
Abstract
Objectives
To biotransform rutin into isoquercitrin.
Results
A α-l-rhamnosidase from Bifidobacterium breve was produced by using Escherichia coli BL21 for biotransformation of rutin to isoquercitrin. The enzyme was purified by Ni2+-NTA chromatography to yield a soluble protein with a specific activity of 56 U protein mg−1. The maximum enzyme activities were at pH 6.5, 55 °C, 20 mM rutin, and 1.2 U enzyme ml−1. Under optimal conditions, the half-life of the enzyme was 96 h. The K m and V max values were 2.2 mM, 56.4 μmol mg−1 min−1 and 2.1 mM, 57.5 μmol mg−1 min−1 using pNP-Rha and rutin as substrates, respectively. The kinetic behavior indicated that the recombinant α-l-rhamnosidase has good catalytic performance for producing isoquercitrin. 20 mM rutin was biotransformed into 18.25 and 19.87 mM isoquercitrin after 60 and 240 min.
Conclusion
The specific biotransformation of rutin to isoquercitrin using recombinant α-l-rhamnosidase from B. breve is a feasible method for use in industrial processes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rutin, a quercetin rutinoside, exists in medicinal herbs, fruits, vegetables as well as many plant-derived foods and beverages (Awatsuhara et al. 2010; Chua 2013). It exhibits various activities including antioxidative, anti-hyperglycemic and neuroprotective effects (Chua 2013; You et al. 2010). The flavonoid, isoquercitrin (quercetin-3-β-d-glucoside) is a de-rhamnosylation product of rutin; the structural difference between them is only a rhamnose. Although there is a structural similarity to rutin, isoquercitrin, and quercetin, there are some differences in physical, chemical, and biological properties (Valentova et al. 2014). In comparison with rutin and quercetin, isoquercitrin is better absorbed, suggesting that conjugation with glucose enhances its absorption in small intestine (Arts et al. 2004). Isoquercitrin has a more potent antiproliferative effect than quercetin or rutin (Paulke et al. 2012; You et al. 2010). Isoquercitrin, due to its specific property as an antioxidant, has an important function in the anti-aging and anti-allergic effects with a high pharmacological activity than that of rutin (Paulke et al. 2012; Valentova et al. 2014). Isoquercitrin is also better than rutin in protecting injured cells by scavenging free radical (Paulke et al. 2012).
Although isoquercitrin has many important and special biological activities, its content in nature is low. Therefore, finding an efficient way to produce it is of significance. Isoquercitrin is produced by chemical and enzymatic methods. Experiments have shown that the aglycon of rutin is easily deglycosylated under mild acidic hydrolysis conditions at appropriate temperatures, but its secondary glucoside (isoquercitrin) was difficult to obtain. So, the preparation of isoquercitrin by enzymatic hydrolysis of rutin is preferable to the acidic hydrolysis.
α-l-Rhamnosidase (EC3.2.1.40) is a glycosyl hydrolase that cleaves the terminal α-l-rhamnose from a large number of natural glycosides, e.g., naringin, rutin, hesperidin, and terpenyl glycoside (Beekwilder et al. 2009). The enzyme is also used industrially for debittering citrus fruits by releasing rhamnose from naringin and employed in enhancing the grape juice aroma by hydrolysis of terpenyl glycosides (Spagna et al. 2000). Based on the current research, α-l-rhamnosidase activity allows the production of expensive flavonoid glycosides, isoquercitrin, in an easy and cheap bioprocess starting from rutin. Thus, the biotransformation of mono-glycosylated isoquercitrin from rutin by the enzymatic hydrolysis method seems to be a good alternative for obtaining compounds with enhanced functional properties.
In this study, a gene coding for α-l-rhamnosidase from the probiotic bacteria, Bifidobacterium breve ATCC 15700, was cloned and expressed in Escherichia coli BL21. The biochemical properties of the recombinant α-l-rhamnosidase were determined, and isoquercitrin was produced from rutin using the enzyme. The enzyme has a high efficiency of transforming rutin to isoquercitrin. It can provide a new method for the production of isoquercitrin in the industry.
Materials and methods
Bacterial strains, plasmid and culture conditions
Bifidobacterium breve ATCC 15700 was grown anaerobically at 37 °C in MRS medium containing 0.05 % (w/v) l-cysteine. E. coli strains DH5α and BL21 (DE3) were employed for gene cloning and expression, respectively, with the strains grown in lysogeny broth (LB) medium. The plasmids pGEM-T easy and pET28a (+) were used as cloning and expression vectors respectively.
Cloning of α-l-rhamnosidase gene
For cloning of gene sequence encoding a putative α-l-rhamnosidase, B. breve was boiled in water for 10 min. After centrifugation, the DNA-containing supernatant was diluted and used as a template for amplification. The primers were designed according to the DNA sequence of the putative α-l-rhamnosidase from B. breve (GenBank accession number, CP006715.1). Forward (5′-CGC GGA TCC ATG CTC GAT GAT AGT GAA CTG C-3′) and reverse primers (5′-CCC AAG CTT TAC GGA CCT CAT TTC AAT CAC-3′) were added the BamHI and HindIII restriction sites (underlined), respectively. To obtain C-terminal His-tag sequence, the stop codon of the gene was removed. The amplified PCR product with restriction sites was cloned into pGEM-T easy vector and then transformed into E. coli DH5α. After identifying and sequencing, the sequence was subcloned into the pET28a (+) plasmid with the same restriction enzymes and then transformed into E. coli BL21.
Expression and purification of α-l-rhamnosidase
Escherichia coli cells containing the pET28a (+)/α-l-rhamnosidase gene from B. breve were grown in LB medium containing 100 μg kanamycin/ml at 37 °C. IPTG was added at 1 mM when the OD600 of the culture reached to 0.5. Induction was carried out for another 3 h before the cells were harvested. The induced cells were subsequently harvested and disrupted via sonication in 50 mM citrate buffer (pH 5.5). The recombinant protein was purified by Ni2+ column chromatography. The protein was eluted with 50 mM phosphate buffer/300 mM NaCl/250 mM imidazole, pH 7. The purified protein was analyzed by 10 % SDS-PAGE. The protein concentration was determined using Pierce BCA Protein Assay Kit (Thermo Scientific) following the manufacturer’s protocol.
Assays for enzyme activity
α-l-Rhamnosidase activity was assayed using 10 mM p-nitrophenyl-α-l-rhamnopyranoside (pNP-Rha) as substrate at various temperatures (20–80 °C) and pH values (3–9). After incubation, the reaction was stopped by adding an equal volume of 200 mM Na2CO3. The released p-nitrophenol (pNP) was determined at 405 nm. One unit of the enzyme activity (U) was defined as the amount of enzyme releasing 1 μmol pNP from the substrate per min (De Winter et al. 2013). All assays were performed in triplicate.
Enzyme kinetics
The kinetic constants K m and V max of the enzyme were calculated by fitting the activity data at different substrate concentrations ranging from 0.2 to 4 mM to a linear regression on Lineweaver–Burk double-reciprocal plots. All assays were performed in triplicate.
Analysis of biotransformed products by TLC and HPLC
Rutin was dissolved in methanol and incubated in 50 mM sodium phosphate buffer (pH 6.5) containing 20 mM rutin, 5 % (v/v) DMSO, and 1.2 U enzyme ml−1 at 55 °C. After incubation, the reaction was stopped by heating at 100 °C for 30 min. The biotransformed products were subsequently extracted and freeze-dried, and then dissolved in methanol, filtered through 0.45 μm syringe filter, and used for TLC and HPLC analysis. TLC analysis was conducted on silica gel G plates with ethyl acetate/methanol/water (30:5:4, by vol). The product was visualized by heating the plate at 110 °C for 5 min after spraying with 10 % (v/v) H2SO4 in ethanol. HPLC was performed with an XTerra C18 column (250 mm × 4.6 mm, 5 μm), at 30 °C with a Prep LC controller (LCD), and a dual absorbance detector. The injection volume of standards and samples was 10 μl. The mobile phase at 0.5 ml/min was 0.1 % (v/v) trifluoroacetic acid (TFA) in water, pH 2.5, and acetonitrile with the following gradient: 0–20 min, linear gradient from 20 to 40 %; 20–30 min, 40 %. The eluate was monitored at 350 nm. All HPLC analyses were performed in triplicate.
Results and discussion
Gene cloning and recombinant α-l-rhamnosidase purification
A full-length gene encoding the α-l-rhamnosidase of B. Breve ATCC 15700 was 2328 bp and encoded a 775-amino acid protein. The α-l-rhamnosidase gene has the same sequence as that reported in GenBank (accession number, CP006715.1). The amplified gene was cloned into pET28a (+) vector at the BamHI and HindIII restriction sites. A recombinant protein, α-l-rhamnosidase was successfully expressed using the T7 expression system in E. Coli. The expressed enzyme was purified from the crude extract by Ni2+-NTA agarose affinity column to yield a soluble protein with a specific activity of 56 U protein mg−1. The molecular mass of the purified enzyme determined by SDS-PAGE was approx. 87 kDa (Fig. 1), which is consistent with the calculated value based on the amino acids. The molecular mass of the B. Breve α-l-rhamnosidase is similar to the molecular mass that has been reported for the extracellular α-l-rhamnosidases from fungi, including Aspergillus aculeatus (87 kDa) (Manzanares et al. 2003), Aspergillus niger (85 kDa) (Manzanares et al. 1997), and Aspergillus kawachii (90 kDa) (Koseki et al. 2008).

SDS-PAGE analysis of recombinant α-l-rhamnosidase from B. breve. M protein molecular weight marker. Lane 1, 2 purified enzyme by Ni2+-NTA agarose affinity column
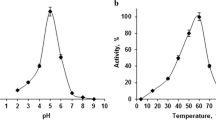
Effects of temperature and pH on the activity of α-l-rhamnosidase from B. breve
The effects of pH and temperature on recombinant enzyme from E. coli were investigated by pNP assay. The activities of α-l-rhamnosidase increased up to 55 °C, and then decreased dramatically (Fig. 2a). The enzyme had maximal activity at 55 °C towards pNP-Rha as substrate, which is similar to those of Aspergillus Rhases (You et al. 2010). The optimum pH for α-l-rhamnosidase was 5.5–7, with 90.5, 95.8, 97.8, and 91.2 % of maximum activity at pH 5.5, 6, 6.5, and 7, respectively (Fig. 2b). The pH range for α-l-rhamnosidase activity was neutral to slightly acid with greater than 90 % of the activity at pH values of 5.5–7. Maximal activity at acid pH values was similar to the enzymes isolated from diverse microbial systems described in the literature when using pNP-Rha as substrate. The optimum pH of rutin hydrolysis by α-l-rhamnosidase from the Bacillus sp. GL1 (Hashimoto et al. 1999) was 5.5. An alkaline pH is preferable only for a few Rhases, such as maximal activity of A. luteoalbus Rhase at pH 9.5. A neutral to slightly acid condition is preferable for applications of Rhase in the food and pharmaceutical industries. Therefore, it has a potential application for the production of the corresponding glycosyl derivatives.

Effects of temperature and pH on the activity of recombinant α-l-rhamnosidase determined using pNP-Rha as a substrate. Data represent the means of three experiments and error bars represent standard deviation. a Effects of temperature on the activity of recombinant α-l-rhamnosidase. Enzyme activity was assayed in sodium phosphate buffer (pH 6.5) for 2 h from 20 to 80 °C. b Effects of pH on the activity of recombinant α-l-rhamnosidase. Enzyme activity was assayed after incubation at 55 °C for 2 h in following buffers (50 mM): glycine/HCl buffer (pH 3), citric acid/sodium citrate buffer (pH 4–5), sodium phosphate buffer (pH 6–7), and Tris/HCl buffer (pH8–9). In both cases, 100 % relative activity = 1.2 U enzyme ml−1
The thermal stability of recombinant α-l-rhamnosidase was assayed at 50, 60, 70, and 80 °C for 90 min (Fig. 3). It was stable at 60 °C for more than 16 h and retained more than 90 % activity after 20 h. However, the stability of the enzyme decreased drastically at 70 and 80 °C with half-life duration of 25 and 10 min, respectively, and the half-life of the enzyme was 96 h under the optimal conditions (55 °C, pH 6.5) (data not shown). The optimal temperature for the activity of the α-l-rhamnosidase from various species of Aspergillus is from 40 to 65 °C. Enzymes from A. niger and A. aculeatus retained 85 and 87 % of their original activity following heating for 4 h at 50 and 55 °C, respectively (Manzanares et al. 1997, 2003). The enzyme from A. nidulans is extremely unstable at 60 °C and loses 90 % of its activity for 1 h (Koseki et al. 2008). Most of the characterized Rhases are slightly lower thermal stabilities (Beekwilder et al. 2009).

Thermal stability of recombinant α-l-rhamnosidase determined using pNP-Rha as a substrate. Data represent the means of three experiments and error bars represent standard deviation. Thermal stability profiles of the enzyme were assayed at 50 °C (filled cycle), 60 °C (open triangle), 70 °C (open cycle), and 80 °C (filled triangle) in sodium phosphate buffer (pH 6.5)
Kinetic parameters of the recombinant α-l-rhamnosidase from B. breve
The Michaelis–Menten kinetics K m , V max , and k cat values of the B. breve α-l-rhamnosidase were determined using pNP-Rha and rutin as substrates respectively. K m , V max , and k cat values were much closer for both substrates. The K m , V max , and k cat values were found to be 2.2 mM, 56.4 μmol mg−1 min−1, and 2.5 s−1 using pNP-Rha as substrate, and the values were 2.01 mM, 57.2 μmol mg−1 min−1, and 2.5 s−1 using rutin as substrate, respectively. Kinetics and mechanism of α-l-rhamnosidase catalyzed reactions have rarely been studied. Lower K m values ranging from 1.5 to 2.9 mM have been described for Aspergillus Rhases (Manzanares et al. 1997). The kinetic property of an enzyme is affected by many factors such as temperature, pH, solvent, metal ions or cofactors, and the presence of surfactants. Metal ions, Na+, K+, Ca2+, Mg2+, Mn2+, and Fe3+, had no effect on enzyme activity which was similar to the α-l-rhamnosidase from Acrostalagmus luteo albus (Rojas et al. 2011). The kinetic behavior here has clearly indicated that the recombinant α-l-rhamnosidase has good catalytic performance and environmental compatibility. However, the kinetics and catalytic mechanisms of α-l-rhamnosidases from B. breve are required to be further studied.
Substrate specificity of the recombinant α-l-rhamnosidase from B. breve
To identify the specificity of the α-l-rhamnosidase, rutin (α-1,6 linkage), hesperidin (α-1,6 linkage), and naringin (α-1,2-linkage) were tested. As shown in Table 1, α-l-rhamnosidase released α-l-rhamnose from three substrates. It is thus able to hydrolyze both α-1,2 and α-1,6 glucosidic linkages. However, the enzyme was more active towards rutin. Substrate specificity of the α-l-rhamnosidase from different species was tested with various α-l-rhamnose-containing natural compounds (flavonoids, terpenoids, and saponins). The substrate specificity of α-l-rhamnosidases towards α-1,2, α-1,3, α-1,4, and α-1,6 glucosidic linkages are found in different species. The majority of α-l-rhamnosidases are active on α-1,2 glucosidic linkages whereas the number of α-l-rhamnosidases active on α-1,6 linkages comes second (Yadav et al. 2010). In Aspergillus, only an α-l-rhamnosidase from A. niger, and two α-l-rhamnosidases from A. aculeatus were active towards all types of linkage (Manzanares et al. 1997, 2003). The α-l-rhamnosidase from B. breve displays broad substrate specificity which may allow its application for derhamnosylation of natural products.
Production of isoquercitrin from rutin by recombinant B. breve α-l-rhamnosidase
The optimal α-l-rhamnosidase concentration for producing isoquercitrin was investigated by varying the enzyme concentration from 0.1 to 2 U enzyme ml−1. Isoquercitrin production increased with increasing concentrations of enzyme until reaching a plateau at 1.2 U enzyme ml−1 (data not shown). Isoquercitrin production from rutin was assessed by varying the rutin concentration from 1 to 25 mM with a constant 1.2 U enzyme ml−1 (data not shown). Increasing the substrate concentration up to 20 mM led to an increase of isoquercitrin with Biotransformation yield decreased above 20 mM. Thus, the improved reaction conditions for producing isoquercitrin from rutin by B. breve α-l-rhamnosidase were: 0.5 % (v/v) DMSO, pH 6.5, 55 °C, 1.2 U enzyme ml−1, and 20 mM rutin.
Under these conditions, the time-course experiment was performed and the biotransformed crude product of rutin by α-l-rhamnosidase was identified by TLC with an authentic sample. A new spot of isoquercitrin was observed on TLC after 10 min. The isoquercitrin spot increased obviously after 60 min, and it reached to a high level at 90 min (data not shown). Finally, initial and final amounts of substrates and products were also confirmed quantitatively by HPLC analysis (Fig. 4), it was almost the same as the results of the TLC analysis. The conversion rates were calculated under the standard assay conditions. As shown in Fig. 5, 20 mM rutin was transformed into 18.25 mM isoquercitrin after 60 min which is a molar conversion of 91 %. 20 mM isoquercitrin was transformed into 19.87 mM after 240 min with a corresponding molar conversion yield of 99 %. This preparatory reaction clearly demonstrated that the recombinant enzyme is able to biotransform rutin selectively to isoquercitrin, quantitatively within 240 min without any trace of the unwanted quercetin. The productivity of isoquercitrin in the present study using B. breve α-l-rhamnosidase was obviously high. To sum up, the enzyme was a suitable biocatalyst for the efficient and selective biotransformation of rutin. However, the scale-up production will be strongly dependent on specific technological conditions. Therefore, the process need to be further improved for scale-up production.

HPLC analysis of the products obtained from the biotransformation of rutin with B. breve α-l-rhamnosidase. The reaction was performed in sodium phosphate buffer (pH 6.5) containing 20 mM rutin, 5 % (v/v) DMSO, and 1.2 U enzyme ml−1 at 55 °C. a standard. b 0 min. c 30 min, d 90 min

Isoquercitrin production from rutin by recombinant α-l-rhamnosidase from B. breve under the optimal conditions. Isoquercitrin (filled triangle) production from rutin (filled circle). The reaction, final volume 10 ml, was performed in sodium phosphate buffer (pH 6.5) containing 20 mM rutin, 5 % (v/v) DMSO, and 200 µl enzyme preparation (giving a final activity of 1.2 U ml−1) at 55 °C for 90 min. Data represent the means of three experiments and error bars represent standard deviation
Biotransformation pathway of isoquercitrin by α-l-rhamnosidase
There are various flavonoid glycosides with rhamnosyl moieties, such as isoquercitrin, rutin, hesperidin, and naringin. For rutin, α-l-rhamnose is bound to the β-d-glucosidic residue of isoqueratrin via an α-1,6 linkage. Based on the above results, it could be inferred that the rhamnosidase selectively hydrolyzes the α-1,6 linkage of rutin to give isoquercitrin. Similar substrate specificity has been described for the α-l-rhamnosidases from fungi (De Winter et al. 2013; Manzanares et al. 1997) and yeasts (Yanai and Sato 2000). The biotransformation pathway of isoquercitrin by α-l-rhamnosidase from B. breve is shown in Fig. 6. Some enzymes can selectively remove rhamnosyl moieties. Two α-l-rhamnosidases from Asp. aculeatus have been used for preparing isoquercitrin, but possessed potential pathogenicity. A potential candidate rhamnosidase from Lactobacillus plantarum was used for biotransformation rutin, but its transformation efficacy was only 13.2 % after 24 h (Beekwilder et al. 2009). α-l-Rhamnosidase and hesperidinase from A. niger and A. aculeatus, respectively, were used for the successful removal of rhamnose from rutin; however, they had high β-d-glucosidase activities that decreased the specificity of the rutin biotransformation (Manzanares et al. 1997, 2003). In our results, the B. breve strain is a food-grade microorganism showing an outstanding transformation rate of rutin to isoquercitrin of 97 % in 2 h. Our recombinant α-l-rhamnosidase was expressed as completely void of α-d-glucosidase activity which predestines its use for highly selective derhamnosylation and the production of such compounds as isoquercitrin.

Biotransformation pathway of rutin to isoquercitrin by α-l-rhamnosidase from B. breve
In conclusion, although α-l-rhamnosidases have several biotechnological applications, only a limited have been characterized. Therefore, the development of new commercially viable processes is desirable for the production of this enzyme by fermentation. The study demonstrates that only isoquercitrin is produced from the rutin by the recombinant α-l-rhamnosidase from B. breve. The recombinant α-l-rhamnosidase completely converted rutin to isoquercitrin with high productivity and specificity. Considering the improved bioavailability of isoquercitrin compared to rutin and quercetin, the α-l-rhamnosidase is considered potentially useful tool for the practical preparation of isoquercitrin from rutin.
References
Arts IC, Sesink AL, Faassen-Peters M, Hollman PC (2004) The type of sugar moiety is a major determinant of the small intestinal uptake and subsequent biliary excretion of dietary quercetin glycosides. Br J Nutr 91:841–847
Awatsuhara R, Harada K, Maeda T, Nomura T, Nagao K (2010) Antioxidative activity of the buckwheat polyphenol rutin in combination with ovalbumin. Mol Med Rep 3:121–125
Beekwilder J, Marcozzi D, Vecchi S, de Vos R, Janssen P, Francke C, van Hylckama Vlieg J, Hall RD (2009) Characterization of rhamnosidases from Lactobacillus plantarum and Lactobacillus acidophilus. Appl Environ Microbiol 75:3447–3454
Chua LS (2013) A review on plant-based rutin extraction methods and its pharmacological activities. J Ethnopharmacol 150:805–817
De Winter K, Simcikova D, Schalck B, Weignerova L, Pelantova H, Soetaert W, Desmet T, Kren V (2013) Chemoenzymatic synthesis of alpha-l-rhamnosides using recombinant alpha-l-rhamnosidase from Aspergillus terreus. Bioresour Technol 147:640–644
Hashimoto W, Nankai H, Sato N, Kawai S, Murata K (1999) Characterization of alpha-l-rhamnosidase of Bacillus sp. GL1 responsible for the complete depolymerization of gellan. Arch Biochem Biophys 368:56–60
Koseki T, Mese Y, Nishibori N, Masaki K, Fujii T, Handa T, Yamane Y, Shiono Y, Murayama T, Iefuji H (2008) Characterization of an alpha-l-rhamnosidase from Aspergillus kawachii and its gene. Appl Microbiol Biotechnol 80:1007–1013
Manzanares P, de Graaff LH, Visser J (1997) Purification and characterization of an alpha-l-rhamnosidase from Aspergillus niger. FEMS Microbiol Lett 157:279–283
Manzanares P, Orejas M, Gil JV, De Graaff LH, Visser J, Ramon D (2003) Construction of a genetically modified wine yeast strain expressing the Aspergillus aculeatus rhaA gene, encoding an alpha-l-rhamnosidase of enological interest. Appl Environ Microbiol 69:7558–7562
Paulke A, Eckert GP, Schubert-Zsilavecz M, Wurglics M (2012) Isoquercitrin provides better bioavailability than quercetin: comparison of quercetin metabolites in body tissue and brain sections after six days administration of isoquercitrin and quercetin. Pharmazie 67:991–996
Rojas NL, Voget CE, Hours RA, Cavalitto SF (2011) Purification and characterization of a novel alkaline alpha-l-rhamnosidase produced by Acrostalagmus luteo albus. J Ind Microbiol Biotechnol 38:1515–1522
Spagna G, Barbagallo RN, Martino A, Pifferi PG (2000) A simple method for purifying glycosidases: alpha-l-rhamnopyranosidase from Aspergillus niger to increase the aroma of Moscato wine. Enz Microb Technol 27:522–530
Valentova K, Vrba J, Bancirova M, Ulrichova J, Kren V (2014) Isoquercitrin: pharmacology, toxicology, and metabolism. Food Chem Toxicol 68:267–282
Yadav V, Yadav PK, Yadav S, Yadav KDS (2010) α-l-Rhamnosidase: a review. Process Biochem 45:1226–1235
Yanai T, Sato M (2000) Purification and characterization of an alpha-l-rhamnosidase from Pichia angusta X349. Biosci Biotechnol Biochem 64:2179–2185
You HJ, Ahn HJ, Ji GE (2010) Transformation of rutin to antiproliferative quercetin-3-glucoside by Aspergillus niger. J Agric Food Chem 58:10886–10892
Acknowledgments
The study was supported by the Natural Science Foundation of Hunan Province (11JJ6009), the Scientific Research Fund of Hunan Provincial Education Department (11C0329) and the National Training Programs of Innovation and Entrepreneurship for Undergraduates (201411342004).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, R., Zhang, BL., Xie, T. et al. Biotransformation of rutin to isoquercitrin using recombinant α-l-rhamnosidase from Bifidobacterium breve . Biotechnol Lett 37, 1257–1264 (2015). https://doi.org/10.1007/s10529-015-1792-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-015-1792-6