
Abstract
Nutrient cations can limit plant productivity in highly weathered soils, but have received much less attention than phosphorus and nitrogen. The reduction of iron (Fe) in anaerobic microsites of surface soils can solubilize organic matter and P sorbed or occluded with short-range-ordered (SRO) Fe phases. This mechanism might also release occluded cations. In the Luquillo Experimental Forest, Puerto Rico, we measured cation release during anaerobic laboratory incubations, and assessed patterns of cation availability in surface soils spanning ridge-slope-valley catenas. During anaerobic incubations, potassium (K), calcium (Ca) and magnesium (Mg) significantly increased with reduced Fe (Fe(II)) in both water and 0.5 M HCl extractions, but did not change during aerobic incubations. In the field, 0.5 M HCl-extractable Fe(II) and Fe(III) were the strongest predictors of K, Mg, and Ca on ridges (R2 0.57–0.75). Here, both Ca and Mg decreased with Fe(III), while K, Ca, and Mg increased with Fe(II), consistent with release of Fe-occluded cations following Fe reduction. Manganese in ridge soils was extremely low, consistent with leaching following reductive dissolution of Mn(IV). On slopes, soil C was the strongest cation predictor, consistent with the importance of organic matter for cation exchange in these highly weathered Oxisols. In riparian valleys, cation concentrations were up to 16-fold greater than in other topographic positions but were weakly or unrelated to measured predictors, potentially reflecting cation-rich groundwater. Predictors of cation availability varied with topography, but were consistent with the potential importance of microsite Fe reduction in liberating occluded cations, particularly in the highly productive ridges. This mechanism may explain discrepancies among indices of “available” soil cations and plant cation uptake observed in other tropical forests.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rock-derived nutrients often limit plant productivity in highly weathered soils characteristic of the humid tropics. Although much research has focused on the importance of phosphorus (P), observations and experiments have also demonstrated the importance of one or more nutrient cations—potassium (K), calcium (Ca), and magnesium (Mg)—in limiting or co-limiting plant and microbial activity in tropical forests (Vitousek and Sanford 1986; Cuevas and Medina 1988; Kaspari et al. 2008; Wright et al. 2011; Baribault et al. 2011; Lloyd et al. 2015). However, less is known about the biogeochemical mechanisms that sustain plant cation demand in soils that have largely been depleted of primary minerals (Chadwick et al. 1999; Markewitz et al. 2001; Porder et al. 2015; Russell et al. 2017). Understanding soil cation availability in tropical forests remains a critical knowledge gap for assessing biogeochemical feedbacks of these ecosystems with climate change.
Iron (Fe) and aluminum (Al) (hydr)oxides are major constituents of weathered tropical forest soils, forming a continuum of short-range-ordered (SRO) to increasingly crystalline phases (Sanchez 1976; Schwertmann 1991). These minerals directly and indirectly influence soil structure and plant access to mineral nutrients. Primary minerals are the dominant ecosystem sources of cations and P in young soils. As these minerals and 2:1 aluminosilicates are progressively depleted during soil development, mineral nutrients can become adsorbed, co-precipitated, or otherwise occluded with Fe and Al (hydr)oxide secondary mineral phases (Walker and Syers 1976). Iron and Al phases and their organic co-precipitates are well known to strongly sorb P, as well as a broad range of trace metals and contaminants (Cornell and Schwertmann 1996). However, their role in sequestering the major plant nutrient cations—K, Ca, and Mg—has received less attention from an ecological perspective. These cations can be occluded with Fe phases as a consequence of several mechanisms, including: isomorphous substitution in minerals (McBride 1989; Giovanoli and Cornell 1992; Fabris et al. 1997), selective adsorption or co-precipitation (Taylor and Graley 1967; McBride 1978), adsorption following charge screening by anions (Rietra et al. 2001). Organic matter occluded within Fe and/or Al phases (Grybos et al. 2007; Kleber et al. 2015) could also harbor cations on ion exchange sites. Thus, as observed for P (Johnson et al. 2003), a significant portion of total soil cations could potentially be occluded by secondary minerals and poorly available for plant uptake. For simplicity in this study, we will refer to non-exchangeable soil cations as “occluded,” irrespective of the precise mechanism.
Dissolution of Fe phases serving a connective or structural role in the soil matrix could release occluded cations for plant or microbial assimilation. Short-range-ordered Fe phases are often abundant in surface horizons of tropical soils, and can be readily dissolved following microbial reduction under anaerobic conditions (Chacon et al. 2006; Dubinsky et al. 2010; Thompson et al. 2011; Ginn et al. 2017). Iron reduction frequently occurs within anaerobic microsites in relatively well-drained soils where biological O2 demand exceeds supply (Hall et al. 2013; Hall and Silver 2015). Iron-associated colloids can be released to suspension or solution following Fe reduction (Thompson et al. 2006; Henderson et al. 2012; Buettner et al. 2014), thus enhancing P uptake by plants and microbes (Peretyazhko and Sposito 2005; Chacon et al. 2006; Liptzin and Silver 2009). A similar mechanism may also influence cation availability, as illustrated by Fig. 1. Under aerobic conditions, a portion of soil cations may be trapped within SRO Fe phases (other dominant minerals in highly weathered soils are omitted from Fig. 1 for simplicity). In the presence of a suitable reductant and/or microbial catalyst, portions of these Fe phases may be dissolved, releasing associated organic matter and cations and thus making them available for assimilation. Manganese (Mn) oxide reduction may play a similar role (McKenzie 1989). However, although Mn(IV) is a more favorable electron acceptor than Fe(III), it is often much less abundant in soil.

Conceptual model describing the occlusion of cations within Fe-organic coprecipitates, expanding on ideas from Fig. 12 in Kleber et al. (2015). Short-range-ordered Fe phases are vulnerable to reductive dissolution, which can release organic matter in soluble and colloidal forms. Previously occluded cations may thus become available for plant assimilation
Here, we tested the hypothesis that soil cation availability increases with Fe reduction in a humid tropical forest, using laboratory incubations and extensive soil sampling in the Luquillo Experimental Forest (LEF), Puerto Rico. This ecosystem is well suited for testing this hypothesis: on volcaniclastic-derived soils in the LEF, cations have been depleted by 99, 97, and 92% (for Ca, Mg, and K, respectively) relative to parent material (Porder et al. 2015). Yet, spatial heterogeneity in soil cation availability remains very high. Within a single forest type in the LEF, spatial variation in soil cation stocks exceeded the range of mean values measured among six other tropical forests (Silver et al. 1994). In that study, base cation concentrations increased along a topographic catena from ridges to slopes to valleys. As cations increased along this gradient, exchangeable Fe decreased, leading the authors to postulate that Fe adsorption had reduced the effective cation exchange capacity on ridge soils (Silver et al. 1994). Iron reduction is also highly significant in surface soils across this catena (Hall and Silver 2015). We built on these previous findings by testing whether spatial variation in Fe(II) and Fe(III) could explain cation concentrations in accordance with the conceptual framework shown in Fig. 1. We also assessed traditional factors thought to influence cation availability: organic C (which dominates cation exchange capacity in weathered soils); clay content; and, live fine root biomass as an index of plant cation demand. A laboratory incubation experiment provided a proof of concept for the importance of anaerobiosis as a control on cation availability in this system.
Methods
Study site
Soils were sampled from the Luquillo Experimental Forest in northwest Puerto Rico, an NSF Long Term Ecological Research site and Critical Zone Observatory. This study focused on a lower montane tropical forest with mean annual precipitation of 3800 mm year−1 and temperature of 23 °C located within the Bisley Watershed (18.31°N, −65.75°W). A detailed geomorphological description of the watershed is provided by Scatena and Lugo (1995). Vegetation is a diverse evergreen forest known locally as “tabonuco” forest after the dominant species, Dacryodes excelsa Vahl. Soils on ridges, slopes, and valleys include Ultisols, Oxisols, and Inceptisols, respectively, in the USDA taxonomy (Soil Survey Staff 2002), but Oxisols dominate at the watershed scale (Johnson et al. 2015). Parent material is andesitic to basaltic volcaniclastic sedimentary rock.
Soil characterization
Soils at our study site are dominated by kaolinite, quartz, Fe oxyhydroxides, and gibbsite, with small amounts of chlorite (Tishchenko et al. 2015; Porder et al. 2015). Carbonates are negligible in these acidic soils (pH < 6; pH data from samples in this experiment are reported below). A valley soil at our site was characterized in detail by 57Fe Mössbauer spectroscopy, showing that nano-goethite and ferrihydrite were the dominant Fe phases (Tishchenko et al. 2015; Ginn et al. 2017). Plagioclase feldspar was the dominant primary mineral source of cations in this ecosystem, and accounts for < 2% of soil mass according to quantitative X-ray diffraction (Porder et al. 2015). A representative profile of a ridge soil at our site is described in detail by Yi-Balan et al. (2014). There is a sparse O horizon, underlain by A and B horizons with a yellow to orange matrix punctuated by Fe-depleted (gleyed) regions of low chroma. The cross section of a representative soil core illustrating this mottling pattern is shown in Fig. 2. A clay-rich aquitard at 30–50 cm largely restricts infiltration to greater depths, channeling subsurface flow to the stream through the variably oxidized rooting zone (McDowell et al. 1992).

Cross-sectional photo of the bottom of a soil core (6 cm diameter, 0–10 cm depth) collected from a slope position in the Luquillo Experimental Forest. The pronounced gleyed regions at the center and bottom of the image, present within a ferric orange matrix, visually illustrate the spatial heterogeneity of reduced and oxidized Fe within surface soils in this ecosystem
Previous work has characterized O2 dynamics and Fe redox cycling in detail in surface soils from this site. Although surface soils are rarely completely saturated with moisture due to their high macroporosity (Hall et al. 2013), they are suitable environments for Fe reduction due to the combination of high clay and organic matter content, well-developed aggregate structure, warm temperatures that stimulate heterotrophic activity and decrease O2 solubility, and high plant productivity and associated labile C inputs. Key findings include: labile C often controls Fe reduction, leading to higher rates in surface horizons than at depth (Chacon et al. 2006; Hall et al. 2016); Fe(III) reducing bacteria are highly abundant in surface soil (0.8 × 108 cells g−1; Dubinsky et al. 2010); net Fe(II) production can occur within aggregates under ambient aerobic conditions (Liptzin and Silver 2009); soil Fe(II) concentrations (0.5 M HCl extractions) can vary by a factor of two or more within hours to days following rainfall events (Hall et al. 2013); soil O2 dynamics vary with precipitation over timescales of days to months (Liptzin et al. 2011). Soil data including root biomass, texture, C, and Fe content of samples from this site were reported previously (Hall and Silver 2015). Briefly, soil C (0–20 cm) averaged 4.8% on ridges, 4.0% on slopes, and 3.8% in valleys. Texture was similar on ridges and slopes (silt and clay content of 50 and 40%, respectively). Valleys had similar silt content but lower clay content (28%).
Sampling design
Four replicate catenas, each spanning a knife-like ridge, steep slope, and riparian valley, were selected for sampling in the Bisley watershed. Ridges were 5–10 m wide. Slopes measured 70–125 m from ridge to valley with an average slope of 35%, and the relatively straight middle sections were selected for sampling. Riparian valleys were adjacent to third-order streams, with a typical distance of 10–20 m from the slope break to the edge of the perennial stream. Along each catena, five sampling locations were established along transects in each topographic position at 5–10 m intervals. Ridge and valley transects were parallel to the contour, and slope transects were perpendicular. Valley transects were at least 5 m away from the edge of the perennial stream channel.
Surface leaf litter was removed where present, and soils were quantitatively sampled at depths of 0–10 and 10–20 cm at each sampling location using a 6 cm diameter stainless steel cylinder (total n = 118; samples at 10–20 cm depth could not be collected in two valley plots because of rocks). These depths were selected because they represent the dominant rooting zone in this forest (Silver and Vogt 1993), have much greater cation stocks than deeper soils (Silver et al. 1994), and are the dominant zones of Fe reduction due to C limitation at depth (Hall et al. 2016). Replicate cores were sampled at each location for soil chemical analyses and fine root biomass measurements. For the laboratory incubation experiment, a 10 kg block of soil from 0 to 10 cm was sampled from a slope near the El Verde field station.
Soil analyses
Soil subsamples (~3 g dry mass equivalent) were immersed in a 1:10 ratio with 0.5 M HCl in the field within two minutes of sampling to inhibit Fe(II) oxidation. Soils were then transported to a field lab within 30 min, vortexed for 60 s, shaken for 1 h, and filtered to 0.22 µm. This extraction is a classic method used to characterize labile Fe(II) in soils and sediments (Lovley and Phillips 1987), and its low pH effectively inhibits Fe(II) oxidation following extraction. It yields soluble and adsorbed Fe(II), in addition to a portion of the Fe (hydr)oxide surface layers (Tishchenko et al. 2015). However, the Fe pool solubilized by 0.5 M HCl accounts for only 10–20% of the SRO Fe pool quantified by citrate-ascorbate (Hall and Silver 2015).
Acid extractions are also useful for assessing nutrient cation availability (Jenny and Leonard 1934), enabling cation quantification on the same supernatant solutions used for Fe(II) and Fe(III) determination. The 0.5 M HCl extraction (pH < 1) entirely displaces cations from exchange sites. Acidic extractions typically yield slightly higher cation concentrations than ammonium acetate, and may provide an improved index of long-term cation availability to plants (Ramirez Romero 1950; Singh et al. 1983). Concentrations of Fe(II) and Fe(III) were measured colorimetrically using an optimized ferrozine method that accounted for Fe(III) interference during Fe(II) determination (Huang and Hall 2017). Other cations were measured via inductively coupled plasma optical emission spectroscopy. Soil pH was measured with an electrode in 1:2 slurries of soil and deionized water.
Particle size fractions (sand, silt, clay) were analyzed on representative soil subsamples using the hydrometer method (Gee and Bauder 1986). Additional subsamples were sieved, dried, and ground to a fine powder for analysis of total C via combustion on an elemental analyzer. Fine root biomass was measured on a separate intact core from each sampling location by wet sieving, and live and dead fine root fractions were separated by turgor and tensile strength (Silver and Vogt 1993).
Anaerobic incubation experiment
The soil sample used for laboratory incubations was stored at ambient temperature (20–25 °C) prior to the experiment (Ginn et al. 2014). Coarse roots were removed by hand and replicate 5 g soil subsamples (dry mass equivalent) were placed in 50 ml polypropylene centrifuge tubes inside glass jars. Previous experiments with soil from this site demonstrated that C availability limited Fe reduction during short-term (days–weeks) incubations (Chacon et al. 2006). Thus, we amended each replicate soil sample with 0.5 g of dried and ground leaf litter (Andropogon gerardii). Samples were incubated inside glass jars sealed with Viton gaskets and aluminum lids with butyl septa. Jars were flushed with N2 at two-day intervals to maintain anaerobic conditions over a 12-day period. Controls were flushed with air. At 0, 4, 8, and 12 days, three replicates of each headspace treatment (total n = 40) were destructively sampled. Samples were quickly homogenized and separate aliquots were extracted in N2-flushed deionized water and 0.5 M HCl for measurement of Fe(II), Fe(III), and cations. Extractions were conducted as described above: for both the water and 0.5 M HCl extractions, a 1:10 ratio of soil to solution was vortexed for 60 s and then shaken for 1 h. Water extractions were filtered through 0.7 µm pre-combusted Whatman GF-F filters.
Statistical analyses
For the field samples, we assessed relationships between K, Ca, Mg, and Mn concentrations and potential predictors using multiple linear regression and mixed effects models in R with the functions lm and lme (Pinheiro et al. 2014). Blocks and plots were included as random effects to account for spatial autocorrelation. Preliminary analyses showed significant interactions among cations, topographic position and predictor variables, such that separate models were fit for each topographic position for clarity of interpretation. Including random effects to account for correlations among samples from individual plots did not improve model fit, as indicated by AIC. Thus, we proceeded with linear regression for clarity of interpretation.
The variables clay content, soil C, live fine root biomass, Fe(II), and Fe(III) were included as potential predictor variables in multiple linear regressions of K, Ca, Mg, and Mn for each topographic position. Model selection for each cation was performed by sequentially eliminating non-significant parameters from the saturated model. A conservative significance criterion of α = 0.01 was used to reduce type I error due to fitting a total of 12 models (four cations × three topographic positions). Impacts of topographic position on cation concentrations were assessed using mixed effects models that included random effects for plots. For the incubation experiment, we calculated linear regressions among Fe(II) and cations.
Results
Incubation experiment
Significant Fe and Mn reduction occurred during the 12-day anaerobic incubation. Concentrations of Fe(II) in 0.5 M HCl extractions increased > 10-fold after 12 days, while Mn (presumably Mn(II)) increased by ~ 50% (Fig. 3). The increase in Fe(II)HCl corresponded with a significant linear decrease in Fe(III)HCl, and almost all cations showed significant linear increases in both the 0.5 M HCl and water extractions (the only exception being CaHCl). Regression coefficients and p values are shown in the legend of Fig. 3. Water-extractable cations increased more than HCl-extractable cations following Fe reduction. An increase in Fe(II)HCl from approximately 10–150 µmol g−1 corresponded with approximately 5–10 fold increases in cations measured in the water extractions, and 0–26% increases in cations in the HCl extractions (Fig. 3). Soil pH increased slightly under anaerobic conditions, measuring 5.82 ± 0.04 at the beginning of the experiment and 5.98 ± 0.02 after 12 days. In the aerobic treatment, cations in the water and HCl extractions did not vary significantly between 0 and 12 days (linear regressions of K, Ca, and Mg versus time; R2 < 0.12, p > 0.1 for all cations).

Pairwise relationships between cations and Fe(II) from samples incubated under anaerobic conditions over 0, 4, 8, and 12 days. Open and solid triangles denote 0.5 M HCl and water extractions, respectively. Solid and dashed lines represent significant linear regressions between Fe(II) and cations measured in 0.5 M HCl and water extractions, respectively. Fe(III)HCl = −0.3x + 54.7, R2 = 0.92, p < 0.0001. MnHCl = 0.05x + 15.6, R2 = 0.59, p < 0.0001. \({\text{Mn}}_{{{\text{H}}_{2} {\text{O}}}}\) = 0.03x + 0.02, R2 = 0.57, p < 0.01. KHCl = 0.005x + 0.146, R2 = 0.28, p < 0.05. \({\text{K}}_{{{\text{H}}_{2} {\text{O}}}}\) = 0.924x + 0.001, R2 = 0.52, p < 0.01. \({\text{Ca}}_{{{\text{H}}_{2} {\text{O}}}}\) = 0.06x + 0.95, R2 = 0.70, p < 0.0001. MgHCl = 0.02x + 17.77, R2 = 0.45, p < 0.01. \({\text{Mg}}_{{{\text{H}}_{2} {\text{O}}}}\) = 0.04x + 0.48, R2 = 0.61, p < 0.001
Topographic variation in cation concentrations in the field
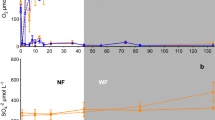
Concentrations of Ca, Mg, and Mn in 0.5 M HCl extractions were 6-, 3- and 13-fold greater (p < 0.05) in valleys than on ridges (Fig. 4). Slope soils generally had intermediate cation concentrations relative to ridges and valleys, but only significantly differed from ridges in the case of Mn (Fig. 4). Soil pH showed the same topographic trends as these cations, and was greatest in valleys (5.28 ± 0.4), intermediate in slopes (4.69 ± 0.4), and lowest on ridges (4.34 ± 0.4). Concentrations of K did not vary among topographic positions.

Boxplots of cation concentrations (0–10 and 10–20 cm samples) plotted by topographic position. Lines represent medians and the first and third quartiles; whiskers extend to the most extreme values. Different letters indicate significant differences at p < 0.01
Relationships among cations and Fe(II)
The cations K, Ca, and Mg generally increased with Fe(II), but the strength of these relationships differed by topographic position (Fig. 5). Pairwise relationships between K and Fe(II) were strongest on ridges and slopes (R2 = 0.69 and 0.57, respectively; p < 0.0001), and weaker in valleys (R2 = 0.23, p < 0.01). Pairwise relationships between Ca and Mg and Fe(II) were strongest on ridges (R2 = 0.38 and 0.35 for Ca and Mg, respectively; p < 0.0001), weaker on slopes (R2 = 0.24 and 0.16; p < 0.001 and < 0.01), and non-significant in valleys.

Pairwise relationships between cations and Fe(II) by topographic position. Circles, triangles, and crosses denote ridge, slope, and valley samples, respectively. Regression lines significant at p < 0.01 are shown in grey
Optimal regression models for cations
Optimal regression models for K, Ca, and Mg were generally similar within a given topographic position, shown in Table 1. In the ridge soils, Fe(II) and Fe(III) were the most important cation predictors. On ridges, K increased with Fe(II) (R2 = 0.69), and Ca and Mg increased with Fe(II) and decreased with Fe(III) (R2 = 0.57 and 0.75, respectively). Concentrations of Mn in ridge soils were consistently low and not significantly related to any measured predictor.
In the slope soils, K, Mg, and Ca increased with soil C, with R2 values of 0.66, 0.44, and 0.32, respectively, and highly significant p values (< 0.0001). Concentrations of Mn also increased with soil C, and decreased with live fine root biomass (R2 = 0.31, p < 0.001). In valley soils, K and Ca increased with soil C but displayed weaker relationships with C than in the slope soils (R2 = 0.32 and 0.39, respectively; p < 0.001). Concentrations of Mg and Mn were unrelated to measured predictors in the valley soils.
Discussion
Our data support the importance of Fe reduction within a hierarchy of biogeochemical and physical factors that influence surface soil cation concentrations across this humid tropical forest. Results from the anaerobic incubation (Fig. 3) were consistent with our conceptual model of occluded cation release following Fe reduction (Fig. 1). The field measurements further supported the role of Fe reduction as a potential control on cation availability (Fig. 4, Table 1). This was particularly true on ridges, which are hotspots of Fe reduction at the landscape scale due to high plant C inputs and associated microbial activity (Hall et al. 2014). On ridges, cations were most strongly predicted by Fe(II) (a positive relationship) and Fe(III) concentrations (a negative relationship). These patterns are consistent with occlusion of cations in association with SRO Fe(III) phases, which can be released following Fe reduction. Ridges had the highest soil C concentrations and SRO Fe content of any topographic position in this forest (Hall and Silver 2015). On slopes, cations were most strongly predicted by soil organic C, consistent with the importance of organic matter in providing cation exchange capacity in highly weathered soils (Silver et al. 1994; Unger et al. 2010). In valleys, cations were poorly related to measured covariates. This may reflect hydrologic interaction of riparian surface soils with stream water, which is comparatively rich in cations (relative to soil water) as a consequence of cation inputs from deep subsurface mineral weathering (McDowell and Asbury 1994; McDowell 1998).
To our knowledge, the influence of Fe redox cycling on the availability of major nutrient cations (K, Ca, Mg) has not been previously addressed in terrestrial soils. This mechanism may be particularly important in humid tropical systems, which are often rich in SRO Fe (Thompson et al. 2011; Tishchenko et al. 2015), and where cations may limit or co-limit biological processes (Vitousek and Sanford 1986; Cuevas and Medina 1988; Kaspari et al. 2008; Cleveland et al. 2011; Wright et al. 2011; Baribault et al. 2011; Lloyd et al. 2015). Our present data expand upon previous investigations of redox-mediated cation dynamics in wetland soils. Brinkman (1970) proposed the mechanism of “ferrolysis,” whereby Fe reduction produces exchangeable Fe2+, which displaces base cations on cation exchange sites and increases solution cation concentrations. This mechanism could lead to cation loss if leaching occurs. Similarly, Phillips and Greenway (1998) found an increase in soluble base cations following soil flooding. They attributed increased cation solubility to displacement of exchangeable cations by newly formed Mn2+ and Fe2+, and found no overall change in exchangeable cations. A previous study in the LEF also hypothesized that displacement of base cations by exchangeable Fe may contribute to cation loss (Silver et al. 1994), and the increase in water-soluble cations during our anaerobic incubation is consistent with this phenomenon.
However, our data suggest that an additional mechanism beyond simple cation displacement by Fe2+ is also likely involved. Population of cation exchange sites by Fe2+ and Mn2+ and displacement of exchangeable K, Ca, and Mg to solution following Fe and Mn reduction and may have occurred in our anaerobic incubation experiment, contributing to the large observed increases in water-extractable cations. However, cation displacement cannot explain the fact that HCl-extractable cations also increased following Fe reduction. By protonating all of the cation exchange sites, the 0.5 M HCl extraction solubilizes the entire exchangeable cation pool (Jenny and Leonard 1934), as well as the surface layer of Fe oxyhydroxide phases while leaving most SRO phases and aluminosilicates intact (Tishchenko et al. 2015; Hall and Silver 2015). Thus, any increase in 0.5 M HCl-extractable cations over time in the same soil indicates that the total size of the exchangeable pool has increased, not simply that exchangeable cations have been released to solution. Consequently, the observed increase in 0.5 M HCl-extractable cations over time in the incubation experiment indicated that additional cations were mobilized under anaerobic conditions, but not in the aerobic controls. Although the precise mechanism remains unclear, this finding is consistent with the release of Fe-occluded cations following Fe reduction. We note that Fe reduction could liberate cations directly due to the reductive dissolution of Fe coprecipitates or adsorbents, or indirectly by increasing pH and promoting colloid dispersion. Iron reduction is often responsible for increased pH under anaerobic conditions, and both of these mechanisms have been documented with respect to the solubilization of organic C (Thompson et al. 2006; Grybos et al. 2007; Pan et al. 2016), and are plausible in the case of increased cations.
Our field data also point to availability of a “new” cation pool following Fe reduction, as opposed to simple shifts in partitioning between adsorbed and solution phases. We observed strong positive relationships between Fe(II) and HCl-extractable cations, which would have been neutral or negative if Fe2+ had simply replaced cations on exchange sites. Thus, we argue that the classic “ferrolysis” hypothesis for periodically flooded wetland soils (Brinkman 1970) is insufficient to explain relationships between Fe and cations in our dataset. Differences in Fe(III) mineral abundance and hydrology between wetland and upland systems (i.e., our study site) may be critical in explaining the observed differences. Hydrologic flowpaths are dramatically different in lowland wetlands (e.g. rice paddies) vs. montane forest soils. The predominance of shallow macropore flow through well-aggregated soils in the Bisley Watershed (McDowell et al. 1992) may lead to lower cation leaching losses following Fe reduction, which dominantly occurs within anaerobic microsites in soils with typical O2 mixing ratios of 10–20% (Hall et al. 2013, 2016).
The dynamic retention and release of occluded cations, whether by Fe reduction or other biogeochemical mechanisms, could contribute to sustaining plant cation uptake in ecosystems where cations—particularly K—may limit plant productivity. The cation concentrations we measured on moist soils extracted in the field using 0.5 M HCl were very similar to those measured ~ 25 years earlier in the same forest using ammonium chloride extractions of moist soil (Silver et al. 1994), despite several severe hurricanes in the intervening period. We measured median concentrations of 1.5, 5.6, and 7.6 µmol g−1 for K, Ca, and Mg, respectively, vs. 1.0, 5.0, and 7.5 µmol g−1 in the Silver et al. (1994) study. Potassium has been shown to co-limit NPP in tropical forests in Panama and Costa Rica, where median K concentrations measured 2.7 µmol g−1 and 2.4 µmol g−1, respectively (Wright et al. 2011; Baribault et al. 2011). The median soil K concentration measured in our study was even lower than in these studies, pointing to possible impacts of K deficits on plant growth. The likely importance of K in the LEF is further illustrated by the fact that moist-soil extractable K from 0 to 35 cm measured < 10% of total plant biomass K stocks on ridges (Silver et al. 1994). In this context, the significant observed release of K in anaerobic microsites could be ecologically relevant.
Mass balance studies provide a key future opportunity for assessing the potential role of Fe reduction in releasing occluded cations. For example, Russell et al. (2017) found that neither uptake of soil exchangeable cations, atmospheric deposition, nor primary mineral weathering could plausibly account for cation accrual in biomass during forest regrowth on a degraded pasture. Their study showed that a “missing” K source of 10–35 kg ha−1 year−1 was needed to explain plant K uptake. Our present data highlight the potential importance of occluded cations in sustaining plant nutrient uptake in highly weathered soils, and provide one potential mechanism—cation release following Fe reduction and associated colloid dispersion—that may contribute to the dynamic cycling of occluded cations. We hypothesize that these occluded cations act as a bank for biotic cation demand that is variably withdrawn and replenished on timescales of hours–months in concert with redox fluctuations. The size of the bank is a function of the soil redox history and consequent impacts on Fe mineral composition over daily to monthly timescales (Hall et al. 2013; Ginn et al. 2017), which control cation “deposits” from decaying organic matter and co-precipitating Fe mineral phases, and cation “withdrawals” from biotic uptake and leaching.
References
Baribault TW, Kobe RK, Finley AO (2011) Tropical tree growth is correlated with soil phosphorus, potassium, and calcium, though not for legumes. Ecol Monogr 82:189–203. doi:10.1890/11-1013.1
Brinkman R (1970) Ferrolysis, a hydromorphic soil forming process. Geoderma 3:199–206. doi:10.1016/0016-7061(70)90019-4
Buettner SW, Kramer MG, Chadwick OA, Thompson A (2014) Mobilization of colloidal carbon during iron reduction in basaltic soils. Geoderma 221–222:139–145. doi:10.1016/j.geoderma.2014.01.012
Chacon N, Silver WL, Dubinsky EA, Cusack DF (2006) Iron reduction and soil phosphorus solubilization in humid tropical forest soils: the roles of labile carbon pools and an electron shuttle compound. Biogeochemistry 78:67–84. doi:10.1007/s10533-005-2343-3
Chadwick OA, Derry LA, Vitousek PM et al (1999) Changing sources of nutrients during four million years of ecosystem development. Nature 397:491–497. doi:10.1038/17276
Cleveland CC, Townsend AR, Taylor P et al (2011) Relationships among net primary productivity, nutrients and climate in tropical rain forest: a pan-tropical analysis. Ecol Lett 14:939–947. doi:10.1111/j.1461-0248.2011.01658.x
Cornell RM, Schwertmann U (1996) The Iron Oxides: Structure, Properties, Reactions. Occurrences and Uses. John Wiley & Sons, Hoboken
Cuevas E, Medina E (1988) Nutrient dynamics within Amazonian forests. II. Fine root growth, nutrient availability and leaf litter decomposition. Oecologia 76:222–235
Dubinsky EA, Silver WL, Firestone MK (2010) Tropical forest soil microbial communities couple iron and carbon biogeochemistry. Ecology 91:2604–2612. doi:10.1890/09-1365.1
Fabris JD, de Jesus Filho MF, Coey JMD et al (1997) Iron-rich spinels from Brazilian soils. Hyperfine Interact 110:23–32. doi:10.1023/A:1012619331408
Gee G, Bauder J (1986) Particle size analysis. In: Klute A (ed) Methods of soil analysis, Part 1, physical and mineralogical methods, 2nd edn. American Society of Agronomy, Madison, pp 383–411
Ginn BR, Habteselassie MY, Meile C, Thompson A (2014) Effects of sample storage on microbial Fe-reduction in tropical rainforest soils. Soil Biol Biochem 68:44–51. doi:10.1016/j.soilbio.2013.09.012
Ginn B, Meile C, Wilmoth J et al (2017) Rapid iron reduction rates are stimulated by high-amplitude redox fluctuations in a tropical forest soil. Environ Sci Technol 51:3250–3259. doi:10.1021/acs.est.6b05709
Giovanoli R, Cornell RM (1992) Crystallization of metal substituted ferrihydrites. Z Für Pflanzenernähr Bodenkd 155:455–460. doi:10.1002/jpln.19921550517
Grybos M, Davranche M, Gruau G, Petitjean P (2007) Is trace metal release in wetland soils controlled by organic matter mobility or Fe-oxyhydroxides reduction? J Colloid Interface Sci 314:490–501. doi:10.1016/j.jcis.2007.04.062
Hall SJ, Silver WL (2015) Reducing conditions, reactive metals, and their interactions can explain spatial patterns of surface soil carbon in a humid tropical forest. Biogeochemistry 125:149–165. doi:10.1007/s10533-015-0120-5
Hall SJ, McDowell WH, Silver WL (2013) When wet gets wetter: decoupling of moisture, redox biogeochemistry, and greenhouse gas fluxes in a humid tropical forest soil. Ecosystems 16:576–589. doi:10.1007/s10021-012-9631-2
Hall SJ, Treffkorn J, Silver WL (2014) Breaking the enzymatic latch: impacts of reducing conditions on hydrolytic enzyme activity in tropical forest soils. Ecology 95:2964–2973. doi:10.1890/13-2151.1
Hall SJ, Liptzin D, Buss HL et al (2016) Drivers and patterns of iron redox cycling from surface to bedrock in a deep tropical forest soil: a new conceptual model. Biogeochemistry 130:177–190. doi:10.1007/s10533-016-0251-3
Henderson R, Kabengi N, Mantripragada N et al (2012) Anoxia-induced release of colloid- and nanoparticle-bound phosphorus in grassland soils. Environ Sci Technol 46:11727–11734. doi:10.1021/es302395r
Huang W, Hall SJ (2017) Optimized high-throughput methods for quantifying iron biogeochemical dynamics in soil. Geoderma 306:67–72. doi:10.1016/j.geoderma.2017.07.013
Jenny H, Leonard C (1934) Functional relationships between soil properties and rainfall. Soil Sci 38:363–381
Johnson AH, Frizano J, Vann DR (2003) Biogeochemical implications of labile phosphorus in forest soils determined by the Hedley fractionation procedure. Oecologia 135:487–499. doi:10.1007/s00442-002-1164-5
Johnson AH, Xing HX, Scatena FN (2015) Controls on soil carbon stocks in El Yunque National Forest, Puerto Rico. Soil Sci Soc Am J 79:294. doi:10.2136/sssaj2014.05.0199
Kaspari M, Garcia MN, Harms KE et al (2008) Multiple nutrients limit litterfall and decomposition in a tropical forest. Ecol Lett 11:35–43. doi:10.1111/j.1461-0248.2007.01124.x
Kleber M, Eusterhues K, Keiluweit M et al (2015) Mineral–organic associations: formation, properties, and relevance in soil environments. In: Advances in agronomy. Elsevier, pp 1–140
Liptzin D, Silver WL (2009) Effects of carbon additions on iron reduction and phosphorus availability in a humid tropical forest soil. Soil Biol Biochem 41:1696–1702
Liptzin D, Silver WL, Detto M (2011) Temporal dynamics in soil oxygen and greenhouse gases in two humid tropical forests. Ecosystems 14:171–182. doi:10.1007/s10021-010-9402-x
Lloyd J, Domingues TF, Schrodt F et al (2015) Edaphic, structural and physiological contrasts across Amazon Basin forest–savanna ecotones suggest a role for potassium as a key modulator of tropical woody vegetation structure and function. Biogeosciences 12:6529–6571. doi:10.5194/bg-12-6529-2015
Lovley DR, Phillips EJP (1987) Rapid assay for microbially reducible ferric iron in aquatic sediments. Appl Environ Microbiol 53:1536–1540
Markewitz D, Davidson EA, de O Figueiredo R et al (2001) Control of cation concentrations in stream waters by surface soil processes in an Amazonian watershed. Nature 410:802–805. doi:10.1038/35071052
McBride MB (1978) Retention of Cu2+, Ca2+, Mg2+, and Mn2+ by amorphous alumina. Soil Sci Soc Am J 42:27. doi:10.2136/sssaj1978.03615995004200010007x
McBride MB (1989) Reactions controlling heavy metal solubility in soils. In: Stewart BA (ed) Advances in Soil Science. Springer, New York, pp 1–56
McDowell WH (1998) Internal nutrient fluxes in a Puerto Rican rain forest. J Trop Ecol 14:521–536
McDowell WH, Asbury CE (1994) Export of carbon, nitrogen, and major ions from three tropical montane watersheds. Limnol Oceanogr 39:111–125. doi:10.4319/lo.1994.39.1.0111
McDowell WH, Bowden WB, Asbury CE (1992) Riparian nitrogen dynamics in two geomorphologically distinct tropical rain forest watersheds: subsurface solute patterns. Biogeochemistry 18:53–75
McKenzie RM (1989) Manganese oxides and hydroxides. In: Dixon JB, Weed SB (eds) Minerals in soil environments, pp 439–465
Pan W, Kan J, Inamdar S et al (2016) Dissimilatory microbial iron reduction release DOC (dissolved organic carbon) from carbon-ferrihydrite association. Soil Biol Biochem 103:232–240. doi:10.1016/j.soilbio.2016.08.026
Peretyazhko T, Sposito G (2005) Iron(III) reduction and phosphorous solubilization in humid tropical forest soils. Geochim Cosmochim Acta 69:3643–3652. doi:10.1016/j.gca.2005.03.045
Phillips IR, Greenway M (1998) Changes in water-soluble and exchangeable ions, cation exchange capacity, and phosphorus max in soils under alternating waterlogged and drying conditions. Commun Soil Sci Plant Anal 29:51–65. doi:10.1080/00103629809369928
Pinheiro J, Bates D, DebRoy S et al (2014) nlme: linear and nonlinear mixed effects models
Porder S, Johnson AH, Xing HX et al (2015) Linking geomorphology, weathering and cation availability in the Luquillo Mountains of Puerto Rico. Geoderma 249–250:100–110. doi:10.1016/j.geoderma.2015.03.002
Ramirez Romero G (1950) Exchangeable cations extracted by 0.1 N hydrochloric acid and ammonium acetate in soils of the Valle. Acta Agron 1:51–56
Rietra RPJJ, Hiemstra T, van Riemsdijk WH (2001) Interaction between calcium and phosphate adsorption on goethite. Environ Sci Technol 35:3369–3374. doi:10.1021/es000210b
Russell AE, Hall SJ, Raich JW (2017) Tree species impact cation dynamics in a tropical rainforest: a new conceptual framework. Ecol Monogr. doi:10.1002/ecm.1274
Sanchez PA (1976) Properties and management of soils in the tropics. Wiley, New York
Scatena FN, Lugo AE (1995) Geomorphology, disturbance, and the soil and vegetation of two subtropical wet steepland watersheds of Puerto Rico. Geomorphology 13:199–213. doi:10.1016/0169-555X(95)00021-V
Schwertmann U (1991) Solubility and dissolution of iron oxides. Plant Soil 130:1–25
Silver WL, Vogt KA (1993) Fine-root dynamics following single and multiple disturbances in a subtropical wet forest ecosystem. J Ecol 81:729–738
Silver WL, Scatena FN, Johnson AH et al (1994) Nutrient availability in a montane wet tropical forest—spatial patterns and methodological considerations. Plant Soil 164:129–145
Singh KD, Goulding KWT, Sinclair AH (1983) Assessment of potassium in soils. Commun Soil Sci Plant Anal 14:1015–1033. doi:10.1080/00103628309367429
Soil Survey Staff (2002) Soil survey of Caribbean National Forest and Luquillo Experimental Forest, Commonwealth of Puerto Rico. United States Department of Agriculture, Natural Resources Conservation Service
Taylor RM, Graley AM (1967) The influence of ionic environment on the nature of iron oxides in soils. J Soil Sci 18:341–348. doi:10.1111/j.1365-2389.1967.tb01512.x
Thompson A, Chadwick OA, Boman S, Chorover J (2006) Colloid mobilization during soil iron redox oscillations. Environ Sci Technol 40:5743–5749. doi:10.1021/es061203b
Thompson A, Rancourt D, Chadwick O, Chorover J (2011) Iron solid-phase differentiation along a redox gradient in basaltic soils. Geochim Cosmochim Acta 75:119–133. doi:10.1016/j.gca.2010.10.005
Tishchenko V, Meile C, Scherer MM et al (2015) Fe2 + catalyzed iron atom exchange and re-crystallization in a tropical soil. Geochim Cosmochim Acta 148:191–202. doi:10.1016/j.gca.2014.09.018
Unger M, Leuschner C, Homeier J (2010) Variability of indices of macronutrient availability in soils at different spatial scales along an elevation transect in tropical moist forests (NE Ecuador). Plant Soil 336:443–458. doi:10.1007/s11104-010-0494-z
Vitousek P, Sanford R (1986) Nutrient cycling in moist tropical forest. Annu Rev Ecol Syst 17:137–167
Walker TW, Syers JK (1976) The fate of phosphorus during pedogenesis. Geoderma 15:1–19. doi:10.1016/0016-7061(76)90066-5
Wright SJ, Yavitt JB, Wurzburger N et al (2011) Potassium, phosphorus, or nitrogen limit root allocation, tree growth, or litter production in a lowland tropical forest. Ecology 92:1616–1625. doi:10.1890/10-1558.1
Yi-Balan SA, Amundson R, Buss HL (2014) Decoupling of sulfur and nitrogen cycling due to biotic processes in a tropical rainforest. Geochim Cosmochim Acta 142:411–428. doi:10.1016/j.gca.2014.05.049
Acknowledgements
This work was supported by NSF grant DEB-1457805, by the NSF Luquillo Critical Zone Observatory, and by Iowa State University. SJH gratefully acknowledges mentorship by W. Silver on related research at this site. We thank A. Russell for discussion about the conceptual model, S. Rathke and S. Bakshi for assistance with ICP analyses, the USFS International Institute of Tropical Forestry for logistical support, and O. Gutierrez del Arroyo for collecting soil for the incubation experiment.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: James Sickman.
Rights and permissions
About this article

Cite this article
Hall, S.J., Huang, W. Iron reduction: a mechanism for dynamic cycling of occluded cations in tropical forest soils?. Biogeochemistry 136, 91–102 (2017). https://doi.org/10.1007/s10533-017-0383-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-017-0383-0