
Abstract
Objective
Brassica juncea, a major oilseed crop, suffers substantial yield losses due to infestation by mustard aphids (Lipaphis erysimi). Unavailability of resistance genes within the accessible gene pool underpins significance of the transgenic strategy in developing aphid resistance. In this study, we aimed for the identification of an aphid-responsive promoter from B. juncea, based on the available genomic resources.
Results
A monosaccharide transporter gene, STP4 in B. juncea was activated by aphids and sustained increased expression as the aphids colonized the plants. We cloned the upstream intergenic region of STP4 and validated its stand-alone aphid-responsive promoter activity. Further, deletion analysis identified the putative cis-elements important for the aphid responsive promoter activity.
Conclusion
The identified STP4 promoter can potentially be used for driving high level aphid-inducible expression of transgenes in plants. Use of aphid-responsive promoter instead of constitutive promoters can potentially reduce the metabolic burden of transgene-expression on the host plant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among the rapeseed-mustard group of crops, Indian mustard [Brassica juncea (L.) Czern.] is the predominant oil yielding crop in India. It alone occupies 85% of the total rapeseed-mustard growing area in India (DRMR 2015). Because of intrinsic tolerance to salinity and moisture deficit, mustard cultivation fits well across the diverse agroclimatic regimes, including marginal lands of resource poor farmers. However, the productivity of this crop is severely affected due to infestation by mustard aphid [Lipaphis erysimi (Kalt)] (Bakhetia 1987; Rohilla et al. 1987; Shekhawat et al. 2012). The severity of infestation may cause 35.4–91.3% yield loss which is equivalent to 11–32% loss in oil (Singh and Sachan 1994). In financial terms, the average loss imposed by aphids may extend to Rs. 1575 crores annually (derived). Aphids are hemipteran, phloem-feeding insects. While feeding, the nymphs and the adults divert excessive photo-assimilates from the host and exudate sugar-rich honeydews. Production of honeydews hinders photosynthetic activities and alters defense response of the host (Schwartzberg and Tumlinson 2014). The salivary components of aphids potentially attenuate host-defense response and establish uninterrupted feeding (Miles 1999; Ilarduya et al. 2003; Zhu-Salzman et al. 2004; Park et al. 2006). Aphids also serve as potential vectors of plant luteo viruses (Hogenhout et al. 2008; Lu et al. 2016).
The gene(s) for genetic resistance is either obscure among the Brassica germplasms or even if reported in a few wild accessions, largely remains uncharacterized (Kumar et al. 2011; Atri et al. 2012; Sarkar et al. 2016). Thus, the scope for developing resistance through conventional breeding is limited (Bhadoria et al. 1995; Dutta et al. 2005). For overcoming such bottleneck, transgenic expression of insecticidal genes from distant sources has been considered as a potential avenue for developing the aphid resistant plant types (reviewed in Bhatia et al. 2011; Das et al. 2018). For example, several plant lectin genes have been expressed in B. juncea for developing transgenic-mediated aphid resistance (Kanrar et al. 2002; Sharma et al. 2004; Dutta et al. 2005; Hossain et al. 2006; Saha et al. 2006; Sadeghi 2007). However, none of these transgenics could be advanced to field trials or released as cultivar. This clearly advocated the lack of field-applicable resistance in these transgenics. In these studies, either the constitutive promoter CaMV 35S or, only in few cases, a phloem-specific promoter had been used for expressing the transgene (Kanrar et al. 2002; Dutta et al. 2005; Hossain et al. 2006). Constitutive expression of the transgene leads to various disadvantages such as metabolic payoffs (Cipollini et al. 2003; Walters and Heil 2007; Garrido et al. 2017) and pleiotropic effects on the plants (Li et al. 2002; Liu et al. 2008; Brini et al. 2011). Thus, use of specific promoters for tissue and stress specific expression of the transgene will be more desirable.
Interestingly, aphids have evolved to bypass the host-defense while feeding on the host plants. It secretes effector proteins such as COO2, MP1, VPS52 etc. into the host cells which inactivate defense signaling in the surrounding tissues (Pitino and Hogenhout 2012; Jaouannet et al. 2014; Rodriguez et al. 2017). Consequently, several independent studies have led to hypothesize countering of host-defense suppression by aphid-inducible expression of endogenous defense genes (Ellis et al. 2002; Boughton et al. 2006; Koramutla et al. 2014). However, validation of such possibility as well as precise temporal expression of aphid deterring genes, will require aphid responsive promoters. Transcriptome data on several plant–aphid interaction studies are available which can be analysed for identifying the host-genes activated due to colonization and feeding by aphids (Voelckel et al. 2004; Smith and Boyko 2007; Kusnierczyk et al. 2008). Mining on these transcripts and their upstream intergenic regions will be the most relevant assignment in identification of the aphid-responsive promoters.
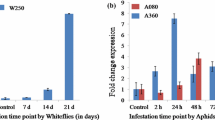
Recently, white fly- and aphid-specific promoters have been described in Arabidopsis (Dubey et al. 2018). However, an aphid responsive promoter is not known in rapeseed-mustard group of crops including B. juncea. In the present study, we have isolated and characterized a potential aphid-responsive promoter in B. juncea by screening the host genes which are transcriptionally activated by aphid infestation. We have also attempted to delineate the important regions of the promoter for the aphid-responsive promoter-activity through deletion analysis.
Materials and methods
Planting materials and growth conditions
Seeds of B. juncea cv. Varuna and Nicotiana benthamiana were available at ICAR-National Institute for Plant Biotechnology, New Delhi. The plants were grown in 8-inch plastic pots filled with a mixture of field soil, soilrite and leaf compost. The plants were maintained in glass house at 24 ± 2 °C with 65–70% relative humidity and with 16/8 h light/dark cycle. The plants were watered twice a week with Hoagland solution (Hi-media, India).
Aphid infestation, sample collection and cDNA preparation
Maintenance of aphid population, aphid inoculation on the experimental plants, collection of leaves for the gene expression studies, isolation of RNA and cDNA synthesis were performed according to the methods as described in Ram et al. (2017).
Mining of aphid-inducible genes from plant-aphid interaction studies
Transcriptome and microarray data of plant-aphid interaction studies (Table 1) were retrieved from GEO database (https://www.ncbi.nlm.nih.gov/gds) and analyzed with GEO2R online tool of NCBI (https://www.ncbi.nlm.nih.gov/geo/geo2r). The top 250 genes from each data set were selected and further filtered for > 2.0-fold change in expression. Additionally, the genes validated for the up-regulation during aphid infestation in plant-aphid interaction studies were also included. Annotation clustering and categorization for the biological functions of the identified genes were carried out using The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 bioinformatic resources (https://david.abcc.ncifcrf.gov) with ease threshold at p-value < 0.05 and Gene Ontology enrichment anaLysis and visuaLizAtion (GORILLA; https://geneontology.org/page/go-enrichment-analysis) tool, respectively. The Gene Ontology (GO) analysis of selected genes was performed using Blast2GO software (https://www.blast2go.com).
PCR and qRT-PCR assay
For designing of gene-specific primers, homologs of the selected genes were identified in Arabidopsis database (https://www.arabidopsis.org). The gene-specific primers (Supplementary Table 1) were designed from intron spanning regions of the genes, using PrimerQuest tool of Integrated DNA Technology (https://eu.idtdna.com/PrimerQuest/Home/Index). PCR was performed in 20 µl reaction volume containing 2.0 µl of 10X Taq DNA polymerase buffer, 0.5 µl of dNTP mix (10 mM), 1.0 µl each of the forward and reverse primer (10 µM), 1 U of Taq DNA polymerase (TAKARA Bio Inc., Japan) and 1.0 µl of template DNA (50 ng/µl). The final volume was made up by nuclease-free water. PCR reaction was performed in a thermal cycler (Applied Biosystems, USA) with the following PCR programme: initial denaturation at 95 °C for 4 min followed by 30 repeated cycles of denaturation at 95 °C for 1 min, primer annealing at 55 °C for 30 s and primer extension at 72 °C for 1 min followed by final extension at 72 °C for 7 min. The amplicons were analysed on 1.5% agarose gel. The desired amplicons were validated by sequencing (SciGenom Labs Pvt. Ltd., India).
The qRT-PCR analysis was performed using the SYBR Premix Ex Taq II (Tli RNaseH Plus) kit (TAKARA Bio Inc., Japan) in a StepOnePlus Real-Time PCR system (Applied Biosystems, USA). CAC gene has been identified as the best reference gene for B. juncea samples treated with aphid stress (Ram et al. 2017). Thus, in case of B. juncea, the gene expression data was normalized using CAC. For analysing GUS gene expression in tobacco samples, GAPDH was taken as the internal control. A 10 µl reaction cocktail contained the following components: 5 µl of 2X SYBR Premix Ex Taq II (Tli RNaseH Plus), 0.2 µl of 50X ROX dye, 0.4 mM of each forward and reverse primers, 1 µl of diluted cDNA and 3.3 µl of nuclease-free water. PCR was carried out with one cycle of initial denaturation at 95 °C for 2 min followed by 40 cycles at 95 °C for 10 s, 58–60 °C for 30 s and 72 °C for 30 s. Amplification specificity was confirmed by dissociation curve analysis with gradual increase in melting temperature from 60 to 95 °C. All the qRT-PCR experiments were performed independently in three biological replicates with minimum three technical replicates each time. Fold-change in gene-expression was calculated using 2−ΔΔCt method (Pfaffl et al. 2004).
Identification and cloning of STP4 promoter
For identifying upstream region of the STP4 gene, the homologous sequence of STP4 CDS was identified in B. rapa database (https://Brassicadb.org) through BLAST analysis. The full length STP4 gene was identified in BAC clone KBrH046K16 using FGENESH software (https://www.softberry.com/fgenesh). The transcription start site (TSS) of the STP4 gene was predicted using TSSP online tool (https://www.softberry.com/berry.phtml? topic = promoter). The corresponding locus in B. rapa, was searched in Genome Browse of B. rapa database (https://Brassicadb.org/cgi-bin/gbrowse/Brassica/). Based on the translation start site (ATG) of the locus Bra017776, an upstream region (2 kb approx.) was retrieved as the putative promoter of the STP4 gene.
The putative promoter was validated in silico by various promoter prediction tools and amplified from B. juncea DNA using sequence-specific primers (proBjSTP4F and proBjSTP4R). Restriction sites for BamHI and NcoI were incorporated at the 5′ end of the forward and reverse primers, respectively. Thus, the forward primer was designed 2.0 kb upstream from the translation initiation codon (ATG) and the reverse primer was designed at the 5′UTR region immediately upstream to the translation initiation codon. PCR amplification was performed in 20 µl of reaction volume containing 18.0 µl of Platinum PCR Super Mix High Fidelity (Invitrogen, USA), 0.4 µM each of the forward and reverse primer and 1.0 µl of DNA (50 ng/µl). The PCR reaction was performed in a thermocycler (Applied Biosystems, USA) with following conditions: one cycle of initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, primer annealing at 54 °C for 30 s and primer extension at 68 °C for 4 min and one cycle of final extension at 68 °C for 15 min. The PCR amplified fragment (proBjSTP4) was cloned in pGEM-T Easy cloning vector (Promega, USA) and validated through sequencing. In silico analysis of the putative promoter for identifying cis-regulatory elements was carried out using Plant CARE (https://bioinformatics.psb.ugent.be/webtools/plantcare).
Development of proBjSTP4-GUS fusion construct and assay of the promoter activity
The proBjSTP4 fragment was sub-cloned in BamHI–NcoI sites of the binary vector pCAMBIA1305.1 by substituting the CaMV 35S promoter of the parent vector. Subsequently, the binary construct was mobilized into a C58 type Agrobacterium strain GV3101. For verifying any bacterial expression of GUS, the Agrobacterium cells harbouring the pCAMBIA-proBjSTP4 construct was incubated with all the components of histochemical assay at 37 °C for 1 h. Agrobacterium strains harbouring pBI121 and pORER2, as they show bacterial expression of GUS, was used as positive controls in the experiment.
Development of deletion constructs
Four deletions, starting from 5′ end of the STP4 upstream sequence, named as proSTP4DC1, proSTP4DC2, proSTP4DC3 and proSTP4DC4 were created by PCR amplifications using 4 set of forward primers and a common reverse primer proBjSTP4R (Supplementary Table 2). All the deletion fragments were cloned in pGEM-T easy vector and validated by sequencing. All these deletion fragments were further cloned in pCAMBIA1305.1, substituting the CaMV 35S promoter upstream to GUS.
Agroinfiltration of B. juncea and N. benthamiana leaves
Agroinfiltration of leaves were carried out according to Xu et al. (2008) with some modifications. The detached leaves of B. juncea were completely dipped into the infiltration culture and subjected to vacuum at 30 psi for 30 min. Density of the Agrobacterium cells were kept uniform every time based on OD600 reading. The vacuum infiltrated leaves were stabilized for 24 h with their petioles immersed in Hoagland solution before further treatment and histochemical assay. The Agroinfiltration in N. benthamiana leaves were performed according to Sparkes et al. (2006) with minor modifications. The N. benthamiana plants at 4–6 leaf stage were Agroinfiltrated at 3–4 spots using 2 ml syringe with 100 μl infiltration suspension containing the Agrobacterium cells. The infiltrated plants were kept for 24 h at normal growth conditions before wound or hormonal treatment and histochemical assay.
Wound and hormonal treatment of Agroinfiltrated N. benthamiana
Wound was inflicted towards petiole of the infiltrated tobacco leaf across the mid vein of at least 40% leaf area and samples were collected in a time course manner at 2 and 6 h. Infiltrated leaves without wounding were collected as control for each time point. In hormonal treatments, 2 mM methyl jasmonate (MJ) and 5 mM salicylic acid (SA), prepared in water with Triton X-100, were sprayed on infiltrated tobacco plants kept in desiccators in independent experiments. Leaf samples were collected at 2 and 6 h time points following the treatments. Mock treated (water with Triton X-100) infiltrated leaves were taken as control for each time point. The collected samples were immediately frozen in liquid nitrogen and stored at − 80 °C until further use. The experiments were repeated three times. Significant difference in mean values was evaluated by Student's t-test at p-value < 0.05.
Aphid bioassay on detached leaves
Forty-to-fifty wingless aphids of assorted life stages were released on each Agroinfiltrated leaf and kept in a desiccator in moist condition. Leaf samples were collected at 0, 24 and 48 h post infestation along with parallel controls sans infestation. The samples were collected in duplicates for histochemical as well as qRT-PCR analysis. The experiments were repeated three times as biological replicates with three technical replicates at each time. Significant difference in means was evaluated by Student's t-test at p-value < 0.05.
Histochemical and gene expression analysis of GUS
Histochemical staining of GUS activity was conducted according to the method described by Jefferson et al. (1987) with minor modifications. The samples were incubated in GUS staining solution containing 10 mM sodium phosphate buffer at pH 7.0, 10 mM Na2-EDTA, 0.1% Triton X-100, 1 mg/mL X-Gluc, 2 mM potassium ferricyanide, 2 mM potassium ferrocyanide at 37 °C for 10–12 h after 30 min of vacuum infiltration at 30 psi pressure. After GUS staining, the samples were incubated in 70% ethanol for removing the chlorophylls. Relative abundance of GUS transcripts was assayed by qRT-PCR of RNA from the leaves, as described earlier.
Results
Mining of aphid-inducible genes
Studies on gene-expression in case of B. juncea-aphid interaction are limited (Bandopadhyay et al. 2013; Koramutla et al. 2014). Thus, for identifying the genes transcriptionally activated by aphids we mined the transcriptome and microarray data from other plant-aphid interaction studies (Table 1). The genes up-regulated by > 2.0-fold due to aphid infestation were identified and thus, 261 up-regulated genes were selected. A flow diagram representing the pipeline used for the identification of the aphid-inducible genes is given in Fig. 1.

A flow diagram of the pipeline used for the identification of aphid inducible genes. The aphid inducible genes were identified from different plant-aphid interaction studies on the basis of > 2.0 fold-change in expression due to treatment. The selected genes were clustered based on their functions using DAVID v6.7 software at threshold p value < 0.05
The selected 261 transcripts were subjected to functional clustering which revealed 14 annotation clusters (named A to N), each representing 1–22% of the transcripts at p-value < 0.05 (Fig. 2a). The cluster A represented 57 (22%) defence responsive genes followed by cluster C representing 47 (18%) metabolic genes. Many of the earlier reports suggested that, the defence genes transiently activated by aphid probing were eventually suppressed in susceptible hosts (De Vos et al. 2005; Koramutla et al. 2014; Schwartzberg and Tumlinson 2014). Therefore, expression of such host-genes may not be persistent under aphid colonization (Gao et al. 2007). Thus, the defence pathway related genes were excluded. The remaining 204 genes were screened for identifying their involvement in various biological processes such as cell wall modification, water transport, vitamin biosynthesis, carbon and nitrogen metabolism and mobilization. We hypothesized that the genes involved in such metabolic processes may be modulated by aphids for generating a favourable metabolic pool supportive to rapid colonization of the host plants. Based on this hypothesis, 41 genes (Table 2) were identified. Gene ontology (GO) analysis of the selected 41 genes revealed majority of them (60.53%) being associated with transporter activity followed by a few (20%) being DNA binding proteins (Fig. 2b). Similarly, when categorized based on involvement in biological processes, majority of the genes were involved in cellular processes (74%) followed by transport (70%, Fig. 2c). Further, based on cell components, majority of them (75%) were found residing on the cell membrane followed by cytoplasmic components (Fig. 2d).

Clustering and GO categorization of aphid-responsive genes. a Functional clustering of 261 aphid responsive genes. b–d GO analysis of the selected 41 aphid-responsive genes for molecular function, biological processes and cellular components. The analysis was performed using DAVID v6.7 at p value of ≤ 0.05 and Blast 2 GO programme. Value at X-axis represents percentage of gene ontology
Gene-expression study of the aphid-inducible genes in B. juncea
Heterologous primers based on the Arabidopsis sequences could amplify 39 of the selected genes in B. juncea DNA and cDNA. Amplicons with desired specificity in length were sequenced for validation and designing of the gene-specific qPCR primers (Supplementary Table 1). In qRT-PCR based gene expression study, 18 genes revealed differential expression in response to aphid infestation over the time course of 6–96 h (Fig. 3). Based on the expression pattern, these genes were grouped into three categories: (a) significantly up-regulated genes with sustained expression which included CAT2, CAT9, STP4, β-fruct, NRT2, MSAM3, ERD6 and MIPS, (b) genes showing early response, but down regulated within 96 h or later time points viz. PINV, PME, EXPA1, EXPA2, LHT7, THI1 and MT and (c) genes showing insignificant activation viz. SDH, GS and MS. Eight significantly up-regulated genes were further compared based on three criteria namely, minimum basal level of expression, significant level of transcriptional activation in response to aphids and sustained level of activated expression at least more than 96 h of treatment. The candidate genes STP4, IPS and β-fruct were found to be the most appropriate in terms of the above criteria. Among the three, STP4 gene was selected for isolation of its promoter. Aphid mediated transcriptional activation of the STP4 gene was also analysed across the major tissues such as leaf, stem, flower and siliques, which are highly infested by aphids. The study validated transcriptional activation of the STP4 promoter, albeit at variable level, across the tissues upon aphid infestation. The highest transcriptional activation of more than fivefold was recorded in the flowers whereas, other tissues such as leaf, stem and siliques showed an activation ranging from 1.6 to 2.5-fold compared to their counterparts from non-infested plants (Fig. 4).

Gene expression studies of the aphid-inducible genes in B. juncea. The fold-change in expression of the genes across the samples was determined by qRT-PCR and calculated using Pfaffl equation (Pfaffl et al. 2004). Different lower-case alphabets indicate statistically significant difference between the samples. The samples were collected in three biological replicates and each sample assayed in three technical replicates. Significant difference in mean was evaluated by Student’s t test at P < 0.05 and represented as mean ± SE (n = 3)

Gene-expression of STP4 in different tissues of B. juncea under aphid infestation. The fold-change in gene-expression was determined by qRT-PCR and calculated using Pfaffl equation (Pfaffl et al. 2004). The different lower-case alphabets indicate significant difference in mean derived from three biological replicates with three technical replicates each. Significant difference in mean was evaluated by Student’s t test at P < 0.05 and represented as mean ± SE (n = 3)
Isolation and in silico analysis of STP4 promoter in B. juncea
The STP4 coding sequence of B. juncea shared 89% and 95% homology to AtSTP4 (At3g19930) gene and the BAC clone of B. rapa subsp. Pekinensis (KbrH046K16), respectively. Since B. rapa is one of the progenitors of B. juncea, the BAC clone KbrH046K16 was used as genomic resource for the identification of upstream sequences of STP4. The STP4 gene in the BAC clone spanned over 1.9 kb consisting of 4 exons and was mapped on chromosome 3 (A03) of B. rapa. Based on the defined TSS and ATG of the STP4 gene of B. rapa (Bra001766), around 2.0 kb upstream region was retrieved from B. rapa BAC clone. The identified upstream sequence was PCR amplified in B. juncea using a specific set of primers. The PCR amplified 2.0 kb fragment was cloned in pGEMT Easy vector and upon sequencing showed 99% homology with the upstream region of Bra001766. The sequence cloned as the putative promoter and named as proBjSTP4, was analysed by Plant CARE software for identification of the cis-regulatory elements (Fig. 5). The basal promoter elements like TATA-box (TATAAATT) and CAAT-box (GACCAA) was found at − 36 and − 94 bp upstream to the TSS, respectively. In silico analysis also revealed the presence of other important cis-regulatory elements, which have been listed in Supplementary Table 3.

Mapping of cis-regulatory elements in proBjSTP4. The putative cis-regulatory elements in proBjSTP4 upstream sequence was predicted by using PlantCare software and indicated in boxes and circles. The + 1 site and italicized bold letters indicate the TSS and translation initiation codon of the STP4 gene, respectively
Functional assay of proBjSTP4
Prior to Agroinfiltration in B. juncea leaves, it was imperative to ensure absence of any bacterial expression of the reporter cassette proBjSTP4::GUS. In histochemical assay, the Agrobacterium cells harbouring proBjSTP4::GUS cassette did not show any GUS activity ruling out any bacterial expression, whereas pBI121 and pORER2 generated blue colour due to bacterial expression of the GUS gene (Supplementary Fig. 1). This is due to the presence of an intron in the GUS gene of pCAMBIA1305.1. The proBjSTP4::GUS construct was Agroinfiltrated into the N. benthamiana leaves and in histochemical assay after 24 h the treated leaves produced blue colour which demonstrated stand-alone promoter activity of the proBjSTP4.
Wound and hormone responsive activity of proBjSTP4 in N. benthamiana
In order to assay the activation of proBjSTP4 promoter in response to wounding, N. benthamiana leaves Agroinfiltrated with pCAMBIA (proBjSTP4::GUS), were wounded across the midrib after 24 h of Agroinfiltration. Histochemical assay of GUS expression in the wounded leaves showed higher intensity of GUS activity in the wounded leaves compared to the unwounded leaves. The results suggested activation of proBjSTP4 promoter activity by wound response (Fig. 6a). The intensity of blue colour also varied between samples collected at 2 and 6 h post wounding. In parallel, qRT-PCR based assessment of GUS transcript level across the samples reaffirmed the wound mediated activation of the proBjSTP4 promoter activity (Fig. 6b). The quantitative analysis revealed a twofold and sixfold increase of GUS transcript level in wounded samples at 2 h and 6 h time points, respectively, compared to the samples from unwounded plants.

Activation of proBjSTP4 promoter activity. Histochemical analysis of GUS-activity and qRT-PCR based quantification of GUS-transcripts following the treatment of wounding (a, b), MJ (c, d) and SA (e, f) in Agroinfiltrated leaves of N. benthaminana. A positive (pCAMBIA1305.1) and negative (Agrobacterium without vector) control were included for validation of the experimental procedure. Different lower-case alphabets indicate significant difference in mean, derived from three biological replicates with three technical replicates each. Significant difference in mean was evaluated by student’s t-test at P < 0.05 and represented as mean ± SE (n = 3)
For assessing effect of the defense related hormones viz. methyl jasmonate (MJ) and salicylic acid (SA) on proBjSTP4 promoter activity, the leaves of N. benthamiana prior Agroinfiltrated with pCAMBIA (proBjSTP4::GUS) were treated with either MJ or SA in independent experiments. In each experiment, the treated and mock-treated leaf samples were collected at 2 and 6 h after the treatment. Histochemical analysis of GUS expression demonstrated significant increase in GUS expression due to MJ (Fig. 6c, d) and SA treatments (Fig. 6e, f). Transcript analysis by qRT-PCR indicated significant and continuous increase in GUS transcript levels from 2 to 6 h due to treatment with MJ and SA. Without any treatment, basal transcript level of GUS driven by the constitutive promoter CaMV 35S was significantly higher compared to the basal transcript level of GUS driven by proBjSTP4 in mock-treated samples. As expected, there was no GUS activity observed in case of plants infiltrated with empty Agrobacterium cells.
Aphid-induced activity of proBjSTP4 in B. juncea
The promoter activity of proBjSTP4 in response to infestation by mustard aphids was assayed in Agroinfiltrated B. juncea leaves. Forty-to-fifty wingless aphids were released on detached leaves of B. juncea after 24 h of Agroinfiltration with the pCAMBIA (proBjSTP4::GUS) construct. Histochemical analysis for GUS was performed in a time course manner on the infested samples. The GUS assay revealed higher accumulation of blue colour in the infested leaves compared to the uninfested leaves (Fig. 7a). A time course experiment indicated increasing level of GUS activity with increased duration of aphid-feeding. qRT-PCR based quantification of GUS transcripts across the samples corroborated the results obtained in histochemical assay (Fig. 7b). Gradual activation in transcript level of GUS at 24 and 48 h following the aphid infestation unambiguously demonstrated aphid mediated activation of the proBjSTP4 promoter activity. Also, aphid-induced expression of GUS gene under proBjSTP4 promoter at 48 h of feeding was on par with the constitutive expression of GUS gene under the CaMV 35S promoter.

Aphid-responsive promoter activity of proBjSTP4 (a, b) and its deletion constructs (c, d). The promoter activity was assessed through histochemical analysis of GUS-activity and qRT-PCR based quantification of GUS-transcripts. Mean was derived from three biological replicates with three technical replicates each time and different lower-case letters indicate significant difference in mean, evaluated by student’s t-test at P < 0.05
Deletion analysis of proBjSTP4 for aphid-responsive promoter activity in B. juncea
Based on distribution of the putative cis-regulatory elements in proBjSTP4 region, four deletion constructs were designed and named as proSTP4DC1-4. For determining promoter activity, the deletion constructs were Agroinfiltrated in detached leaves of B. juncea, followed by the release of aphids. Histochemical analysis revealed that any decrease in the promoter activity in deletion construct proSTP4DC1 (with deletion of − 2017 to − 1618) was insignificant and the promoter activity remained on par with proBjSTP4 and CaMV35S::GUS. However, in deletion construct proSTP4DC2 (with deletion of − 2017 to − 1377) and deletion construct proSTP4DC3 (with deletion of − 2017 to − 918), reduced promoter activity due to the deletions were evident (Fig. 7c). In case of deletion construct proSTP4DC4 (with deletion of − 2017 to − 308), the promoter activity was further reduced significantly compared to the promoter activities in other deletion constructs and proBjSTP4. Transcript analysis by qRT-PCR empirically supported the reduced pattern of expression in the deletion constructs (Fig. 7d). No significant difference in GUS transcript level was observed between deletion construct 2 and deletion construct 3. Thus, the deletion analysis revealed indispensable association of the sequence elements up to -1617 upstream region in aphid mediated activation of proBjSTP4 promoter.
Discussion
Aphids create strong sink around the host-feeding sites and modulate the source-to-sink relationship of the host towards its own favour (Will et al. 2007). Thus, the majority of genes up-regulated during the early stages of aphid infestation were in the category of multi facilitator super family. These genes are essentially involved in facilitating the transport of carbon and nitrogen assimilates such as sugars, amino acids, etc. along with metabolites to the growing tissues of the plants (Lemoine et al. 2013; Tegeder 2014). Many of the up-regulated transcripts also represent early activated genes as a part of induced defense response against insects. In a susceptible plant-aphid interaction, expressions of defense response genes are short-lived and subjected to host-defense suppression by aphids (De Vos and Jander 2009; War et al. 2012). Thus, up-regulated transcripts which were related to early defense response were not taken into account in this study. Aphids secrete honeydews which attract moulds and bacterial growth on the leaf surface. Therefore, the genes known for early induction due to pathogen attack were also discarded in further narrowing down to the possible candidate genes (Zust and Agrawal 2016). Thus, the screening pipeline primarily considered host metabolic genes related to resource allocation phenomena as these genes have been hypothesized to be activated by aphids with sustained expression (Dorschner et al. 1987; Sandström et al. 2000; Rehill and Schultz 2003; Girousse et al. 2005; Goggin 2007). However, their tissue and temporal specificity in gene-expression might differ from one species to another (Sunilkumar et al. 2002; Zhu-Salzman et al. 2004; Heidel and Baldwin, 2004; Divol et al. 2005; Appel et al. 2014). Therefore, gene-expression of the in silico identified genes were validated for their temporal expression pattern upon aphid infestation in B. juncea (Fig. 3). In further comparison among the identified genes, three criteria: minimal basal level of expression, rapid induction in response to aphids and sustained expression have been adhered to identify the most appropriate candidate gene for the promoter isolation. Minimal basal level of expression in case of transgene expression by such promoter is likely to minimize metabolic burden on the host plant in absence of the stress.
The monosaccharide transporter gene STP4 showed gradual increase in gene-expression as the aphids rapidly multiplied on the B. juncea plants. The uptake of carbon assimilates from the source tissues (reservoir) and loading it towards the growing tissues (sink) is the key role of sugar transporter proteins (Truernit et al. 1996; Fotopoulos et al. 2003). Earlier, the STP4 gene was characterized during wounding and bacterial infection in Arabidopsis (Truernit et al. 1996). Subsequently, its expression was also analysed during aphid infestation in Arabidopsis which showed tenfold increase in transcript levels due to aphid feeding (Moran and Thompson 2001). Thus, multi-fold increase in STP4 expression during aphid colonization in B. juncea as shown in this study was consistent with the previous results. Additionally, transcriptional activation of STP4 gene along with the gene encoding invertase enzyme during growth and development in Arabidopsis strongly supported the involvement of STP4 in maintaining the source-to-sink relationship in plants (Sherson et al. 2003). In the present investigation, induced expression of STP4 gene was observed in leaves, stem, flowers and siliques of aphid infested B. juncea plants. Since leaves, flowers and siliques constitute the major feeding sites for the aphids, regulation of STP4 gene-expression was expected to be more evident in these tissues.
With the availability of genome sequences of various plant species, it has become advantageous to predict upstream promoter sequence of a gene (Kim et al. 2005). During the course of this investigation, genome sequence of B. juncea was not available. B. rapa is one of the progenitors in amphidiploid genome of B. juncea (Kaur et al. 2014). Therefore, the available genomic resources of B. rapa was utilized for identification of the upstream sequence of STP4. Based on the genomic organization of STP4 in B. rapa, a pair of primers were designed and the homologous upstream counterpart in B. juncea was cloned. Though, the genome of B. juncea is amphidiploid the pair of primers led to the amplification of a single amplicon possibly targeting the STP4 paralogue which was descended from B. rapa and conserved the sequence homology.
The cis-regulatory elements in the promoter sequence play pivotal role in interactions with the transcriptional factors for tissue and stress specific expression of a gene (Davuluri et al. 2003; Kaur et al. 2017). The putative cis-regulatory elements identified on the promoter proBjSTP4 were found to be mostly associated with gene-expression during growth and development, biotic stresses including pathogen infection, elicitor treatment, treatments by defense hormones, wounding and insect-infestation. For functional analysis of the putative promoter, Agroinfiltration mediated transient assay was used. Transient assay has been preferred because of its technical simplicity which led to rapid assay of a large number of constructs. Also, as it did not involve any plant transformation or gene integration event, confounding effect of the site of integration or positional effect on the promoter activity could be eliminated. In functional assays, proBjSTP4 promoter activity was found to be activated by wound, MJ and SA treatment in a gradual manner from 2 to 6 h of the treatments and peaked higher compared to the CaMV 35S. However, the basal promoter activity of proBjSTP4 in control plants was significantly lower compared to the CaMV 35S promoter. It empirically demonstrated the potential applicability of proBjSTP4 in reducing metabolic burden of constitutive transgene-expression and at the same time high inducibility of proBjSTP4 under aphid-stress in the host plant.
Conventionally, for elucidating the minimal functional region of a promoter, deletion analysis is carried out in promoter analysis (Sugaya and Uchimiya 1992; Ijaz et al. 2020). It is carried out to shorten the promoter length required for stand-alone function of the promoter and also for explicating specific role of the cis-regulatory elements of the promoter (Mahoney et al. 2016). In the present study, four deletion constructs of proBjSTP4 were generated in order to identify the indispensable regions of the promoter for aphid-responsive promoter-activity. Histochemical analysis of GUS activity in conjunction with qRT-PCR based transcript analysis suggested that, deletion of 5′ end (− 2017) up to − 1618 (deletion 1) did not result into any significant decrease in aphid-responsive promoter activity. Thus, the identified elements over this region viz. CCAAT box, Box-W1, 5′UTR Py-rich repeat, ACE motif, Box-4, circadian-regulated elements, LTRE and skn-1 presumably were not very significant to proBjSTP4 activity; though these elements were found to be crucial for the inducible activity of many promoters (Sun et al. 2003; Xu et al. 2006; Mikkelsen and Thomashow 2009; Gao et al. 2016; Kaur et al. 2017). However, in case of deletion construct proSTP4DC2, with further deletion of − 1616 to − 1377, the aphid-responsive promoter activity was significantly diminished indicating indispensability of this region (Fig. 5). A further deletion to − 916 bp in deletion construct proSTP4DC3 led to similar reduced promoter activity with no further significant decrease compared to proSTP4DC2. Thus, the deletion analysis suggested that, the putative cis-elements viz CAAT-box, Box-W1, 5′UTR Py-rich repeat, ACE motif, Box-4, circadian-regulated elements, LTRE, skn-1 along with some unknown cis-elements present in -1616 to -1377 region of the STP4 were associated with the aphid-responsive promoter activity of proBjSTP4. However, further study based on a greater number of deletions, tetramerization of putative motifs, use of synthetic promoters, etc. needs to be carried out for identifying the minimal promoter region associated with the aphid responsive activity (Ali and Kim 2019).
Conclusions
Developing varietal resistance against aphids is constrained due to unavailability of resistance genes within the crossable germplasms in case of many of the major crop species including B. juncea. Thus, transgenic strategy has been considered as a potential alternative for developing aphid resistance. Use of an inducible promoter with temporal specificity instead of constitutive promoters for transgene expression can potentially decrease metabolic burden on the host plants and pleiotropic effects. Gene expression pattern of STP4 conformed its aphid-inducible expression in the major aphid-feeding tissues of the oilseed crop, B. juncea. Cloning of the upstream intergenic region of STP4 and further deletion analysis demonstrated that the 1617 bp upstream region of STP4 can be potentially used as a promoter for driving aphid-responsive transgene-expression. With much lower basal activity, this promoter can potentially reduce undesirable constitutive expression of the transgene and can be activated by aphid-attack leading to high level gene-expression, comparable to constitutive gene-expression by CaMV 35S. The promoter shall remain useful for engineering inducible expression of plant immunity genes in reversing host-defense suppression by aphids in susceptible hosts.
References
Aguayo MF, Ampuero D, Mandujano P, Parada R, Munoz R, Gallart M, Altabella T, Cabrera R, Stange C, Handford M (2013) Sorbitol dehydrogenase is a cytosolic protein required for sorbitol metabolism in Arabidopsis thaliana. Plant Sci 205:63–75
Ali S, Kim W-C (2019) A fruitful decade using synthetic promoters in the improvement of transgenic plants. Front Plant Sci 10:1433
Aluri S, Büttner M (2007) Identification and functional expression of the Arabidopsis thaliana vacuolar glucose transporter 1 and its role in seed germination and flowering. Proc Natl Acad Sci USA 104:2537–2542
Appel HM, Fescemyer H, Ehlting J, Weston D, Rehrig E, Joshi T, Xu D, Bohlmann J, Schultz J (2014) Transcriptional responses of Arabidopsis thaliana to chewing and sucking insect herbivores. Front Plant Sci 5:565
Ascencio-Ibáñez JT, Sozzani R, Lee TJ, Chu TM, Wolfinger RD, Cella R, Hanley-Bowdoin L (2008) Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during gemini virus infection. Plant Physiol 148:436–454
Atri C, Kumar B, Kumar H, Kumar S, Sharma S, Banga SS (2012) Development and characterization of Brassica juncea-fruticulosa introgression lines exhibiting resistance to mustard aphid (Lipaphis erysimiKalt). BMC Genet 13:104
Bakhetia DRC (1987) Insect pests of rapeseed-mustard and their management. In: Rao MV, Sithanathan S (eds) Plant protection in field crops. Rajendranagar, PPAI, Irving, pp 249–259
Bandopadhyay L, Basu D, Sikdar SR (2013) Identification of genes involved in wild crucifer Rorippa indica resistance response on mustard aphid Lipaphis erysimi challenge. PLoS ONE 8:e73632
Barah P, Winge P, Kusnierczyk A, Tran DH, Bones AM (2013) Molecular signatures in Arabidopsis thaliana in response to insects’ attack and bacterial infection. PLoS ONE 8:e58987
Bhadoria NS, Jakhmola SS, Dhamdhere SV (1995) Relative susceptibility of mustard cultivars to Lipaphis erysimi in North West Madhya Pradesh (India). J Entomol Res 19:143–146
Bhatia V, Uniyal P, Bhattacharya R (2011) Aphid resistance in Brassica crops: challenges, biotechnological progress and emerging possibilities. Biotechnol Adv 29:879
Boughton AJ, Hoover K, Felton GW (2006) Impact of chemical elicitor applications on greenhouse tomato plants and population growth of the green peach aphid, Myzus persicae. Entomol Exp Appl 120:175–188
Brini F, Yamamoto A, Jlaiel L, Takeda S, Hobo T, Dinh HQ (2011) Pleiotropic effects of the wheat dehydrin DHN-5 on stress responses in Arabidopsis. Plant Cell Physiol 52:676–688
Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR (2012) Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335:207–211
Cho MH, Lim H, Shin DH, Jeon JS, Bhoo SH, Park YI, Hahn TR (2011) Role of the plastidic glucose translocator in the export of starch degradation products from the chloroplasts in Arabidopsis thaliana. New Phytol 190:101–112
Cipollini D, Purrington CB, Bergelson J (2003) Costs of induced responses in plants. BAS Appl Ecol 4:79–85
Couldridge C, Newbury HJ, Ford-Lloyd B, Bale J, Pritchard J (2007) Exploring plant responses to aphid feeding using a full Arabidopsis microarray reveals a small number of genes with significantly altered expression. Bull Entomol Res 97:523–532
Das A, Ghosh P, Das S (2018) Expression of Colocasia esculenta tuber agglutinin in Indian mustard provides resistance against Lipaphis erysimi and the expressed protein is non-allergenic. Plant Cell Rep 37:849
Davuluri RV, Sun H, Palaniswamy SK, Matthews N, Molina C, Kurtz M, Grotewold E (2003) AGRIS: Arabidopsis gene regulatory information server, an information resource of Arabidopsiscis-regulatory elements and transcription factors. BMC Bioinform 4:25
de Ilarduya MO, Xie QG, Kaloshian I (2003) Aphid-induced defence responses in Mi-1-mediated compatible and incompatible tomato interactions. Mol Plant Microbe Interact 16:699–708
De Vos M, Jander G (2009) Myzus persicae (green peach aphid) salivary components induce defense responses in Arabidopsis thaliana. Plant Cell Environ 32:1548–1560
De Vos M, Van-Oosten VR, Van-Poecke RMP, Van-Pelt JA, Pozo MJ, Mueller MJ (2005) Signal signature and transcriptome changes of Arabidopsis during pathogen and insect attack. Mol Plant Microbe Interact 18:923–937
Divol F, Vilaine F, Thibivilliers S, Amselem J, Palauqui JC, Kusiak C, Dinant S (2005) Systemic response to aphid infestation by Myzus persicae in the phloem of Apium graveolens. Plant Mol Biol 57:517–540
Donahue JL, Alford SR, Torabinejad J, Kerwin RE, Nourbakhsh A, Ray WK, Hernick M, Huang X, Lyons BM, Hein PP, Gillaspy GE (2010) The Arabidopsis thaliana Myo-Inositol 1-phosphate synthase1 gene is required for Myo-inositol synthesis and suppression of cell death. Plant Cell 22:888–890
Dorschner KW, Ryan JD, Johnson RC, Eikenbary RD (1987) Modification of host nitrogen levels by the greenbug (Homoptera: Aphididae): its role in resistance of winter wheat to aphids. Environ Entomol 16:1007–1011
DRMR (2015) Vision 2050. https://www.drmr.res.in/publication/DRMR%2520Rajasthan-(Vision2050)-Final.pdf
Dubey NK, Mishra DK, Idris A, Nigam D, Singh PK, Sawant SV (2018) Whitefly and aphid inducible promoters of Arabidopsis thaliana L. J Genet 97:109–119
Dündar E, Bush DR (2009) BAT1, a bidirectional amino acid transporter in Arabidopsis. Planta 229:1047–1056
Dutta I, Majumder P, Saha K, Ray P, Das S (2005) Constitutive and phloem specific expression of Allium sativum leaf agglutinin (ASAL) to engineer aphid (Lipaphis erysimi) resistance in transgenic Indian mustard (Brassica juncea). Plant Sci 169:996–1007
Ellis C, Karafyllidis I, Turner JG (2002) Constitutive activation of jasmonate signalling in an Arabidopsis mutant correlates with enhanced resistance to Erysiphe cichoracearum, Pseudomonas syringae, and Myzus persicae. Mol Plant Microbe Interact 15:1025–1030
Fotopoulos V, Gilbert MJ, Pittman JK, Marvier AC, Buchanan AJ, Sauer N (2003) The monosaccharide transporter gene, AtSTP4, and the cell-wall invertase, Atβfruct1, are induced in Arabidopsis during infection with the fungal biotroph Erysiphe cichoracearum. Plant Physiol 132:821–829
Gallardo K, Job C, Groot SP, Puype M, Demol H, Vandekerckhove J, Job D (2002) Importance of methionine biosynthesis for Arabidopsis seed germination and seedling growth. Physiol Plant 116:238–247
Gao LL, Anderson JP, Klingler JP, Nair RM, Edwards OR, Singh KB (2007) Involvement of the octadecanoid pathway in bluegreen aphid resistance in Medicago truncatula. Mol Plant Microbe Interact 20:82–93
Gao Y, Jia S, Wang C, Wang F, Wang F, Zhao K (2016) BjMYB1, a transcription factor implicated in plant defence through activating BjCHI1 chitinase expression by binding to a W-box-like element. J Exp Bot 67:4647–4658
Garcia AF, Dyszy F, Munte CE, Demarco R, Beltramini LM, Oliva G, Costa-Filho AJ, Araujo AP (2014) THI1, a protein involved in the biosynthesis of thiamin in Arabidopsis thaliana: structural analysis of THI1 (A140V) mutant. Biochim Biophys Acta 1844:1094–1103
Garrido E, Díaz MF, Bernal H, Nustez CE, Thaler J, Jander G, Poveda K (2017) Costs and trade-offs of resistance and tolerance to belowground herbivory in potato. PLoS ONE 12:e0169083
Girousse C, Moulia B, Silk W, Bonnemain JL (2005) Aphid infestation causes different changes in carbon and nitrogen allocation in alfalfa stems as well as different inhibitions of longitudinal and radial expansion. Plant Physiol 137:1474–1484
Goggin FL (2007) Plant-Aphid Interactions: molecular and ecological perspectives. Curr Opin Plant Biol 10:399–408
Hanada K, Sawada Y, Kuromori T, Klausnitzer R, Saito K, Toyoda T, Shinozaki K, Li WH, Hirai MY (2011) Functional compensation of primary and secondary metabolites by duplicate genes in Arabidopsis thaliana. Mol Biol Evol 28:377–382
Heidel AJ, Baldwin IT (2004) Microarray analysis of salicylic acid and jasmonic acid signalling in responses of Nicotiana attenuata to attack by insects from multiple feeding guilds. Plant Cell Environ 27:1362–1373
Hogenhout SA, Ammar ED, Whitfield AE, Redinbaugh MG (2008) Insect vector interactions with persistently transmitted viruses. Annu Rev Phytopathol 46:327–359
Hossain MA, Maiti MK, Basu A, Sen S, Ghosh AK, Sen SK (2006) Transgenic expression of onion leaf lectin gene in Indian mustard offers protection against aphid colonization. Crop Sci 46:2022–2032
Hu W, Wang Y, Bowers C, Ma H (2003) Isolation, sequence analysis, and expression studies of florally expressed cDNAs in Arabidopsis. Plant Mol Biol 53:545–563
Ijaz U, Pervaiz T, Ahmed T, Seemab R, Shahid M, Noman M, Nadeem M, Azeem F (2020) Plant cis-regulatory elements: methods of identification and applications. Asian J Agric Biol 8:207–222
Jaouannet M, Rodriguez PA, Thorpe P, Lenoir CJ, MacLeod R, Escudero-Martinez C et al (2014) Plant immunity in plant-aphid interactions. Front Plant Sci 5:663
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Johanson U, Karlsson M, Johansson I, Gustavsson S, Sjövall S, Fraysse L, Weig AR, Kjellbom P (2001) The complete set of genes encoding major intrinsic proteins in Arabidopsis provides a framework for a new nomenclature for major intrinsic proteins in plants. Plant Physiol 126:1358–1369
Jost R, Pharmawati M, Lapis-Gaza HR, Rossig C, Berkowitz O, Lambers H, Finnegan PM (2015) Differentiating phosphate-dependent and phosphate-independent systemic phosphate-starvation response networks in Arabidopsis thaliana through the application of phosphite. J Exp Bot 66:2501–2514
Kanrar S, Venkateswari J, Kirti P, Chopra VL (2002) Transgenic Indian mustard (Brassica juncea) with resistance to the mustard aphid (Lipaphis erysimi Kalt.). Plant Cell Rep 20:976–981
Kaur A, Pati PK, Pati AM, Nagpal AK (2017) In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PLoS ONE 12:e0184523
Kaur P, Banga S, Kumar N, Gupta S, Akhtar J, Banga S (2014) Polyphyletic origin of Brassica juncea with B. rapa and B. nigra (Brassicaceae) participating as cytoplasm donor parents in independent hybridization events. Am J Bot 101:1157–1166
Kim TH, Barrera LO, Qu C, Van Calcar S, Trinklein ND, Cooper SJ, Luna RM, Glass CK, Rosenfeld MG, Myers RM, Ren B (2005) Direct isolation and identification of promoters in the human genome. Genome Res 15:830–839
Kiyosue T, Abe H, Yamaguchi-Shinozaki K, Shinozaki K (1998) ERD6, a cDNA clone for an early dehydration-induced gene of Arabidopsis, encodes a putative sugar transporter. Biochim Biophys Acta 1370:187–191
Koramutla MK, Kaur A, Negi M, Venkatachalam P, Bhattacharya R (2014) Elicitation of jasmonate-mediated host defense in Brassica juncea (L.) attenuates population growth of mustard aphid Lipaphis erysimi (Kalt.). Planta 240:177–194
Krapp A, David LC, Chardin C, Girin T, Marmagne A, Leprince AS, Chaillou S, Ferrario-Méry S, Meyer C, Daniel-Vedele F (2014) Nitrate transport and signalling in Arabidopsis. J Exp Bot 65:789–798
Kumar S, Atri C, Sangha M, Banga S (2011) Screening of wild crucifers for resistance to mustard aphid, Lipaphis erysimi (Kaltenbach) and attempt at introgression of resistance gene(s) from Brassica-fruticulosa to Brassica juncea. Euphytica 179:461–470
Kuśnierczyk A, Winge P, Jørstad TS, Troczyńska J, Rossiter JT, Bones AM (2008) Towards global understanding of plant defence against aphids–timing and dynamics of early Arabidopsis defence responses to cabbage aphid (Brevicoryne brassicae) attack. Plant Cell Environ 31:1097–1115
Ladwig F, Stahl M, Ludewig U, Hirner AA, Hammes UZ, Stadler R, Harter K, Koch W (2012) Siliques are Red1 from Arabidopsis acts as a bidirectional amino acid transporter that is crucial for the amino acid homeostasis of siliques. Plant Physiol 158:1643–1655
Lee YH, Tegeder M (2004) Selective expression of a novel high-affinity transport system for acidic and neutral amino acids in the tapetum cells of Arabidopsis flowers. Plant J 40:60–74
Lemoine R, Camera SL, Atanassova R, Dédaldéchamp F, Allario T, Pourtau N, Bonnemain JJ, Laloi M, Coutos-Thévenot P, Maurousset L, Faucher M, Girousse C, Lemonnier P, Parrilla J, Durand M (2013) Source-to-sink transport of sugar and regulation by environmental factors. Front Plant Sci 4:272
Lezhneva L, Kiba T, Feria-Bourrellier AB, Lafouge F, Boutet-Mercey S, Zoufan P, Hitoshi Sakakibara H, Daniel-Vedele F, Krapp A (2014) The Arabidopsis nitrate transporter NRT2.5 plays a role in nitrate acquisition and remobilization in nitrogen-starved plants. Plant J 80:230–241
Li J, Shan L, Zhou JM, Tang X (2002) Overexpression of Pto induces a salicylate-independent cell death but inhibits necrotic lesions caused by salicylate-deficiency in tomato plants. Mol Plant Microbe Interact 15:654–661
Liu H, Zhang H, Yang Y, Li G, Yang Y, Wang X, Basnayake BM, Li D, Song F (2008) Functional analysis reveals pleiotropic effects of rice RING-H2 finger protein gene OsBIRF1 on regulation of growth and defence responses against abiotic and biotic stresses. Plant Mol Biol 68:17–30
Liu TY, Aung K, Tseng CY, Chang TY, Chen YS, Chiou TJ (2011) Vacuolar Ca2+/H+ transport activity is required for systemic phosphate homeostasis involving shoot-to-root signalling in Arabidopsis. Plant Physiol 156:1176–1189
Lothier J, Gaufichon L, Sormani R, Lemaître T, Azzopardi M, Morin H, Chardon F, Reisdorf-Cren M, Avice JC, Masclaux-Daubresse C (2011) The cytosolic glutamine synthetase GLN1;2 plays a role in the control of plant growth and ammonium homeostasis in Arabidopsis rosettes when nitrate supply is not limiting. J Exp Bot 62:1375–1390
Louvet R, Cavel E, Gutierrez L, Guénin S, Roger D, Gillet F, Guerineau F, Pelloux J (2006) Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana. Planta 224:782–791
Lu H, Yang P, Xu Y, Luo L, Zhu J, Cui N, Kang L, Cui F (2016) Performances of survival, feeding behavior, and gene expression in aphids reveal their different fitness to host alteration. Sci Rep 6:19344
Mahoney AK, Anderson EM, Bakker RA, Williams AF, Flood JJ, Sullivan KC, Pillitteri LJ (2016) Functional analysis of the Arabidopsis thaliana MUTE promoter reveals a regulatory region sufficient for stomatal-lineage expression. Planta 243:987–998
Mikkelsen MD, Thomashow MF (2009) A role for circadian evening elements in cold-regulated gene expression in Arabidopsis. Plant J 60:328–339
Miles PW (1999) Aphid saliva. Biol Rev 74:41–85
Moran PJ, Thompson GA (2001) Molecular responses to aphid feeding in Arabidopsis in relation to plant defence pathways. Plant Physiol 125:1074–1085
Moran PJ, Cheng YF, Cassell JL, Thompson GA (2002) Gene expression profiling of Arabidopsis thaliana in compatible plant-aphid interactions. Arch Insect Biochem Physiol 51:182–203
Park SJ, Huang Y, Ayoubi P (2006) Identification of expression profiles of sorghum genes in response to greenbug phloem feeding using cDNA subtraction and microarray analysis. Planta 223:932–947
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Pitino M, Hogenhout SA (2012) Aphid protein effectors promote aphid colonization in a plant species-specific manner. Mol Plant Microbe Interact 26:130–139
Ram C, Koramutla MK, Bhattacharya R (2017) Identification and comprehensive evaluation of reference genes for RT-qPCR analysis of host gene-expression in Brassica juncea-aphid interaction using microarray data. Plant Physiol Biochem 116:57–67
Reddy ASN, Takezawa D, Fromm H, Poovaiah BW (1993) Isolation and characterization of two cDNAs that encode for calmodulin-binding proteins from corn root tips. Plant Sci 94:109–117
Rehill BJ, Schultz JC (2003) Enhanced invertase activities in the galls of Hormaphisham amelidis. J Chem Ecol 29:2703–2720
Rodriguez PA, Escudero-Martinez C, Bos JIB (2017) An aphid effector targets trafficking protein VPS52 in a host-specific manner to promote virulence. Plant Physiol 173:1892–1903
Rohilla HR, Singh H, Kalra VK, Kharub SS (1987) Losses caused by mustard aphid, Lipaphis erysimi K. in different Brassica genotype. In: Proceedings of the international rapeseed congress, 11–14 May, 1987, Poland, p. 46
Ruta LL, Lin YF, Kissen R, Nicolau I, Neagoe AD, Ghenea S, Bones AM, Farcasanu IC (2017) Anchoring plant metallothioneins to the inner face of the plasma membrane of Saccharomyces cerevisiae cells leads to heavy metal accumulation. PLoS ONE 12:e0178393
Sadeghi A (2007) Expression of garlic leaf lectin under the control of the phloem specific promoter Asus1 from Arabidopsis thaliana protects tobacco plants against the tobacco aphids (Myzus nicotianae). Pest Manag Sci 63:1215–1223
Saha P, Majumder P, Dutta I, Ray T, Roy SC, Das S (2006) Transgenic rice expressing Allium sativum leaf lectin with enhanced resistance against sap sucking insect pests. Planta 223:1329–1343
Sandström J, Telang A, Moran NA (2000) Nutritional enhancement of host plants by aphids—a comparison of three aphid species on grasses. J Insect Physiol 46:33–40
Santiago JP, Tegeder M (2016) Connecting source with sink: the role of Arabidopsis AAP8 in phloem loading of amino acids. Plant Physiol 171:508–521
Sarkar P, Jana J, Chatterjee S, Sikdar SR (2016) Functional characterization of Rorippa indica defensin and its efficacy against Lipaphis erysimi. SpringerPlus 5:511
Schneider S, Schneidereit A, Konrad KR, Hajirezaei MR, Gramann M, Hedrich R, Sauer N (2006) Arabidopsis inositol transporter4 mediates high-affinity H+ symport of myoinositol across the plasma membrane. Plant Physiol 141:565–577
Schuler M, Rellán-Álvarez R, Fink-Straube C, Abadía J, Bauer P (2012) Nicotianamine functions in the phloem-based transport of iron to sink organs, in pollen development and pollen tube growth in Arabidopsis. Plant Cell 24:2380–2400
Schwartzberg EG, Tumlinson JH (2014) Aphid honeydew alters plant defence responses. Funct Ecol 28:386–394
Sharma M, Sahni R, Kansal R, Koundal KR (2004) Transformation of oilseed mustard B. juncea var. PJK with Snowdrop lectin gene. Indian J Biotechnol 3:97–102
Shekhawat K, Rathore SS, Premi OP, Kandpal BK, Chauhan JS (2012) Advances in agronomic management of Indian mustard (Brassica juncea (L.) Czernj. Cosson): an overview. Intern J Agron 2012:1–14
Sherson SM, Alford HL, Forbes SM, Wallace G, Smith SM (2003) Roles of cell-wall invertases and monosaccharide transporters in the growth and development of Arabidopsis. J Exp Bot 54:525–531
Singh CP, Sachan GC (1994) Assessment of yield losses in yellow sarson due to mustard aphid. Lipaphis erysimi (Kalt). J Oilseeds Res 11:179–184
Smith CM, Boyko EV (2007) The molecular bases of plant resistance and defense responses to aphid feeding: current status. Entomol Exp Appl 122:1–16
Sparkes IA, Runions J, Kearns A, Hawes C (2006) Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc 1:2019–2025
Su YH, Frommer WB, Ludewig U (2004) Molecular and functional characterization of a family of amino acid transporters from Arabidopsis. Plant Physiol 136:3104–3113
Sugaya H, Uchimiya S (1992) Deletion analysis of the 5'-upstream region of the Agrobacterium rhizogenes Ri plasmid rolC gene required for tissue-specific expression. Plant Physiol 99:464–467
Sullivan S, Ralet MC, Berger A, Diatloff E, Bischoff V, Gonneau M, Marion-Poll A, North HM (2011) CESA5 is required for the synthesis of cellulose with a role in structuring the adherent mucilage of Arabidopsis Seeds. Plant Physiol 156:1725–1739
Sun C, Palmqvist S, Olsson H, Borén M, Ahlandsberg S, Jansson C (2003) A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell 15:2076–2092
Sunilkumar G, Mohr L, Lopata-Finch E, Emani C, Rathore KS (2002) Developmental and tissue-specific expression of CaMV 35S promoter in cotton as revealed by GFP. Plant Mol Biol 50:463–474
Tegeder M (2014) Transporters involved in source to sink partitioning of amino acids and ureides: opportunities for crop improvement. J Exp Bot 65:1865–1878
Tegeder M, Ward JM (2012) Molecular evolution of plant AAP and LHT amino acid transporters. Front Plant Sci 3:21
Tian S, Wang X, Li P, Wang H, Ji H, Xie J, Qiu Q, Shen D, Dong H (2016) Plant aquaporin AtPIP1;4 links apoplastic H2O2 induction to disease immunity pathways. Plant Physiol 171:1635–1650
Truernit E, Schmid J, Epple P, Illig J, Sauer N (1996) The sink-specific and stress-regulated Arabidopsis STP4 gene: enhanced expression of a gene encoding a monosaccharide transporter by wounding, elicitors, and pathogen challenge. Plant Cell 8:2169–2182
Voelckel C, Weisser W, Baldwin I (2004) An analysis of plant–aphid interactions by different microarray hybridization strategies. Mol Ecol 13:3187–3195
Walters D, Heil M (2007) Costs and trade-offs associated with induced resistance. Physiol Mol Plant Pathol 71:3–17
War AR, Paulraj MG, Ahmad T, Buhroo AA, Hussain B, Ignacimuthu S, Sharma HC (2012) Mechanisms of plant defense against insect herbivores. Plant Signal Behav 7:1306–1320
Wei PC, Zhang XQ, Zhao P, Wang XC (2011) Regulation of stomatal opening by the guard cell expansin AtEXPA1. Plant Signal Behav 6:740–742
Will T, Tjallingii WF, Thönnessen A, van Bel AJ (2007) Molecular sabotage of plant defense by aphid saliva. Proc Natl Acad Sci USA 104:10536–10541
Xu X, Chen C, Fan B, Chen Z (2006) Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 18:1310–1326
Xu H, Wang X, Zhao H, Liu F (2008) An intensive understanding of vacuum infiltration transformation of pakchoi (Brassica rapa ssp. chinensis). Plant Cell Rep 27:1369–1376
Zhu XF, Shi YZ, Lei GJ, Fry SC, Zhang BC, Zhou YH, Braam J, Jiang T, Xu XY, Mao CZ, Pan YJ, Yang JL, Wu P, Zheng SJ (2012) XTH31, Encoding an in vitro xeh/xet-active enzyme, regulates aluminum sensitivity by modulating in vivo xet action, cell wall xyloglucan content, and aluminum binding capacity in Arabidopsis. Plant Cell 24:4731–4747
Zhu-Salzman K, Salzman RA, Ahn JE, Koiwa H (2004) Transcriptional regulation of sorghum defense determinants against a phloem-feeding aphid. Plant Physiol 134:420–431
Zust T, Agrawal AA (2016) Mechanisms and evolution of plant resistance to aphids. Nat Plants 2:15206
Acknowledgements
This work was supported by Post Graduate School, IARI through a Ph.D fellowship to CR and DST-SERB grant (EMR/2017/003463) to RB.
Supporting information
Supplementary Fig. 1 Assay of pCAMBIA-proBjSTP4 construct for bacterial expression. The Agrobacterium cells harbouring the pCAMBIA-proBjSTP4 construct was incubated at 37 °C for 1 h along with GUS assay buffer. Agrobacterium strain harbouring pBI121 (35S::GUS) and pORER2 (35S::GUS) were taken as positive control. Supplementary Table 1 Details of gene-specific primers used for qRT-PCR analysis. Supplementary Table 2 List of primers used for amplification of proBjSTP4 and deletion fragments. Supplementary Table 3 Characteristics features of putative cis-regulatory elements in proBjSTP4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
About this article

{kind=link}
Cite this article
Ram, C., Annamalai, M., Koramutla, M.K. et al. Characterization of STP4 promoter in Indian mustard Brassica juncea for use as an aphid responsive promoter. Biotechnol Lett 42, 2013–2033 (2020). https://doi.org/10.1007/s10529-020-02961-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02961-7