminal damage (Prozialeck and Edwards 2012). By using MT-null mice, Liu et al. (Liu et al. 1998) demonstrated that Cd-induced renal injury is not mediated via Cd2+-MT complex. Fels et al. (Fels et al. 2019) further revealed that the Cd2+-MT complex is likely nontoxic to tubule cells. Instead, the ligand proteins that bind with Cd2+ to form complexes, such as Cd2+-β2M (microglobulin), Cd2+-Alb (albumin) and Cd2+-Lcn2 (lipocalin-2), and are taken up by PT cells via megalin-cubilin endocytosis that eventually damages PT cells. Barrouillet et al. (Barrouillet et al. 2001) revealed potential effects of Cd speciation in renal toxicity in vitro, and showed that in cell culture media with bovine serum albumin (BSA) or fetal calf serum (FCS), both CdCl2 and CdSO4 had EC50 value of approximately 3 × 10−5 M in porcine kidney cells (LLC-PK1). However, in protein-free cell culture media, CdSO4 was more toxic than CdCl2 at pH 7.4 after 24 h incubation. Notably, the amount of protein in the cell media determines the EC50 and Cd sulphate may produce more Cd2+ in LLC-PK1.
Autophagy is considered an early event in the onset of Cd-induced nephrotoxic effects (Lv et al. 2019). Several studies have demonstrated the involvement of autophagy in Cd-induced nephrotoxicity. Gong et al. (Gong et al. 2022) reported that Cd can induce lysosomal dysfunction and block autophagic flow, which exacerbates oxidative stress and acute kidney injury in primary rat proximal tubule cells; however, bromodomain-containing protein 4 (BRD4) blocked autophagy and lysosomal transcriptional function, which attenuated Cd damage to the kidney. Fan et al. (Fan et al. 2021) showed that Cd exposure inhibited autophagy in rat kidney and rat renal cells, and that sustained inhibition of nuclear factor erythroid 2-related factor 2 (Nrf2), an antioxidant signaling factor, led to the restoration of autophagosome-lysosome fusion, thereby attenuating Cd-induced nephrotoxicity. The outcome of Cd-induced autophagy dysfunction often determines cell fate. Chargui et al. (2011) demonstrated that in rats, Cd exposure at a subtoxic concentration enhanced autophagy and caused slight damage to proximal convoluted tubule (PCT) cells. However, the glomerular and tubular function of rat kidneys was not affected, and apoptosis was not detected in both in vivo and in vitro tests. This finding provides further evidence that upregulated autophagy could serve as an adaptive response to protect PTC cells from further damage, such as apoptosis, by environmental Cd doses. Lee et al. (Lee et al. 2017) further revealed that excessive stress triggered by Cd can lead to cell death with a disrupted autophagy flux and lysosomal instability. They also demonstrated that the ER stress pathway PERK-eIF2α was activated by Cd exposure, leading to impaired autophagy in rat kidney proximal tubule cells. The work by Lee et al. (Lee et al. 2017) was pioneering in demonstrating lysosomal dysfunction and decreased autophagosome-lysosome fusion induced by Cd. Similarly, Luo et al. (Luo et al. 2016) also demonstrated that the ER stress eIF2α-ATF4 pathway and autophagy were activated by Cd exposure in both mice kidney tissues and cultured cells. Importantly, they found that interfering with the ER stress eIF2α-ATF4 pathway can block autophagy via cyclooxygenase-2, a rate limiting enzyme for prostaglandin E2 synthesis. Research also has demonstrated that Cd exposure activates the PERK-elf2α-ATF4-CHOP pathway and induces ferroptosis via the PERK-autophagy pathway (Zhao et al. 2021). Lee and Oh (Lee and Oh 2021) demonstrated that Cd induces apoptosis and cellular autophagy through the P53-mediated DRAM-BAX signaling pathway. Beclin-1, a critical regulator of autophagy, can specifically interact with caspase-8 and activate autophagy to protect against Cd-induced apoptosis via Fas/FasL signaling in rat PT cells. These findings suggest that the activation or blockage of autophagy plays an important role in the degree of renal cell impairment and even cell death.
Hepatotoxicity
The liver is another important organ that is susceptible to Cd toxicity (Yuan et al. 2021). Once Cd enters the body, it is transported to the liver via blood circulation, where MTs binds to Cd and sequesters it to buffer its cytotoxic effects (Guo et al. 2022). Approximately 30% of the Cd absorbed by an organism accumulates in the liver, while the rest is distributed to other organs or tissues (Niture et al. 2021). Under physiological conditions, autophagy can maintain the stability of the intracellular environment of hepatocytes, and overactivation of autophagy can lead to damage to intracellular organelles in hepatocytes and even death. Mitophagy is a process that eliminates damaged mitochondria via selective autophagy (Ding and Yin 2012). Mitochondrial loss is a crucial event during hepatotoxicity. Pi et al. (Pi et al. 2013) revealed that exposure to Cd led to mitochondrial fragmentation /loss via the overactivation of mitophagy in normal liver L02 cells. This excessive mitochondrial loss is caused by increased dynamin 1-like (DNM1L) expression and translocation into the mitochondria. Moreover, Liu et al. (Liu et al. 2023) demonstrated that the expression of mitochondrial calcium uniporter (MCU) was upregulated in Cd-induced excessive mitophagy in HepG2 liver cells. This upregulation is attributed to increased cytosolic Ca2+ levels and cAMP response element binding (CREB) protein-induced MUC translocation and accumulation in the mitochondria under Cd exposure conditions. Finally, they demonstrated that VDAC1, a channel protein that mediates mitochondrial metabolism, can directly interact with MCU and overactivate mitophagy, resulting in hepatocytic death. These two studies demonstrated DNM1L and MCU as potential molecular targets for the prevention and treatment of Cd-mediated hepatotoxicity.
Lysosomes are highly dynamic organelles that are distributed rapidly around microtubules (Cabukusta and Neefjes 2018). Yuan et al. (Yuan et al. 2021) found that Cd can disrupt the microtubule network and downregulate the microtubule-associated proteins KIF5B, γ-tubulin, and acetylated α-tubulin in buffalo rat liver 3 A cells (BRL3A). They also demonstrated that Cd induced an increase in endosomal lysosomes by disrupting cellular microtubules, thereby blocking autophagic flux in BRL3A cells. Nrf2 is an oxidative regulator that modulates cellular redox imbalance. As previously mentioned, Cd can trigger oxidative stress and autophagy dysfunction. A recent study showed that puerarin (PU) restored Cd-induced Nrf2 inhibition, thereby preventing Cd-induced cellular autophagy and inflammatory vesicle activation (Yu et al. 2021).
The liver is the major organ involved in lipid metabolism. Autophagy can selectively target lipid droplets in the liver, namely, lipophagy (Shin 2020). Numerous studies have demonstrated that autophagy in hepatocytes is critical for lipolysis and the control of triglyceride homeostasis. In vitro studies have shown that Cd induces cholesterol redistribution from HepG2 cells to the culture medium by inhibiting the autophagy-lysosomal pathway (Rosales-Cruz et al. 2018). In vivo studies confirmed that Cd inhibits hepatic autophagic flux in mice, thereby promoting the accumulation of apolipoprotein ApoE, whereas the inhibition of low-density lipoprotein receptor expression by Cd contributed to the elevation of blood triacylglycerol levels (Liu et al. 2020b). Together, these studies demonstrate that Cd interferes with the normal physiological functions of the liver by inducing autophagy in hepatocytes. The results contribute to our understanding of the biological role of the autophagy pathway in the pathogenesis of Cd-induced hepatotoxicity.
Respiratory system
Cd is a known human carcinogen certified by the International Agency for Research on Cancer (IARC). The link between chronic exposure to Cd and the induction of malignant tumors has been well-established through animal (Waalkes et al. 2000) and epidemiological studies (Humans 2012) including those on lung cancer (Baan et al. 2019). Long-term exposure to low-dose Cd in the respiratory system is likely to cause malignant transformation of respiratory epithelial cells (Humans 2012; Li et al. 2023a; Wang et al. 2021). However, the precise mechanism of action of the malignant transformation into lung cancer by Cd remains unclear. In vitro studies have shown that Cd exposure of human lung epithelial Beas-2B cells generates ROS, which promotes the inflammatory microenvironment, and thus facilitates the malignant transformation of Beas-2B cells. In contrast, the transformed cells exhibit a deficiency in autophagy, characterized by a state of blockage in the fusion of the autophagosome and lysosome. This blockage leads to an increase in autophagosomes and the expression of P62 proteins, which together with Nrf2, form a positive feedback mechanism leading to an increase in the expression of anti-apoptotic proteins Bcl-2 and Bcl-xl, the downstream molecules of Nrf2, thus leading to apoptosis resistance (Wang et al. 2018). Another study on ROS-dependent autophagy (Lv et al. 2018) reported that Cd (2 µM CdCl2) induces the proliferation, migration, and invasion of A549 cells, resulting in a significant increase in ROS levels and autophagy. ROS act as molecular signals to increase the expression of Atg4, an enzyme facility LC3II production, enhances cellular autophagy. Atg4 and autophagy promote proliferation, migration, and invasion of A549 cells. Atg4 expression and cellular autophagy may be valuable in preventing Cd-induced lung toxicity. In vivo experiments have also shown that respiratory Cd exposure in mice induces autophagy in B lymphocytes and promotes apoptosis in immune cells while inhibition of autophagy attenuates Cd-induced apoptosis in splenic and immune cells in mice (Gu et al. 2019).
Environmental Cd exposure is closely associated with the development of several respiratory diseases. In a case-control study, Wang et al. (Wang et al. 2022b) demonstrated that serum Cd levels were higher in patients with COPD than in the control group and the serum Cd level gradually increased with the progression of COPD. They also showed that markers of autophagy (e.g., P62, Beclin-1, LC3 II, and LC3I) and molecules of apoptosis were increased in the lungs of COPD patients. Serum Cd levels were positively correlated with autophagy and apoptosis in the lungs of patients with COPD, and in vitro experiments confirmed that Cd induces autophagy and apoptosis in human lung epithelial cells. Surolia et al. (Surolia et al. 2015) showed that heme oxygenase-1 (HO-1) protects against Cd-induced emphysema in mice and revealed that HO-1(-/-) Cd exposed mice are more susceptible to apoptosis and emphysema. However, cell death was reduced by pretreatment of HO-1(-/-) cells with rapamycin, an autophagy activator. This indicates that the overexpression of heme oxygenase-1 induced autophagy and reduces and delays apoptosis, thereby protecting mice from the development of emphysema.
Other systems
Chronic exposure to Cd can also cause toxic effects on osteoporosis and the neurological, reproductive, and immune systems. Li et al. (Li et al. 2016) and Pi et al. (Pi et al. 2017) found that Cd impedes autophagosome-autophagy lysosome fusion and impairs lysosomal function in mouse neuroblastoma cells by decreasing the level of TFEB, which subsequently leads to neuronal cell death. Although melatonin increases TFEB expression, this study suggests that melatonin could be a potential target for exploring Cd-induced neurotoxicity by controlling the autophagy pathway.
Chronic exposure to Cd can induce human skeletal toxicity, including osteoporosis, bone atrophy, and joint pain, which mostly occurs after Cd-induced nephrotoxicity (Ma et al. 2022). RANKL (RANK) and osteoprotegerin (OPG) receptors are important molecules involved in osteoblast development and bone remodeling. In a study of chronic low-dose CdCl2 exposure, Cd was found to act on rat mesenchymal stem cells (MSCs) through the RANKL/OPG pathway and downregulate the expression of key genes involved in the osteogenic differentiation of MSCs when CdCl2(1 mg/kg bw and 2 mg/kg bw) was continuously fed to rats for 38 weeks. However, Cd did not cause significant damage to the kidney, suggesting that the increased expression of proteins, including autophagy-related proteins LC3B, Beclin-1, P62, and HO-1, was the result of a renal adaptive response induced by Cd chloride (Lv et al. 2019). The toxic effects of Cd on bone may occur simultaneously with renal toxicity. Another study by Tong et al. (Tong et al. 2022) reported that puerarin attenuated Cd-induced oxidative damage in rat bones by attenuating autophagosome and lysosome fusion inhibition.
The toxic effects of Cd exposure on the reproductive system via autophagy have also been reported. For instance, Wang et al. (2020b) found that Cd-exposed testicular cells promote autophagy via the PI3K- and mTOR-independent signaling pathways, which regulate apoptosis in testicular injury and recovery. Autophagy plays a vital role in the occurrence and development of diseases as well as in defenses against the invasion of exogenous pathogens in the immune response induced by Cd. So et al. (So et al. 2018) demonstrated that Cd-induced oxidative stress triggers endoplasmic reticulum (ER) stress, which leads to the activation of Ca2+ proteases and subsequently activates autophagy and apoptosis, leading to immunosuppression.
Molecular mechanism of Cd-induced autophagy
Molecules and cells of living organisms perform their functions through specific signaling pathways. Similarly, Cd exerts its toxic effects through certain molecules and specific signaling pathways, and a network of these molecules has been identified. Autophagy arises from the cellular generation of stress responses, such as oxidative stress, ER stress, and Ca2+disruption (Aki et al. 2013; Yorimitsu et al. 2006; Chen et al. 2008).
Among the toxicological mechanisms associated with autophagy-induced Cd, we review the literature on classical Cd-induced (1) ROS-dependent signaling pathways; (2) ER stress pathways; (3) mTOR pathways; (4) Beclin-1 and Bcl-2 families; and (5) Emerging molecules /pathways of autophagy-mediated Cd toxicity. It should be noted that these Cd-induced signaling pathways are not isolated but rather are interconnected, thus representing the main mechanisms by which Cd triggers toxicity in its target organs (kidneys, liver, lungs, skeleton and nervous system). For example, Cd exposure leads to oxidative stress in hepatocytes by generating large amounts of ROS, which in turn induces autophagy by inhibiting the AKT/mTOR signaling pathway (Niture et al. 2021). By activating or inhibiting signaling molecules at different stages of autophagy, Cd ultimately determines cell fate, that is, whether cells undergo an adaptive response without severe damage or trigger programmed or non-programmed cell death (Fig.1).

Model diagram of autophagy-mediated cadmium toxicity and mechanism
ROS-dependent signaling pathways
Oxidative stress is one of the most important mechanisms by which Cd exerts toxicity (Cuypers et al. 2010). It has long been established that Cd disrupts the electron transport chain in the mitochondria, particularly affecting complexes II/III. Cd binds to the Q0 site of cytochrome b on complex III, leading to the accumulation of semiubiquinones. Semi-ubiquinone is an unstable molecule that readily transfers an electron to molecular oxygen, resulting in the formation of superoxide and, thus, oxidative stress (Wang et al. 2004). Autophagy and apoptosis are triggered by various stress conditions that may be derived from exogenous factors, such as environmental pollutants, or endogenous stimuli, e.g. leakage during the mitochondrial electron transfer process (Dickinson and Chang 2011; Redza-Dutordoir and Averill-Bates 2016). Low levels of ROS are necessary for the maintenance of normal physiological states and adaptive cellular responses. Moderate levels of ROS may induce autophagy, which is considered a cell self-protection strategy (Gibson 2013). Excessive ROS may cause cell damage and activate cell survival or cell death through various signaling pathways (PI3K/Akt, MAPK, Nrf2, and ER stress) (Redza-Dutordoir and Averill-Bates 2016). In an in vivo model of Cd-exposed weaned piglets, Cd was shown to be involved in ROS-mediated autophagy and apoptosis in the liver by promoting ROS production, activating the AMPK/PPAR-γ/NF-κB pathway, and finally inducing autophagy (increasing the expression of Beclin-1, LC3-II/LC3-I and P62) and apoptosis (increasing the expression of Bax, Bak, caspase-9, and caspase-3 and decreasing the expression of Bcl-2) (Wang et al. 2022a). Zhang et al. (Zhang et al. 2019) also reported that Cd-induced ROS activates ER stress, autophagy, and apoptosis in retinal pigment epithelial cells.
The imbalance between ROS production and restriction is a common feature of cancer cells, and ROS have the function of regulating the tumor microenvironment (Cheung and Vousden 2022). As mentioned in "Respiratory system" Section, chronic exposure to Cd leads to the malignant transformation of respiratory epithelial cells. Increased ROS expression induces ER stress in Cd-exposed prostate epithelial cells (RWPE-1), leading to defective autophagy in Cd-induced prostate carcinogenesis (Kolluru et al. 2019). Similarly, Tyagi et al. (Tyagi et al. 2023) found that Cd exposure promotes NOX1 assembly through the activation of the cytoplasmic regulators p47phox and p67phox in RWPE-1 cells, which is essential for Cd-induced persistent ROS production and the control of ER stress-induced autophagy defects in both mice and humans. Further, Fan et al. (Fan et al. 2019) showed that ROS-dependent NUPR1-mediated autophagy plays an important role in repetitive Cd exposure-induced cell growth, migration, and invasion of oral squamous cell carcinoma (OSCC) cells. Cd increases autophagic flux, upregulates LC3-II, and down-regulation P62 in CAL27 cells. Autophagy blockers inhibit Cd-induced cell migration and invasion, suggesting that autophagy plays an oncogenic role in Cd-treated CAL27 cells.
Endoplasmic reticulum stress pathway
The ER is a key organelle involved in protein folding, trafficking, and Ca2+homeostasis maintenance. Approximately 25% ROS in cells is generated by the ER (Tu and Weissman 2004). For example, the majority of ROS in cells is produced via the electron transfer chain of protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductin-1 (ERO1α) during oxidative protein folding in the ER (Malhotra and Kaufman 2007). ROS generation can be further increased by the interaction of PDI with NADPH oxidases, which induces Ca2+ release to the mitochondria and further activates oxidative stress in the mitochondrial respiratory chain, resulting in Ca2+overload and an unfolded protein response (UPR) in the ER (Gorlach et al. 2015). ER stress or persistent protein misfolding can also initiate apoptotic cascades (Malhotra and Kaufman 2007). Once ER function is disrupted, ER stress occurs through three transmembrane proteins: protein kinase R-like endoplasmic reticulum kinase (PERK), human-requiring inosinolase 1 (IRE1), and activating transcription factor 6 (ATF6) (Li et al. 2023b). Activation of PERK/eukaryotic initiation factor 2 alpha (eIF2 alpha)/ATF4/C/EBP (CCAAT-enhanced binding protein) homolog (CHOP), IRE1, and ATF6 has important effects on autophagy-related gene expression and protein phosphorylation, which regulate autophagy (Cybulsky 2017). As mentioned in "Autophagy" Section, the ER is an important organelle for the initiation of phagophores, which is a pre-autophagosome structure. Moreover, ER stress triggers autophagy (Hoyer-Hansen and Jaattela 2007; Qi and Chen 2019; Yorimitsu et al. 2006) and apoptosis (Song et al. 2017). Recent studies have shown that ER stress and autophagy play dual roles in response to Cd toxicity. Mild-to-moderate ER stress or autophagy attenuates cytotoxicity and plays a protective role, whereas excessive ER stress and autophagy trigger a destructive cascade of reactions, resulting in increased cellular damage (Chou et al. 2019; Qi and Chen 2019). Liu et al. (Liu et al. 2020a) used Cd-treated rat NRK-52E cells and found that Nrf2 was activated and puerarin could reduce the damage caused by excessive ER stress; however, puerarin no longer significantly reducedER stress after autophagy-associated protein 7 was knocked down in NRK-52E cells. This suggests that puerarin restores autophagy and reduces endoplasmic reticulum stress by inhibiting the activation of Nrf2.
Ca2+is another important signaling molecule for ER stress-induced autophagy (Hoyer-Hansen and Jaattela 2007); ER stress leads to the release of Ca2+from the ER into the cytoplasm (Biagioli et al. 2008), which activates various kinases, such as mTOR and AMPK, in the autophagy signaling pathway (Hoyer-Hansen et al. 2007). Ca2+transients on the outer surface of the ER membrane are closely associated with the initiation of autophagosome formation (Zheng et al. 2022). Biagioli et al. (2008) demonstrated that ER is another organelle impacted by Cd, in addition to mitochondria. They proposed that Cd induces ER stress and apoptosis by releasing Ca2+ and inhibiting SERCA pumps in the ER. Mitochondria and the ER may act in parallel to evoke caspase-dependent apoptosis. In testing the cytotoxic effects of Cd on Mesangial cells (MES-13), it was found that Cd induced autophagy and apoptosis by elevating cytosolic calcium (Wang et al. 2008). In addition, Cd interferes with the cycling of adhesin (CNX)/calmodulin (CRT) and triggers alterations in ER stress and autophagy-related indices in avian leghorn male hepatoma cells. By regulating intracellular Ca2+homeostasis, selenium intervention inhibits Cd-induced LDH release as well as the interplay between ER stress and autophagy (Zhang et al. 2020). Thus, the interaction between Ca2+ involved ER stress and autophagy is one of the main focuses of Cd toxicity research, with both processes playing dual roles in maintaining cellular homeostasis.
mTOR pathway
mTOR, a 289 kDa serine/threonine protein kinase, is a key regulator of cell growth and proliferation. mTOR participates in the convergence point of the upstream signaling cascade that regulates cellular autophagy. Aberrant regulation of the mTOR signaling pathway is closely related to cellular autophagy status (Wang and Zhang 2019). Three classes of pathways regulate mTOR activity: class I PtdIns3K-protein kinase B (PI3K/AKT), RAS proto-oncogene, and liver kinase B1-AMPK (LKB1/AMPK) pathways (Wang et al. 2012; Wang and Zhang 2019). Autophagosome formation can be suppressed by mTOR signaling, which is negatively regulated by AMPK via LKB1 tumor suppressor kinase and Ca2+/CaMKK-β (Shaw et al. 2004; Woods et al. 2005).
Cd activates the AKT/mTOR pathway and initiates autophagy, thereby inducing various diseases (Arab et al. 2022a; Sun et al. 2023; Wang et al. 2022d). Nrf2 is associated with the regulation of autophagy, as reviewed in "Nephrotoxicity", "Hepatotoxicity", "Respiratory system", "Endoplasmic reticulum stress pathway" Sections. In a study by Dong et al. (Dong et al. 2021), AMPK/AKT/mTOR was identified as a downstream pathway of Cd-induced Nrf2 in primary rat proximal tubular cell (rPT) injury. They further demonstrated that P62 accumulation due to the blockage of autophagic flux contributes to Nrf2 nuclear translocation, which further suppresses AMPK-induced AKT/mTOR signaling in Cd-exposed rPT and rat renal tubular epithelial cells. Environmental Cd exposure has been reported to reduce male fertility rates in humans and animals (Arab et al. 2022b; Ikokide et al. 2022). According to a study by Wang et al. (Wang et al. 2020b), five consecutive weeks of Cd exposure triggered reproductive toxicity in male rats, whereas at eight weeks after cessation of exposure, the toxic effects were significantly ameliorated. mTOR is a vital regulator of autophagy and apoptosis under various pathological conditions (Cayo et al. 2021; Maiese 2020; Wang et al. 2022d). The authors showed that the PI3K inhibitor 3-MA regulates apoptosis by inhibiting autophagy via an mTOR-independent pathway in Cd-treated testicular cells. Ser2448p-mTOR/mTOR protein expression did not change in any of the four testicular cell lines. Similarly, Arab et al. (Arab et al. 2022b) showed that Cd-induced testicular impairment was associated with a decline in autophagy flux with the accumulation of P62. A decrease in AMPK (Ser487) phosphorylation and an increase in the mTOR (Ser2448) phosphorylation cascade were also observed in rats. Furthermore, chronic and low concentration of Cd exposure was reported to promote both osteoclastogenesis (CdCl2 at 0.025 and 0.050 µM in RAW264.7 cells) (Sun et al. 2023) and nephrotoxicity (CdCl2at 5 mg/kg/day via oral gavage in rats) (Arab et al. 2022a) by enhancing autophagy via mTOR signaling. In summary, the AKT/mTOR pathway, especially the activation of upstream PI3K and AMPK, play an important protective role against Cd-induced cellular damage.
Beclin-1 and Bcl-2 families
Beclin-1 is a reliable autophagy marker, and Beclin-1 and its binding ligand class III phosphatidylinositol 3-kinase (PI3KCIII, also known as Vps34) are involved in autophagosome formation and regulate exogenous chemical-induced cellular autophagy (Hill et al. 2019). The Beclin-1/Bcl-2 complex is a key factor linking autophagy and apoptosis (Xu and Qin 2019). Phosphorylated Beclin-1 in the initiation complexes of autophagy can activate autophagy via Ca2+/CaM (calcium/calmodulin)-DAPK (serine/threonine kinase death-associated protein kinase), triggering the dissociation of Beclin-1 from Bcl-2 (Keller et al. 2023). Another study (Arab et al. 2022b) showed that the expression of cellular Beclin-1 and the anti-apoptosis protein Bcl-2 was downregulated in Cd-exposed rat testes; however, linagliptin restored these two proteins and rescued the rebalance of autophagy and apoptosis in the testes of Cd-intoxicated rats. The relative amount of intracellular Beclin-1 and Bcl-2 bound to each other determines the level of cellular autophagy to a certain extent. In a model of duck renal tubular epithelial cells, the addition of the autophagy inhibitor 3-MA to the Cd-treated group significantly increased the rate of apoptosis; increased the mRNA expression of Caspase-3, Cyt C, Bax, and Bak-1; and decreased the mRNA expression of Bcl-2 and the ratio of Bcl-2 to Bax when compared with the control group (Wang et al. 2020a).
Stress can affect autophagy by altering Beclin1/Bcl-2 complex interactions (Lian et al. 2011). In the presence of Cd, increased Ca2+release from the ER lumen leads to the dissociation of Beclin-1 from Bcl-2, thereby activating cellular autophagy. Li et al. (Li et al. 2019) found that Cd induces apoptosis and autophagy in A549 cells and Z-VAD-FMK (an apoptosis inhibitor) inhibits LC3 and Beclin1. Furthermore, they showed that the upregulation of the autophagy-inducing factor Atg4B reduced the expression of Bcl-2 protein and its localization in the mitochondria and influenced mitochondrial function. These changes promoted the dissociation of the Beclin1/Bcl-2 complex and released the autophagy protein Beclin1 to activate the autophagy pathway. The occurrence of Cd-induced cellular autophagy is closely related to the Beclin-1 and Bcl-2 families. Exposure of cells to Cd leads to a decrease in the binding of Beclin-1 to Bcl-2, which activates cellular autophagy.
Emerging molecules and signaling pathways for Cd-induced autophagy
In addition to the classical pathways mentioned above, recent studies (Gong et al. 2022) have shown that Cd exposure can inhibit autophagy and lysosomal transcriptional function by increasing the acetylation of H4K16, which promotes the aggregation of bromodomain-containing protein 4 (BRD4), a novel epigenetic regulator, into the lysosomal gene promoter region. They also demonstrated that pharmacological (JQ1) inhibition of BRD4 can suppress Cd-induced lysosomal gene transcription and function both in vitro and in vivo, thereby inhibiting autophagy and oxidative stress-induced kidney injury. JQ1 selectively suppresses the BRD4 gene and thus, is commonly used with BRD4 for treating various cancers (Jiang et al. 2020). As mentioned above, JQ1 alone or combined with Cd were applied in Gong et al. (Gong et al. 2022) study, to investigate the potential of BRD4 as autophagy regulator on Cd-induced kidney oxidative damage. It should be noted that the link between Cd (as a potential carcinogen) and JQ1 (as anti-tumor agent) is still unclear, the combined effects of JQ1 and Cd warrant for further investigation. In addition, Zou et al. (Zou et al. 2021) reported that Cd negatively regulates the Rheb/mTOR signaling pathway by upregulating miR-155 in BRL3A cells, which induces Cd-induced autophagy in rat hepatocytes. Moreover, Rheb knockdown downregulates Cd-induced autophagy. Thus, Cd exposure may regulate autophagy through epigenetic regulatory mechanisms in both liver and renal injury. In a recent publication, Guo et al. (Guo et al. 2022) reviewed epigenetic mechanisms as key functional targets of Cd-mediated damage and covered a broad range of literature about epigenetic processes impacted due to Cd toxicity in the kidney.
In a study published in 2023 on the effects of Cd on spermatogenesis, Ali et al. (Ali et al. 2023) demonstrated that spermatozoa from 12-week Cd-contaminated mice showed aberrations, vacuolization of the sperm cytoplasm, a decrease in the production of multivesicular vesicles (MVBs) supporting the cytoplasm, and reduction in the biological secretion of exosomes. Thus, a new mechanism by which Cd reduces the exosome-MVB pathway by over-activation of the autophagy pathway has been revealed, and it triggers sperm developmental disorders.
Summary and perspectives
Autophagy is an important regulatory mechanism that maintains homeostasis in organisms. Under physiological conditions, autophagy occurs at a low level to maintain the recycling of cellular contents. When the organism is stimulated by harmful substances such as Cd, autophagy can be activated or blocked to adapt to the alteration of the external environment. If adaptation fails to maintain homeostasis in the internal environment, then inflammation, proliferation, apoptosis, necrosis, or other defensive reactions occur, which lead to cellular injury or even organ failure.
This review summarizes and updates the literature on the origin, transport, and distribution of Cd in the external environment as well as the physicochemical characteristics of Cd distribution after entering the body through the respiratory and digestive pathways. We reviewed and discussed the basic regulatory mechanism of autophagy and the target organs and possible health effects of Cd exposure, with a focus on recently updated molecules/signaling pathways that are mediated by autophagy under Cd toxicity. Moreover, the subsequent fate of the cells following Cd exposure were described. Although the mediation of Nrf2 by the autophagy-lysosome pathway is a promising anti-toxicity molecule for Cd, the specific mechanisms of Cd-induced autophagy that cause damage to the organism are not yet fully understood. Effective therapeutic drugs, tools and sensitive biomarkers for the assessment of Cd toxicity are still lacking (Guo et al. 2022). The interactions or cascades between the reviewed molecules and signaling with autophagy as the core warrant further investigation and the correlation between autophagy and the indicators of exogenous chemical-induced organ-based toxicity and mechanism is necessary to be identified. Furthermore, prevention and treatment strategies focused on the mechanisms underlying Cd toxicity must be developed to prevent the negative effects of autophagy in pathological conditions. Last, although many studies have investigated how Cd species are distributed and transported in the environment, such as in soil, crops and marine organisms, only few studies have focused on the effects of different Cd species on human health. More precise methods for detecting inorganic and organic forms of Cd, particularly in the human matrix, and determining the behavior of different organic complexes of Cd in the human body warrant further research.
References
Abbasabadi MK, Shirkhanloo H (2020) Speciation of cadmium in human blood samples based on Fe(3)O(4)-supported naphthalene-1-thiol- functionalized graphene oxide nanocomposite by ultrasound-assisted dispersive magnetic micro solid phase extraction. J Pharm Biomed Anal 189:113455. https://doi.org/10.1016/j.jpba.2020.113455
Aki T, Funakoshi T, Unuma K, Uemura K (2013) Impairment of autophagy: from hereditary disorder to drug intoxication. Toxicology 311(3):205–215. https://doi.org/10.1016/j.tox.2013.07.001
Ali W, Bian Y, Ali H, Sun J, Zhu J, Ma Y, Liu Z, Zou H (2023) Cadmium-induced impairment of spermatozoa development by reducing exosomal-MVBs secretion: a novel pathway. Aging 15(10):4096–4107. https://doi.org/10.18632/aging.204675
Arab HH, Ashour AM, Eid AH, Arafa EA, Khabbaz A, H. J., El-Aal A, S. A (2022a) Targeting oxidative stress, apoptosis, and autophagy by galangin mitigates cadmium-induced renal damage: role of SIRT1/Nrf2 and AMPK/mTOR pathways. Life Sci 291:120300. https://doi.org/10.1016/j.lfs.2021.120300
Arab HH, Elhemiely AA, El-Sheikh AAK, Khabbaz HJA, Arafa EA, Ashour AM, Kabel AM, Eid AH (2022b) Repositioning linagliptin for the mitigation of cadmium-induced testicular dysfunction in rats: targeting HMGB1/TLR4/NLRP3 axis and autophagy. Pharmaceuticals (Basel). https://doi.org/10.3390/ph15070852
Baan RA, Stewart BW, Straif K (2019) Tumour site concordance and mechanisms of carcinogenesis. https://www.ncbi.nlm.nih.gov/pubmed/33979073
Barrouillet MP, Ohayon-Courtes C, Dubus I, L’Azou B, Nguyen Ba C (2001) Influence of cadmium speciation for the evaluation of in vitro cadmium toxicity on LLC-PK(1) cells. Toxicol In Vitro 15(4–5):525–529. https://doi.org/10.1016/s0887-2333(01)00072-8
Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, Pinton P (2008) Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 43(2):184–195. https://doi.org/10.1016/j.ceca.2007.05.003
Cabukusta B, Neefjes J (2018) Mechanisms of lysosomal positioning and movement. Traffic 19(10):761–769. https://doi.org/10.1111/tra.12587
Cayo A, Segovia R, Venturini W, Moore-Carrasco R, Valenzuela C, Brown N (2021) mTOR activity and autophagy in senescent cells, a complex partnership. Int J Mol Sci. https://doi.org/10.3390/ijms22158149
Chandler JD, Hu X, Ko EJ, Park S, Fernandes J, Lee YT, Orr ML, Hao L, Smith MR, Neujahr DC, Uppal K, Kang SM, Jones DP, Go YM (2019) Low-dose cadmium potentiates lung inflammatory response to 2009 pandemic H1N1 influenza virus in mice. Environ Int 127:720–729. https://doi.org/10.1016/j.envint.2019.03.054
Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, Duranton C, Tauc M, Hofman P, Poujeol P, May E, M. V., Mograbi B (2011) Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 121(1):31–42. https://doi.org/10.1093/toxsci/kfr031
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2008) Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15(1):171–182. https://doi.org/10.1038/sj.cdd.4402233
Chen D, Ran D, Wang C, Liu Y, Ma Y, Song R, Gao Y, Liu Z (2021) Role of mitochondrial dysfunction and PINK1/Parkin-mediated mitophagy in Cd-induced hepatic lipid accumulation in chicken embryos. Life Sci 284:119906. https://doi.org/10.1016/j.lfs.2021.119906
Chen F, Cai X, Kang R, Liu J, Tang D (2023) Autophagy-dependent ferroptosis in Cancer. Antioxid Redox Signal 39(1–3):79–101. https://doi.org/10.1089/ars.2022.0202
Cheung EC, Vousden KH (2022) The role of ROS in tumour development and progression. Nat Rev Cancer 22(5):280–297. https://doi.org/10.1038/s41568-021-00435-0
Chiarelli R, Martino C, Roccheri MC (2019) Cadmium stress effects indicating marine pollution in different species of sea urchin employed as environmental bioindicators. Cell Stress Chaperones 24(4):675–687. https://doi.org/10.1007/s12192-019-01010-1
Chou X, Ding F, Zhang X, Ding X, Gao H, Wu Q (2019) Sirtuin-1 ameliorates cadmium-induced endoplasmic reticulum stress and pyroptosis through XBP-1s deacetylation in human renal tubular epithelial cells. Arch Toxicol 93(4):965–986. https://doi.org/10.1007/s00204-019-02415-8
Cong L, Bai Y, Guo Z (2022) The crosstalk among autophagy, apoptosis, and pyroptosis in cardiovascular disease. Front Cardiovasc Med 9:997469. https://doi.org/10.3389/fcvm.2022.997469
Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, Nawrot T, Vangronsveld J, Smeets K (2010) Cadmium stress: an oxidative challenge. Biometals 23(5):927–940. https://doi.org/10.1007/s10534-010-9329-x
Cybulsky AV (2017) Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol 13(11):681–696. https://doi.org/10.1038/nrneph.2017.129
Dickinson BC, Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7(8):504–511. https://doi.org/10.1038/nchembio.607
Ding WX, Yin XM (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393(7):547–564. https://doi.org/10.1515/hsz-2012-0119
Dong W, Liu G, Zhang K, Tan Y, Zou H, Yuan Y, Gu J, Song R, Zhu J, Liu Z (2021) Cadmium exposure induces rat proximal tubular cells injury via p62-dependent Nrf2 nucleus translocation mediated activation of AMPK/AKT/mTOR pathway. Ecotoxicol Environ Saf 214:112058. https://doi.org/10.1016/j.ecoenv.2021.112058
Fan T, Chen Y, He Z, Wang Q, Yang X, Ren Z, Zhang S (2019) Inhibition of ROS/NUPR1-dependent autophagy antagonises repeated cadmium exposure -induced oral squamous cell carcinoma cell migration and invasion. Toxicol Lett 314:142–152. https://doi.org/10.1016/j.toxlet.2019.07.017
Fan RF, Tang KK, Wang ZY, Wang L (2021) Persistent activation of Nrf2 promotes a vicious cycle of oxidative stress and autophagy inhibition in cadmium-induced kidney injury. Toxicology 464:152999. https://doi.org/10.1016/j.tox.2021.152999
Fels J, Scharner B, Zarbock R, Zavala Guevara IP, Lee WK, Barbier OC, Thevenod F (2019) Cadmium complexed with beta2-microglubulin, albumin and lipocalin-2 rather than metallothionein cause megalin:cubilin dependent toxicity of the renal proximal tubule. Int J Mol Sci. https://doi.org/10.3390/ijms20102379
Filippini T, Wise LA, Vinceti M (2022) Cadmium exposure and risk of diabetes and prediabetes: a systematic review and dose-response meta-analysis. Environ Int 158:106920. https://doi.org/10.1016/j.envint.2021.106920
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G (2017) Molecular definitions of autophagy and related processes. EMBO J 36(13):1811–1836. https://doi.org/10.15252/embj.201796697
Ganguly K, Levanen B, Palmberg L, Akesson A, Linden A (2018) Cadmium in tobacco smokers: a neglected link to lung disease? Eur Respir Rev. https://doi.org/10.1183/16000617.0122-2017
Genchi G, Sinicropi MS, Lauria G, Carocci A, Catalano A (2020) The effects of cadmium toxicity. Int J Environ Res Public Health https://doi.org/10.3390/ijerph17113782
Ghasempour A, Dehghan H, Ataee M, Chen B, Zhao Z, Sedighi M, Guo X, Shahbazi MA (2023) Cadmium sulfide nanoparticles: preparation, characterization, and biomedical applications. Molecules. https://doi.org/10.3390/molecules28093857
Gibson SB (2013) Investigating the role of reactive oxygen species in regulating autophagy. Methods Enzymol 528:217–235. https://doi.org/10.1016/B978-0-12-405881-1.00013-6
Gong ZG, Zhao Y, Wang ZY, Fan RF, Liu ZP, Wang L (2022) Epigenetic regulator BRD4 is involved in cadmium-induced acute kidney injury via contributing to lysosomal dysfunction, autophagy blockade and oxidative stress. J Hazard Mater 423(Pt A):127110. https://doi.org/10.1016/j.jhazmat.2021.127110
Gorlach A, Bertram K, Hudecova S, Krizanova O (2015) Calcium and ROS: a mutual interplay. Redox Biol 6:260–271. https://doi.org/10.1016/j.redox.2015.08.010
Gu J, Wang Y, Liu Y, Shi M, Yin L, Hou Y, Zhou Y, Chu Wong CK, Chen D, Guo Z, Shi H (2019) Inhibition of Autophagy alleviates Cadmium-Induced mouse spleen and human B cells apoptosis. Toxicol Sci 170(1):109–122. https://doi.org/10.1093/toxsci/kfz089
Guo AH, Kumar S, Lombard DB (2022) Epigenetic mechanisms of cadmium-induced nephrotoxicity. Curr Opin Toxicol. https://doi.org/10.1016/j.cotox.2022.100372
Hartwig A (2013) Cadmium and cancer. Met Ions Life Sci 11:491–507. https://doi.org/10.1007/978-94-007-5179-8_15
Hill A, Gailer J (2021) Linking molecular targets of cd in the bloodstream to organ-based adverse health effects. J Inorg Biochem 216:111279. https://doi.org/10.1016/j.jinorgbio.2020.111279
Hill SM, Wrobel L, Rubinsztein DC (2019) Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ 26(4):617–629. https://doi.org/10.1038/s41418-018-0254-9
Horn NM, Thomas AL (1996) Interactions between the histidine stimulation of cadmium and zinc influx into human erythrocytes. J Physiol. https://doi.org/10.1113/jphysiol.1996.sp021721
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, Kang R, Tang D (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12(8):1425–1428. https://doi.org/10.1080/15548627.2016.1187366
Hoyer-Hansen M, Jaattela M (2007) Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14(9):1576–1582. https://doi.org/10.1038/sj.cdd.4402200
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M (2007) Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25(2):193–205. https://doi.org/10.1016/j.molcel.2006.12.009
Humans IWG (2012) Arsenic, metals, fibres, and dusts. IARC Monogr Eval Carcinog Risks Hum 100(Pt C):11–465
Ikokide EJ, Oyagbemi AA, Oyeyemi MO (2022) Impacts of cadmium on male fertility: lessons learnt so far. Andrologia 54(9):e14516. https://doi.org/10.1111/and.14516
Jeong J, Yun SM, Kim M, Koh YH (2020) Association of blood cadmium with cardiovascular disease in korea: from the korea national health and nutrition examination survey 2008–2013 and 2016. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph17176288
Jiang G, Deng W, Liu Y, Wang C (2020) General mechanism of JQ1 in inhibiting various types of cancer. Mol Med Rep 21(3):1021–1034. https://doi.org/10.3892/mmr.2020.10927
Jiang YL, Fei J, Cao P, Zhang C, Tang MM, Cheng JY, Zhao H, Fu L (2022) Serum cadmium positively correlates with inflammatory cytokines in patients with chronic obstructive pulmonary disease. Environ Toxicol 37(1):151–160. https://doi.org/10.1002/tox.23386
Keller CW, Adamopoulos IE, Lunemann JD (2023) Autophagy pathways in autoimmune diseases. J Autoimmun 136:103030. https://doi.org/10.1016/j.jaut.2023.103030
Klaassen CD, Liu J, Choudhuri S (1999) Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol 39:267–294. https://doi.org/10.1146/annurev.pharmtox.39.1.267
Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K, Cecconi F, Choi AMK, Choi ME, Chu CT, Codogno P, Colombo MI, Cuervo AM, Deretic V, Dikic I, Elazar Z, Eskelinen EL, Fimia GM, Gewirtz DA, Green DR, Hansen M, Jaattela M, Johansen T, Juhasz G, Karantza V, Kraft C, Kroemer G, Ktistakis NT, Kumar S, Lopez-Otin C, Macleod KF, Madeo F, Martinez J, Melendez A, Mizushima N, Munz C, Penninger JM, Perera RM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Sadoshima J, Santambrogio L, Scorrano L, Simon HU, Simon AK, Simonsen A, Stolz A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Galluzzi L, Pietrocola F (2021) Autophagy in major human diseases. EMBO J 40(19):e108863. https://doi.org/10.15252/embj.2021108863
Kolluru V, Tyagi A, Chandrasekaran B, Ankem M, Damodaran C (2019) Induction of endoplasmic reticulum stress might be responsible for defective autophagy in cadmium-induced prostate carcinogenesis. Toxicol Appl Pharmacol 373:62–68. https://doi.org/10.1016/j.taap.2019.04.012
Kouroumalis E, Tsomidis I, Voumvouraki A (2023) Pathogenesis of hepatocellular carcinoma: the interplay of apoptosis and autophagy. Biomedicines. https://doi.org/10.3390/biomedicines11041166
Latunde-Dada GO (2017) Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj 1861(8):1893–1900. https://doi.org/10.1016/j.bbagen.2017.05.019
Lee HY, Oh SH (2021) Autophagy-mediated cytoplasmic accumulation of p53 leads to apoptosis through DRAM-BAX in cadmium-exposed human proximal tubular cells. Biochem Biophys Res Commun 534:128–133. https://doi.org/10.1016/j.bbrc.2020.12.019
Lee WK, Probst S, Santoyo-Sanchez MP, Al-Hamdani W, Diebels I, von Sivers JK, Kerek E, Prenner EJ, Thevenod F (2017) Initial autophagic protection switches to disruption of autophagic flux by lysosomal instability during cadmium stress accrual in renal NRK-52E cells. Arch Toxicol 91(10):3225–3245. https://doi.org/10.1007/s00204-017-1942-9
Lefojane RP, Sone BT, Matinise N, Saleh K, Direko P, Mfengwana P, Mashele S, Maaza M, Sekhoacha MP (2021) CdO/CdCO(3) nanocomposite physical properties and cytotoxicity against selected breast cancer cell lines. Sci Rep 11(1):30. https://doi.org/10.1038/s41598-020-78720-5
Lener MR, Reszka E, Marciniak W, Lesicka M, Baszuk P, Jablonska E, Bialkowska K, Muszynska M, Pietrzak S, Derkacz R, Grodzki T, Wojcik J, Wojtys M, Debniak T, Cybulski C, Gronwald J, Kubisa B, Pierog J, Waloszczyk P, Scott RJ, Jakubowska A, Narod SA, Lubinski J (2021) Blood cadmium levels as a marker for early lung cancer detection. J Trace Elem Med Biol 64:126682. https://doi.org/10.1016/j.jtemb.2020.126682
Lepine S, Allegood JC, Edmonds Y, Milstien S, Spiegel S (2011) Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J Biol Chem 286(52):44380–44390. https://doi.org/10.1074/jbc.M111.257519
Li Y, Cheng X, Li M, Wang Y, Fu T, Zhou Z, Wang Y, Gong X, Xu X, Liu J, Pan L (2020) Decoding three distinct states of the Syntaxin17 SNARE motif in mediating autophagosome-lysosome fusion. Proc Natl Acad Sci U S A 117(35):21391–21402. https://doi.org/10.1073/pnas.2006997117
Li M, Pi H, Yang Z, Reiter RJ, Xu S, Chen X, Chen C, Zhang L, Yang M, Li Y, Guo P, Li G, Tu M, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Yu Z, Zhou Z (2016) Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J Pineal Res 61(3):353–369. https://doi.org/10.1111/jpi.12353
Li Z, Li Q, Lv W, Jiang L, Geng C, Yao X, Shi X, Liu Y, Cao J (2019) The interaction of Atg4B and Bcl-2 plays an important role in Cd-induced crosstalk between apoptosis and autophagy through disassociation of bcl-2-Beclin1 in A549 cells. Free Radic Biol Med 130:576–591. https://doi.org/10.1016/j.freeradbiomed.2018.11.020
Li M, Chen W, Cui J, Lin Q, Liu Y, Zeng H, Hua Q, Ling Y, Qin X, Zhang Y, Li X, Lin T, Huang L, Jiang Y (2023a) circCIMT silencing promotes Cadmium-Induced Malignant Transformation of Lung epithelial cells through the DNA base excision repair pathway. Adv Sci (Weinh) 10(14):e2206896. https://doi.org/10.1002/advs.202206896
Li N, Yi BJ, Saleem MAU, Li XN, Li JL (2023b) Autophagy protects against Cd-induced cell damage in primary chicken hepatocytes via mitigation of oxidative stress and endoplasmic reticulum stress. Ecotoxicol Environ Saf 259:115056. https://doi.org/10.1016/j.ecoenv.2023.115056
Lian J, Wu X, He F, Karnak D, Tang W, Meng Y, Xiang D, Ji M, Lawrence TS, Xu L (2011) A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ 18(1):60–71. https://doi.org/10.1038/cdd.2010.74
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22(3):367–376. https://doi.org/10.1038/cdd.2014.143
Liu J, Liu Y, Habeebu SS, Klaassen CD (1998) Susceptibility of MT-null mice to chronic CdCl2-induced nephrotoxicity indicates that renal injury is not mediated by the CdMT complex. Toxicol Sci 46(1):197–203. https://doi.org/10.1006/toxs.1998.2541
Liu G, Zhang K, Dong W, Tan Y, Long M, Zou H, Liu Z (2020a) Puerarin restores the autophagic flux to alleviate cadmium–induced endoplasmic reticulum stress in NRK–52E cells. Mol Med Rep 22(3):2551–2563. https://doi.org/10.3892/mmr.2020.11301
Liu H, Wang Y, Ren Z, Ji X, Peprah FA, Zhang X, Dai S, Zhou Y, Gu J, Shi H (2020b) Dietary cadmium exposure causes elevation of blood ApoE with triglyceride level in mice. Biometals 33(4–5):241–254. https://doi.org/10.1007/s10534-020-00247-z
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020c) Autophagy-dependent ferroptosis: Machinery and Regulation. Cell Chem Biol 27(4):420–435. https://doi.org/10.1016/j.chembiol.2020.02.005
Liu C, Li HJ, Duan WX, Duan Y, Yu Q, Zhang T, Sun YP, Li YY, Liu YS, Xu SC (2023) MCU Upregulation overactivates Mitophagy by promoting VDAC1 dimerization and ubiquitination in the hepatotoxicity of Cadmium. Adv Sci (Weinh) 10(7):e2203869. https://doi.org/10.1002/advs.202203869
Lou M, Garay R, Alda JO (1991) Cadmium uptake through the anion exchanger in human red blood cells. J Physiol 443:123–136. https://doi.org/10.1113/jphysiol.1991.sp018826
Luo B, Lin Y, Jiang S, Huang L, Yao H, Zhuang Q, Zhao R, Liu H, He C, Lin Z (2016) Endoplasmic reticulum stress eIF2alpha-ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis 7(6):e2251. https://doi.org/10.1038/cddis.2016.78
Lv W, Sui L, Yan X, Xie H, Jiang L, Geng C, Li Q, Yao X, Kong Y, Cao J (2018) ROS-dependent Atg4 upregulation mediated autophagy plays an important role in Cd-induced proliferation and invasion in A549 cells. Chem Biol Interact 279:136–144. https://doi.org/10.1016/j.cbi.2017.11.013
Lv YJ, Wei QZ, Zhang YC, Huang R, Li BS, Tan JB, Wang J, Ling HT, Wu SX, Yang XF (2019) Low-dose cadmium exposure acts on rat mesenchymal stem cells via RANKL/OPG and downregulate osteogenic differentiation genes. Environ Pollut 249:620–628. https://doi.org/10.1016/j.envpol.2019.03.027
Ma Y, Su Q, Yue C, Zou H, Zhu J, Zhao H, Song R, Liu Z (2022) The effect of oxidative stress-induced autophagy by cadmium exposure in kidney, liver, and bone damage, and neurotoxicity. Int J Mol Sci. https://doi.org/10.3390/ijms232113491
Maiese K (2020) Dysregulation of metabolic flexibility: the impact of mTOR on autophagy in neurodegenerative disease. Int Rev Neurobiol 155:1–35. https://doi.org/10.1016/bs.irn.2020.01.009
Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18(6):716–731. https://doi.org/10.1016/j.semcdb.2007.09.003
Man YH, Liu YH, Xiong CZ, Zhang Y, Zhang L (2023) Non-lethal concentrations of CdCl2 cause marked alternations in cellular stress responses within exposed sertoli cell line. Toxics 11(2):167
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509(7498):105–109. https://doi.org/10.1038/nature13148
Martinez-Garcia GG, Marino G (2020) Autophagy role in environmental pollutants exposure. Prog Mol Biol Transl Sci 172:257–291. https://doi.org/10.1016/bs.pmbts.2020.02.003
McElroy JA, Kruse RL, Guthrie J, Gangnon RE, Robertson JD (2017) Cadmium exposure and endometrial cancer risk: a large midwestern U.S. population-based case-control study. PLoS ONE 12(7):e0179360. https://doi.org/10.1371/journal.pone.0179360
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152(4):657–668. https://doi.org/10.1083/jcb.152.4.657
Nieto-Torres JL, Zaretski S, Liu T, Adams PD, Hansen M (2023) Post-translational modifications of ATG8 proteins - an emerging mechanism of autophagy control. J Cell Sci 136(16). https://doi.org/10.1242/jcs.259725
Niture S, Lin M, Qi Q, Moore JT, Levine KE, Fernando RA, Kumar D (2021) Role of autophagy in cadmium-induced hepatotoxicity and liver diseases. J Toxicol. https://doi.org/10.1155/2021/9564297
Nguyen TG, Honson NS, Arns S, Davis TL, Dhe-Paganon S, Kovacic S, Kumar NS, Pfeifer TA, Young RN (2014) Development of fluorescent substrates and assays for the key autophagy-related cysteine protease enzyme, ATG4B. Assay Drug Dev Technol 12(3):176–189. https://doi.org/10.1089/adt.2013.561
Nordberg M, Nordberg GF (2022) Metallothionein and cadmium toxicology-historical review and commentary. Biomolecules. https://doi.org/10.3390/biom12030360
Nordberg GF, Bernard A, Diamond GL, Duffus JH, Illing P, Nordberg M, Bergdahl IA, Jin T, Skerfving S (2018) Risk assessment of effects of cadmium on human health (IUPAC technical report). Pure Appl Chem 90(4):755–808. https://doi.org/10.1515/pac-2016-0910
Panwar V, Singh A, Bhatt M, Tonk RK, Azizov S, Raza AS, Sengupta S, Kumar D, Garg M (2023) Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther 8(1):375. https://doi.org/10.1038/s41392-023-01608-z
Peana M, Pelucelli A, Medici S, Cappai R, Nurchi VM, Zoroddu MA (2021) Metal toxicity and speciation: a review. Curr Med Chem 28(35):7190–7208. https://doi.org/10.2174/0929867328666210324161205
Phadwal K, Feng D, Zhu DX, MacRae VE (2020) Autophagy as a novel therapeutic target in vascular calcification. Pharmacol Therapeutics 206:107430
Pi H, Xu S, Zhang L, Guo P, Li Y, Xie J, Tian L, He M, Lu Y, Li M, Zhang Y, Zhong M, Xiang Y, Deng L, Zhou Z, Yu Z (2013) Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy 9(11):1780–1800. https://doi.org/10.4161/auto.25665
Pi H, Li M, Tian L, Yang Z, Yu Z, Zhou Z (2017) Enhancing lysosomal biogenesis and autophagic flux by activating the transcription factor EB protects against cadmium-induced neurotoxicity. Sci Rep 7:43466. https://doi.org/10.1038/srep43466
Prozialeck WC, Edwards JR (2012) Mechanisms of cadmium-induced proximal tubule injury: new insights with implications for biomonitoring and therapeutic interventions. J Pharmacol Exp Ther 343(1):2–12. https://doi.org/10.1124/jpet.110.166769
Qi Z, Chen L (2019) Endoplasmic reticulum stress and Autophagy. Adv Exp Med Biol 1206:167–177. https://doi.org/10.1007/978-981-15-0602-4_8
Rahman MA, Rahman MS, Parvez MAK, Kim B (2023) The emerging role of autophagy as a target of environmental pollutants: an update on mechanisms. Toxics 11(2):135
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863(12):2977–2992. https://doi.org/10.1016/j.bbamcr.2016.09.012
Rosales-Cruz P, Dominguez-Perez M, Reyes-Zarate E, Bello-Monroy O, Enriquez-Cortina C, Miranda-Labra R, Bucio L, Gomez-Quiroz LE, Rojas-Del Castillo E, Gutierrez-Ruiz MC, Souza-Arroyo V (2018) Cadmium exposure exacerbates hyperlipidemia in cholesterol-overloaded hepatocytes via autophagy dysregulation. Toxicology. https://doi.org/10.1016/j.tox.2018.02.007
Sabolic I, Breljak D, Skarica M, Herak-Kramberger CM (2010) Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 23(5):897–926. https://doi.org/10.1007/s10534-010-9351-z
Sadeghzadeh H, Dianat-Moghadam H, Bakhshayesh D, Mohammadnejad AR, D., Mehdipour A (2023) A review on the effect of nanocomposite scaffolds reinforced with magnetic nanoparticles in osteogenesis and healing of bone injuries. Stem Cell Res Ther 14(1):194. https://doi.org/10.1186/s13287-023-03426-0
Satarug S (2018) Dietary Cadmium Intake and Its Effects on Kidneys. Toxics. https://doi.org/10.3390/toxics6010015
Satarug S, Baker JR, Urbenjapol S, Haswell-Elkins M, Reilly PE, Williams DJ, Moore MR (2003) A global perspective on cadmium pollution and toxicity in non-occupationally exposed population. Toxicol Lett 137(1–2):65–83. https://doi.org/10.1016/s0378-4274(02)00381-8
Satir S (2022) The relationship between oral cancer and cadmium: a review. Mol Biol Rep 49(3):2413–2419. https://doi.org/10.1007/s11033-021-07000-w
Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6(1):91–99. https://doi.org/10.1016/j.ccr.2004.06.007
Shin DW (2020) Lipophagy: Molecular mechanisms and implications in Metabolic disorders. Mol Cells 43(8):686–693. https://doi.org/10.14348/molcells.2020.0046
So KY, Lee BH, Oh SH (2018) The critical role of autophagy in cadmium-induced immunosuppression regulated by endoplasmic reticulum stress-mediated calpain activation in RAW264.7 mouse monocytes. Toxicology 393:15–25. https://doi.org/10.1016/j.tox.2017.10.016
So KY, Park BH, Oh SH (2021) Cytoplasmic sirtuin 6 translocation mediated by p62 polyubiquitination plays a critical role in cadmium-induced kidney toxicity. Cell Biol Toxicol 37(2):193–207. https://doi.org/10.1007/s10565-020-09528-2
Song J, Luo H, Yin X, Huang G, Luo S, Lin du R, Yuan DB, Zhang W, Zhu J (2015) Association between cadmium exposure and renal cancer risk: a meta-analysis of observational studies. Sci Rep 5:17976. https://doi.org/10.1038/srep17976
Song S, Tan J, Miao Y, Li M, Zhang Q (2017) Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol 232(11):2977–2984. https://doi.org/10.1002/jcp.25785
Sun M, Jiang Z, Gu P, Guo B, Li J, Cheng S, Ba Q, Wang H (2023) Cadmium promotes colorectal cancer metastasis through EGFR/Akt/mTOR signaling cascade and dynamics. Sci Total Environ 899:165699. https://doi.org/10.1016/j.scitotenv.2023.165699
Surolia R, Karki S, Kim H, Yu Z, Kulkarni T, Mirov SB, Carter AB, Rowe SM, Matalon S, Thannickal VJ, Agarwal A, Antony VB (2015) Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am J Physiol Lung Cell Mol Physiol 309(3):L280–292. https://doi.org/10.1152/ajplung.00097.2015
Tai YT, Chou SH, Cheng CY, Ho CT, Lin HC, Jung SM, Chu PH, Ko FH (2022) The preferential accumulation of cadmium ions among various tissues in mice. Toxicol Rep 9:111–119. https://doi.org/10.1016/j.toxrep.2022.01.002
Thevenod F (2003) Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol 93(4):87–93. https://doi.org/10.1159/000070241
Thevenod F, Lee WK (2013) Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Arch Toxicol 87(10):1743–1786. https://doi.org/10.1007/s00204-013-1110-9
Thevenod F, Wolff NA (2016) Iron transport in the kidney: implications for physiology and cadmium nephrotoxicity. Metallomics 8(1):17–42. https://doi.org/10.1039/c5mt00215j
Tinkov AA, Gritsenko VA, Skalnaya MG, Cherkasov SV, Aaseth J, Skalny AV (2018) Gut as a target for cadmium toxicity. Environ Pollut 235:429–434. https://doi.org/10.1016/j.envpol.2017.12.114
Tong X, Yu G, Liu Q, Zhang X, Bian J, Liu Z, Gu J (2022) Puerarin alleviates cadmium-induced oxidative damage to bone by reducing autophagy in rats. Environ Toxicol 37(4):720–729. https://doi.org/10.1002/tox.23437
Tsapras P, Nezis IP (2017) Caspase involvement in autophagy. Cell Death Differ 24(8):1369–1379. https://doi.org/10.1038/cdd.2017.43
Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164(3):341–346. https://doi.org/10.1083/jcb.200311055
Tuffour A, Kosiba AA, Peprah FA, Gu J, Zhou Y, Shi HF (2023) Cadmium-induced stress: a close look at the relationship between autophagy and apoptosis. Toxicol Sci. https://doi.org/10.1093/toxsci/kfad045
Tyagi A, Chandrasekaran B, Navin AK, Shukla V, Baby BV, Ankem MK, Damodaran C (2023) Molecular interplay between NOX1 and autophagy in cadmium-induced prostate carcinogenesis. Free Radic Biol Med 199:44–55. https://doi.org/10.1016/j.freeradbiomed.2023.02.007
Waalkes MP (2000) Cadmium carcinogenesis in review. J Inorg Biochem 79(1–4):241–244. https://doi.org/10.1016/s0162-0134(00)00009-x
Waalkes MP, Rehm S, Cherian MG (2000) Repeated cadmium exposures enhance the malignant progression of ensuing tumors in rats. Toxicol Sci 54(1):110–120. https://doi.org/10.1093/toxsci/54.1.110
Wang Y, Zhang H (2019) Regulation of Autophagy by mTOR Signaling Pathway. Adv Exp Med Biol 1206:67–83. https://doi.org/10.1007/978-981-15-0602-4_3
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36(11):1434–1443. https://doi.org/10.1016/j.freeradbiomed.2004.03.010
Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65(22):3640–3652. https://doi.org/10.1007/s00018-008-8383-9
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012) Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338(6109):956–959. https://doi.org/10.1126/science.1225967
Wang Y, Mandal AK, Son YO, Pratheeshkumar P, Wise JTF, Wang L, Zhang Z, Shi X, Chen Z (2018) Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol Appl Pharmacol 353:23–30. https://doi.org/10.1016/j.taap.2018.06.003
Wang C, Nie G, Zhuang Y, Hu R, Wu H, Xing C, Li G, Hu G, Yang F, Zhang C (2020a) Inhibition of autophagy enhances cadmium-induced apoptosis in duck renal tubular epithelial cells. Ecotoxicol Environ Saf 205:111188. https://doi.org/10.1016/j.ecoenv.2020.111188
Wang M, Wang XF, Li YM, Chen N, Fan Y, Huang WK, Hu SF, Rao M, Zhang YZ, Su P (2020b) Cross-talk between autophagy and apoptosis regulates testicular injury/recovery induced by cadmium via PI3K with mTOR-independent pathway. Cell Death Dis 11(1):46. https://doi.org/10.1038/s41419-020-2246-1
Wang Q, Pan S, Jiang Q, Li L, Tu W, Zhang Q, Zhou X (2021) CircSPAG16 suppresses cadmium-induced transformation of human bronchial epithelial cells by decoying PIP5K1alpha to inactivate Akt. Mol Carcinog 60(8):582–594. https://doi.org/10.1002/mc.23325
Wang H, Wang A, Wang X, Zeng X, Xing H (2022a) AMPK/PPAR-gamma/NF-kappaB axis participates in ROS-mediated apoptosis and autophagy caused by cadmium in pig liver. Environ Pollut 294:118659. https://doi.org/10.1016/j.envpol.2021.118659
Wang L, Fei J, Wang XM, Xie GF, Cao P, Zhang C, Zhao H, Fu L, Cao W (2022b) Environmental cadmium positively correlates with autophagy and apoptosis in chronic obstructive pulmonary disease patients. Atmospheric Pollution Research 13(1):101275. https://doi.org/10.1016/j.apr.2021.101275
Wang S, Li H, Yuan M, Fan H, Cai Z (2022c) Role of AMPK in autophagy. Front Physiol 13:1015500. https://doi.org/10.3389/fphys.2022.1015500
Wang Z, Li D, Mo L, Liang S, Liao X, Guo S, Yang X, Wei Q (2022d) Low-dose cadmium exposure promotes osteoclastogenesis by enhancing autophagy via inhibiting the mTOR/p70S6K1 signaling pathway. Toxicol Lett 367:9–18. https://doi.org/10.1016/j.toxlet.2022.07.005
Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29(11):1792–1802. https://doi.org/10.1038/emboj.2010.74
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D (2005) Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2(1):21–33. https://doi.org/10.1016/j.cmet.2005.06.005
Xu HD, Qin ZH (2019) Beclin 1, Bcl-2 and autophagy. Adv Exp Med Biol 1206:109–126. https://doi.org/10.1007/978-981-15-0602-4_5
Yamamoto H, Zhang S, Mizushima N (2023) Autophagy genes in biology and disease. Nat Rev Genet 24(6):382–400. https://doi.org/10.1038/s41576-022-00562-w
Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281(40):30299–30304. https://doi.org/10.1074/jbc.M607007200
Yu ZM, Wan XM, Xiao M, Zheng C, Zhou XL (2021) Puerarin induces Nrf2 as a cytoprotective mechanism to prevent cadmium-induced autophagy inhibition and NLRP3 inflammasome activation in AML12 hepatic cells. J Inorg Biochem 217:111389
Yuan J, Zhao Y, Bai Y, Gu J, Yuan Y, Liu X, Liu Z, Zou H, Bian J (2021) Cadmium induces endosomal/lysosomal enlargement and blocks autophagy flux in rat hepatocytes by damaging microtubules. Ecotoxicol Environ Saf 228:112993. https://doi.org/10.1016/j.ecoenv.2021.112993
Zhang L, Xia Q, Zhou Y, Li J (2019) Endoplasmic reticulum stress and autophagy contribute to cadmium-induced cytotoxicity in retinal pigment epithelial cells. Toxicol Lett 311:105–113. https://doi.org/10.1016/j.toxlet.2019.05.001
Zhang C, Wang LL, Cao CY, Li N, Talukder M, Li JL (2020) Selenium mitigates cadmium-induced crosstalk between autophagy and endoplasmic reticulum stress via regulating calcium homeostasis in avian leghorn male hepatoma (LMH) cells. Environ Pollut 265(Pt A):114613. https://doi.org/10.1016/j.envpol.2020.114613
Zhao CJ, Yu D, He ZQ, Bao LJ, Feng LJ, Chen LT, Liu ZY, Hu XY, Zhang NS, Wang TJ, Fu YH (2021) Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free Radic Biol Med 175:236–248. https://doi.org/10.1016/j.freeradbiomed.2021.09.008
Zellner S, Schifferer M, Behrends C (2021) Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Mol Cell 81(6):1337–1354. https://doi.org/10.1016/j.molcel.2021.01.009
Zhen Y, Stenmark H (2023) Autophagosome Biogenesis. Cells:12(4). https://doi.org/10.3390/cells12040668
Zheng Q, Chen Y, Chen D, Zhao H, Feng Y, Meng Q, Zhao Y, Zhang H (2022) Calcium transients on the ER surface trigger liquid-liquid phase separation of FIP200 to specify autophagosome initiation sites. Cell 185(22):4082–4098e4022. https://doi.org/10.1016/j.cell.2022.09.001
Zou H, Wang L, Zhao J, Yuan Y, Wang T, Bian J, Liu Z (2021) MiR-155 promotes cadmium-induced autophagy in rat hepatocytes by suppressing Rheb expression. Ecotoxicol Environ Saf 227:112895. https://doi.org/10.1016/j.ecoenv.2021.112895
Funding
This work was supported by the grants from National Natural Science Foundation of China (81973086) and GuangDong Basic and Applied Basic Research Foundation (2023A1515010542).
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shao, Y., Zheng, L. & Jiang, Y. Cadmium toxicity and autophagy: a review. Biometals 37, 609–629 (2024). https://doi.org/10.1007/s10534-023-00581-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-023-00581-y