
Abstract
In recent years, the impact of methylation modifications on Dickkopf-1 (DKK1) in relation to ankylosing spondylitis (AS) has remained elusive. Our objective was to investigate the potential link between DKK1 methylation patterns and transcript levels and AS susceptibility. DNA methylation level of DKK1 was measured in 82 AS and 82 healthy controls (HCs) using targeted bisulfite sequencing. In addition, the transcript level of DKK1 in peripheral blood mononuclear cells from 35 AS patients and 35 HCs was detected using real-time quantitative transcription-polymerase chain reaction. Our study showed that the DKK1 was significantly hypomethylated in AS patients (P < 0.001). The Receiver operating characteristic curve (ROC) showed that DKK1 methylation may be a potential biomarker. The results showed that the difference in DKK1 transcript levels between AS and HCs was not statistically significant. Further analysis showed that DKK1 methylation levels were positively correlated with age and negatively correlated with C-reactive protein levels, neutrophil/lymphocyte ratio (NLR) and platelet/lymphocyte ratio (PLR). The methylation level of DKK1 in PBMC of AS patients was significantly lower than that of HCs, and DKK1 methylation may be associated with susceptibility to AS. In addition, DNA methylation levels of DKK1 were negatively correlated with the level of inflammation in AS patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory autoimmune disease that typically involves the spine and sacroiliac joints (Taurog et al. 2016). In addition to inflammatory back pain, spinal ankylosis and bone fusion caused by systemic inflammation are typical features of AS (Everson et al. 2021). It is estimated that the global prevalence of AS is about 0.1–1.4%, and the prevalence varies among different races (Dean et al. 2014). The prevalence of AS in China is about 0.29%, and the prevalence of AS in males is about 2.8 times higher than that in females, and the peak incidence of AS is between 20 and 30 years old (Zhao et al. 2020). Considering that the affected population is mainly young men, the disability rate for patients with ankylosing spondylitis is 4% after 5 years and 50% after 45 years, causing not only incapacitation but also a huge burden of the disease on society (Dean et al. 2014). Although the pathogenesis of AS is currently unclear, studies have suggested that genetic susceptibility, environmental factors, and infectious factors may trigger the occurrence of AS and be involved in disease progression, with genetics as the most significant determinant among these causative factors (Hanson and Brown 2017; Brown et al. 1997; Zhang et al. 2021). Human leukocyte antigen B27 (HLA-B27), the predominant genetic locus, has been shown to be highly associated with AS, but it only explains 20.1% of the heritability (Chen et al. 2017). Currently, 116 genetic loci have been identified, accounting for 28% of the genetic variation in AS (Brown and Wordsworth 2017), but they also do not fully explain the heritability of AS. In addition, the expression of gene functions is regulated by epigenetic modifications (Yang et al. 2021). Therefore, epigenetics should be further applied to explain the risk of AS and expand the horizon of AS etiology.
In contrast to genetics based on changes in gene expression level due to gene sequence changes, epigenetics is the reversible heritable modification of gene expression without altering the DNA sequence, mainly including DNA methylation, histone modifications, chromatin remodeling, and regulation of non-coding RNAs (Peixoto et al. 2020). DNA methylation is a key epigenetic procedure for the regulation of mammalian gene expression and refers to the process of chemical modification of the cytosine 5 carbon position of genomic CpG dinucleotides by covalent bonding of methyl groups in the presence of DNA methyltransferases (Moore et al. 2013). DNA methylation is implicated in a diverse range of biological processes, including regulation of gene expression, chromatin structure, X chromosome inactivation, genomic imprinting, and DNA stability (Meng et al. 2015). The relationship between promoter methylation and the transcript silencing of genes has been investigated in the pathogenesis of various autoimmune diseases (Sun et al. 2016). For the past few years, DNA methylation has been continuously reported to be involved in the development of AS, implying a potential role in the genetic mechanisms. A study involving genome-wide methylation revealed an association between aberrant methylation levels at over 1900 loci in 1214 genes and AS (Hao et al. 2017a). Previous research has suggested that early changes in gene methylation patterns can be a marker for predicting clinical outcomes (Mishra et al. 2022). Additionally, altered methylation levels of ERAP1, IRF8, and TRAF5 genes in AS patients have been reported (Wang et al. 2021; Chen et al. 2019; Xu et al. 2021). DNA methylation as an important and stable genetic mechanism of epigenetic memory has currently become a new horizon in AS. Nevertheless, the pathogenic mechanisms of DNA methylation in AS deserve deeper investigation and research.
Dickkopf-1 (DKK1) is a secretory glycoprotein consisting of two cysteine-rich structural domains and a signal peptide and has been proven to be one of the inhibitors of the classical Wnt/beta-catenin pathway (Zhu et al. 2021). The Wnt signaling pathway plays an essential role in regulating cell growth and differentiation, while mediating bone formation and bone resorption (Dincel and JøRGENSEN 2023). Moreover, studies have shown that DKK1 is involved in the erosion and fusion of the sacroiliac joint, which can be used as a biomarker of AS radiological injury (Uderhardt et al. 2010; Wu et al. 2018). Several studies have shown that DKK1 plays a vital role in the pathogenesis of autoimmune diseases, such as ankylosing spondylitis, rheumatoid arthritis, systemic sclerosis, and psoriatic arthritis (Liu et al. 2019a; Henderson et al. 2021; Chung et al. 2021; Liao et al. 2018). Some research has revealed that DKK1 is involved in the occurrence and progression of AS (Liao et al. 2018; Di et al. 2018; Zhang et al. 2016). It was found that the rs1569198 polymorphism and haplotype (T-G) of DKK1 gene were significantly associated with susceptibility to AS in females (Liu et al. 2019b). However, the association between the DNA methylation profile of DKK1 and AS is currently unclear. Taken together, case–control research was performed to explore the DNA methylation and transcription of DKK1 in AS patients and healthy controls (HCs).
Materials and Methods
Study Populations
This study was a case–control study. In the first step, a total of 164 subjects were included in the DNA methylation detection assays, including 82 cases of AS and 82 HCs who were matched in sex and age. In the second step, an additional 35 patients with AS and 35 HCs were enrolled for the mRNA transcription detection assays. The patients were from the Rheumatology and Immunology Department in the First Affiliated Hospital of Anhui Medical University. The AS diagnosis was judged by rheumatologists based on the 1984 modified New York criteria (Linden et al. 1984). We excluded patients with other autoimmune diseases, rheumatic diseases, and other chronic diseases. All healthy controls came from the physical examination center, without rheumatism history or family history. This research was granted approval by the Ethics Committee of Anhui Medical University (Approval Number: 20200260). All subjects were informed about the protocol and signed an informed consent form.
Collection of Data and Blood Sample
The patients filled out an epidemiological questionnaire that included basic demographic characteristics and clinical characteristics of AS patients. Specific clinical features included HLA-B27, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), neutrophil/lymphocyte ratio (NLR), platelet/lymphocyte ratio (PLR), lymphocyte count, neutrophil count, platelet count, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), and Ankylosing Spondylitis Disease Activity Score (ASDAS). We collected 5 ml of venous whole blood from each research subject.
DNA Methylation Detection
The CpG islands of DKK1 gene were screened according to three criteria: (1) length of not less than 200 bp; (2) guanine and cytosine content of not less than 50%; and (3) the ratio of observed CpG dinucleotides to expected is not less than 0.6. A total of two CpG islands containing twenty-nine CpG sites for DKK1. Prime3 software (http://primer3.ut.ee/) was used to design the primers for multiplex polymerase chain reaction PCR and then the primers were optimized to confirm the appropriate primers composition and concentration. Details of the primers are given in Table S1. DNA samples were treated with bisulfite to convert genomic DNA unmethylated cytosine into uracil following the manufacturer’s protocols. Multiplex PCR of target gene sequences was conducted with optimized primers. PCR amplicons were separated and purified by agarose electrophoresis and QIAquick Gel Extraction kit (QIAGEN, Germany). The corresponding libraries were sequenced in 2 × 150 bp double-end sequencing mode on the Illumina Hiseq platform. The methylation levels of DKK1 gene and its CpG islands and CpG sites were determined by the proportion of methylated cytosine to total cytosine.
Quantitative Real-Time PCR (qRT-PCR)
The peripheral blood mononuclear cells (PBMCs) were extracted from peripheral venous blood by Ficoll density gradient centrifugation. Total RNA was isolated and purified from PBMCs using TRIzol reagent (Invitrogen, USA). Total RNA was reversely transcribed into complementary DNA (cDNA) by PrimeScriptTM RT reagent Kit with gDNA Eraser. Real-time quantitative PCR of cDNA was performed with SYBR Premix Ex Taq II (Takara Bio, Japan). The internal reference gene of DKK1 was β-actin, and the relative expression level of mRNA was obtained by the 2−ΔΔCt formula. A duplicate well was added to each sample to increase accuracy. The primer sequences of DKK1 and β-actin are shown in Table S1.
Statistical Analysis
Quantitative variables were determined by the Shapiro–Wilk test to determine whether the variables conform to a normal distribution. Variables that conformed to a normal distribution were expressed as mean ± standard deviation (SD), otherwise, they were expressed as median and inter-quartile range. Qualitative variables were expressed as frequencies (%). Student's T-test was used for comparisons between groups of normally distributed variables, and the Mann–Whitney U test was used for comparisons of non-normally distributed variables. Categorical variables were tested with chi-square test. Univariate logistic regression was used to analyze the association of individual CpG sites with AS by odds ratios (OR) and 95% confidence intervals (CIs) and corresponding forest plots. The false discovery rate (FDR) was controlled via Benjamini–Hochberg (BH) method, and the corrected P value (PFDR) of less than 0.05 was considered statistically significant. The Spearman correlation method and the Mann–Whitney U test were used to assess the association of methylation of DKK1 with clinical indicators of continuous and categorical variables. The receiver operating characteristic (ROC) curve analysis method was used to determine the diagnostic power of potentially meaningful indicators for AS. Data analysis was operated in SPSS 23.0 software (SPSS, Chicago, IL, USA). Use MedCalc (Version 20.218) to create ROC graphs and other statistical graphs were produced in GraphPad Prism 7.00 (GraphPad Software Inc., CA, USA) and R software (StataCorp, College Station, TX). Statistical tests were two-sided and P < 0.05 was considered statistically significant.
Results
Characteristics of Study Subjects
The study ultimately included 82 AS patients (63 males and 19 females) and 82 healthy controls (63 males and 19 females) for methylation testing based on the inclusion and exclusion criteria described previously. No statistical differences were found between the cases and controls in either age (Z = − 0.191, P = 0.849) or gender (χ2 = 0.000, P = 1.000). Transcript level detection was performed in 35 AS patients (28 males and 7 females) and 35 HCs (26 males and 9 females). Similarly, there were no statistical differences between the two groups in terms of age (Z = − 1.123, P = 0.261) or gender (χ2 = 0.324, P = 0.569). Other relevant demographic and clinical characteristics are detailed in Table 1.
DNA Methylation Level of DKK1
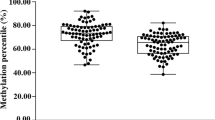
In our study, 29 sites from 2 CpG islands of DKK1 gene were tested for methylation levels. As shown in Fig. 1, The methylation levels of DKK1 gene as well as its two CpG islands were significantly lower compared to the HCs (DKK1, Z = − 5.255, P < 0.001, DKK1-1, Z = − 4.808, P < 0.001, DKK1-2, Z = − 5.173, P < 0.001). In addition, methylation levels at 12 sites in DKK1-1 island and 17 sites in DKK1-2 island were significantly lower than in controls (Fig. 2). The methylation levels of DKK1 gene and its 2 CpG islands were further evaluated for diagnostic value in AS patients with ROC curve analysis. The AUC of ROC curve for DKK1 was 0.738 (95% CI = 0.661, 0.814, P < 0.001), with a sensitivity of 0.622 and a specificity of 0.780 for the optimal cutoff point of 0.040 (Fig. 1, Table S2). The diagnostic values of DKK1-1 and DKK1-2 showed the AUC of 0.717 (95% CI = 0.639, 0.796, P < 0.001) and 0.734 (95% CI = 0.656, 0.812, P < 0.001), respectively. The diagnostic value of methylation of DKK1 and its two CpG islands is more pronounced in males compared to females and details are shown in Table S2. After stratifying for gender, we found significant differences in DKK1 methylation levels between the AS case group and the HC group in both males and females (Fig. 3, Figure S1), and we also calculated OR using univariate logistic regression, which showed similar results to those of the Mann–Whitney U test, with some discrepancies in the results only in the female strata, which were due to the smaller sample sizes of the females as well as to the different principles of the two statistical methods. In addition, we corrected for age and found that age had no effect on the results (Figure S1).

Comparison of DKK1 methylation between AS and HC groups, and receiver operating characteristic curves. A Methylation level of DKK1 gene; B Methylation level of DKK1-1 CpG island; C Methylation level of DKK1-2 CpG island; D The ROC curves of the methylation levels of DKK1, DKK1-1, and DKK1-2 islands. AS ankylosing spondylitis, HC healthy controls, ROC receiver operating characteristic, rs Spearman's rank correlation coefficient

Forest plot of methylation levels at 29 CpG sites of DKK1 in AS patients versus HC groups. A Methylation of 12 CpG sites in the CpG-1 island of DKK1; B Methylation of 17 CpG sites in the CpG-2 island of DKK1. The squares and horizontal lines correspond to the odds ratio (OR) and 95% confidence interval (CI), respectively. Statistical methods: logistic regression. PFDR P value after the correction by Benjamini–Hochberg method. P values with “*” were considered statistically significant differences

Comparison of DKK1 methylation between AS and HC groups by gender stratification. Statistical methods: Mann–Whitney U test
Transcription Level of DKK1
To further explore the transcript levels of DKK1, 35 cases of AS were collected in the second phase with 35 healthy controls. The results showed that the difference in DKK1 transcript levels between the case and control groups was not statistically significant (Z = − 1.280, P = 0.200, Fig. 4). As seen in Fig. 4, the distribution of the data was dispersed, with large individual differences in DKK1 transcript expression in peripheral venous blood. Further, a sex-stratified analysis of DKK1 transcript levels revealed no statistical significance either in males or females (Figure S2).

Comparison of DKK1 transcription levels between AS group and HC group. Statistical methods: Mann–Whitney U test
Correlation Analysis of Methylation Levels and Clinical Characteristics
For quantitative variables, we used Spearman correlation analysis to explore the association of age, ESR, CRP, NLR, PLR, BMI, disease duration, BASFI, BASDAI, and ASDAS with methylation levels. Further, subcomponents of NLR and PLR (lymphocyte count, neutrophil count, and platelet count) were included in the analysis. As shown in Table 2, age was positively correlated with DKK1 and its two islands (DKK1, rs = 0.299, P = 0.006, DKK1−1, rs = 0.311, P = 0.004, DKK1-2, rs = 0.251, P = 0.023). DKK1 DNA methylation levels were negatively correlated with CRP, NLR, and PLR (CRP, rs = − 0.227, P = 0.042; NLR, rs = − 0.306, P = 0.012; PLR, rs = − 0.414, P < 0.001), and similarly, DNA methylation levels in two CpG islands of DKK1 were negatively correlated with CRP, NLR, and PLR (Table 2). Next, we observed that lymphocyte counts, a subcomponent of NLR and PLR, were significantly positively correlated with the methylation levels of DKK1 and its two CpG islands (DKK1, rs = 0.307, P = 0.012, DKK1- 1, rs = 0.331, P = 0.006, DKK1-2, rs = 0.271, P = 0.026), and platelet counts were negatively correlated with DKK1 methylation levels (rs = − 0.235, P = 0.040), while none of the remaining associations were statistically significant, thus the significant negative correlation between NLR and PLR was driven primarily by lymphocytes and secondarily by platelets. More visual correlation plots are shown in Figure S3.
For categorical variables, subgroup analyses were performed separately to investigate the association of gender, HLA-B27, BASFI, BASDAI, and ASDAS with DKK1 methylation levels. We did not find the association between gender, HLA-B27, BASFI, BASDAI, and ASDAS and DKK1 methylation levels (Table S3).
Discussion
AS is an immune-mediated inflammatory disease with a distinct genetic predisposition. Although the involvement of genetics in the onset and development of AS is currently well established, the available genetic evidence can only explain a minority of the pathogenesis, and therefore requires an in-depth exploration of epigenetic aspects. In this study, we investigated the DNA methylation level and corresponding transcriptional level of DKK1 in AS patients from the epigenetic perspective in an attempt to elucidate the pathogenesis of AS and provide new insights for its early diagnosis. Our study showed that the DKK1 gene and its two CpG islands and its 29 loci were significantly hypomethylated in AS patients. Methylation levels of the DKK1 gene and its two CpG islands exhibited significant diagnostic value in AS by ROC curve analysis. Further analysis showed that DKK1 methylation levels were positively correlated with age and negatively correlated with C-reactive protein levels, NLR, and PLR.
In the present study, aberrant hypomethylation of the DKK1 gene was identified for the first time in whole blood cells of AS patients, and all 29 loci of the DKK1 gene showed significant hypomethylation. DNA methylation affects a variety of biological processes, and recent findings suggest that DNA methylation plays a key role in AS by altering gene expression profiles (Wu et al. 2023; Ni et al. 2022). Furthermore, one study found that demethylation of the DKK1 promoter restored DKK1 expression in patients with multiple myeloma (Kocemba et al. 2012). In the present study, there was a weak trend of elevated DKK1 mRNA levels in AS patients, but the association between methylation levels and expression levels is still unknown and needs to be further explored. At present, research on DKK1 protein levels in AS mainly focuses on patient serum, and pooled analysis indicated that there were no significant changes in serum DKK1 concentrations between AS patients and healthy controls (Fang et al. 2023), while studies in recent years have found downregulation of DKK1 levels at hip or femoral head tissues in AS (Di et al. 2018; Daoussis et al. 2022; Zou et al. 2016), suggesting that the pathogenesis of DKK1 at different parts may be different in AS patients. Further ROC curve analysis indicated that methylation of either the entire DKK1 gene or each CpG island can be considered a potential biomarker for the diagnosis of AS. Besides, the diagnostic value of methylation of DKK1 and its two CpG islands is more prominent in males compared to females. Nevertheless, further research is needed for more difficult-to-differentiate diseases, such as osteoarthritis.
AS is a progressive disease with inflammatory abnormalities in the early stages, followed by calcification of the joint attachment points and ankylosis in the later stages (Xie et al. 2016). One of the key functions of DNA methylation is to maintain T-cell regulation (Mazzone et al. 2019). It was shown that inhibition of the DNA demethylases Tet1 and Tet2 induced hypermethylation of the DKK1 promoter, thereby activating the Wnt signaling pathway to enhance the immunomodulatory function of periodontal stem cells and promote T-cell apoptosis (Yu et al. 2019). Furthermore, it has also been proposed that activation of Wnt signaling can block effector T-cell differentiation (Muralidharan et al. 2011). We found no difference in Tet1 between AS and HCs and Tet2 was upregulated in AS by analyzing the transcript levels of Tet1 and Tet2 in the Gene Expression Omnibus database of AS (Gracey et al. 2016), which may induce demethylation of genes (Figure S4). In our study, DKK1 methylation levels were negatively correlated with NLR and PLR, and this association was mainly driven by lymphocytes due to the significant positive correlation of DKK1 methylation levels with lymphocytes. It is well known that T cells are the main component of lymphocytes. It is hypothesized that a similar mechanistic pathway may be involved in AS, where DKK1 hypomethylation may inhibit T-cell apoptosis or interfere with effector T-cell differentiation through the Wnt signaling pathway and thus disrupt immune system homeostasis. Second, DKK1 is a typical inhibitor of the Wnt signaling pathway, which is a key regulatory pathway that stimulates the proliferation and maturation of osteoblasts, and has been shown to have an influential role in the formation of new bone (Haynes et al. 2012). The Wnt signaling pathway has been extensively studied in AS, and current evidence suggests that DKK1 expression is down-regulated in tissues from AS patients and that the expression of genes related to the Wnt signaling pathway, Wnt3a, Wnt4, Wnt5a, Wnt7b, and Wnt10b, is upregulated, which may lead to new bone formation (Li et al. 2018; Sheng et al. 2022; Zeng et al. 2022). Meanwhile, the Wnt pathway may also serve as a bridge between inflammation and abnormal ossification in AS (Li et al. 2018).
In the correlation analysis, a significant positive correlation was found between age and DKK1 methylation levels in both AS patients and HCs, suggesting that this association may be prevalent in the population. A growing body of literature supports this view (Vaidya et al. 2023; Faul et al. 2023). Emerging research has found that levels of methylation may be related to inflammation and disease activity in AS (Wu et al. 2023), and we have conducted similar explorations. It was found that DKK1 DNA methylation levels were negatively correlated with CRP, NLR, and PLR. In a meta-analysis, DNA methylation plays an important role in establishing or balancing CRP levels (Ligthart et al. 2016). In the present study, DKK1 methylation levels were decreased and DKK1 methylation was found to be negatively correlated with CRP, suggesting that DKK1 methylation may be partly responsible for the higher levels of inflammation in AS compared to the general population and that the potential mechanisms need to be explored in more depth. NLR and PLR are inflammatory indicators calculated from complete blood count tests and are usually utilized as auxiliary diagnostic biomarkers in autoimmune diseases (Hao et al. 2017b). Previous studies have found that NLR and PLR can also be used in AS to reflect systemic inflammation and indicate disease severity (Al-Osami et al. 2020; Liang et al. 2021; Xu et al. 2020). In our study, DKK1 methylation was found to be associated with the inflammation level in AS. In addition, inflammatory intensity-dependent expression of osteoinducible Wnt proteins plays a key role between inflammation and ectopic new bone formation in AS (Li et al. 2018). In conclusion, aberrant DNA methylation of the DKK1 gene may be involved in the pathogenesis of AS.
There are several limitations to the current study. First, the transcriptional expression levels of DKK1 are low in the blood, making it difficult to obtain stable results. Since AS is a systemic inflammatory autoimmune disease, the measurement of whole venous blood samples can reflect the condition of the organism, and AS tissues are extremely difficult to obtain, so whole blood samples were chosen for this study. Currently, the association between DKK1 methylation levels and transcriptional levels remains elusive. Second, methylation status varies in different cells or tissues, and multiple cell subtypes and tissues should be covered in subsequent studies. Third, despite the potential diagnostic value of DKK1 and its two CpG islands in AS and HCs, the differentiation of AS from other immune arthropathies requires further study. Finally, protein levels of DKK1 were not measured and the association between methylation and transcript levels and protein levels is unknown and needs further study.
Conclusion
Taken together, our findings identified significant hypomethylation of DKK1 that may be involved in the pathogenesis of AS. We suggest that abnormal DKK1 methylation levels are a potential biomarker to help diagnose AS, and that DKK1 methylation levels may influence the level of inflammation in patients. The above findings need to be further validated in large-scale research.
Abbreviations
- AS:
-
Ankylosing spondylitis
- AUC:
-
Area under curve
- BASDAI:
-
Bath Ankylosing Spondylitis Disease Activity Index
- BASFI:
-
Bath Ankylosing Spondylitis Functional Index
- CI:
-
Confidence interval
- CpG:
-
Cytosine-guanine dinucleotide
- CRP:
-
C-reactive protein
- NLR:
-
Neutrophil/lymphocyte ratio
- PLR:
-
Platelet/lymphocyte ratio
- DKK1:
-
Dickkopf-1
- HLA:
-
Human leukocyte antigen
- OR:
-
Odds ratio
- PBMCs:
-
Peripheral blood mononuclear cells
- PCR:
-
Polymerase chain reaction
- ROC:
-
Receiver operating characteristic
References
Al-Osami MH, Awadh NI, Khalid KB et al (2020) Neutrophil/lymphocyte and platelet/lymphocyte ratios as potential markers of disease activity in patients with ankylosing spondylitis: a case-control study. Adv Rheumatol 60(1):13
Brown MA, Wordsworth BP (2017) Genetics in ankylosing spondylitis-current state of the art and translation into clinical outcomes. Best Pract Res Clin Rheumatol 31(6):763–776
Brown MA, Kennedy LG, Macgregor AJ et al (1997) Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 40(10):1823–1828
Chen B, Li J, He C et al (2017) Role of HLA-B27 in the pathogenesis of ankylosing spondylitis (Review). Mol Med Rep 15(4):1943–1951
Chen M, Wu M, Hu X et al (2019) Ankylosing spondylitis is associated with aberrant DNA methylation of IFN regulatory factor 8 gene promoter region. Clin Rheumatol 38(8):2161–2169
Chung Y, Li ZC, Sun XL et al (2021) Elevated serum dickkopf-1 is a biomarker for bone erosion in patients with psoriatic arthritis. Chin Med J (engl) 134(21):2583–2588
Daoussis D, Kanellou A, Panagiotopoulos E et al (2022) DKK-1 is underexpressed in mesenchymal stem cells from patients with ankylosing spondylitis and further downregulated by IL-17. Int J Mol Sci 23(12):6660
Dean LE, Jones GT, Macdonald AG et al (2014) Global prevalence of ankylosing spondylitis. Rheumatology (oxford) 53(4):650–657
Di G, Kong L, Zhao Q et al (2018) MicroRNA-146a knockdown suppresses the progression of ankylosing spondylitis by targeting dickkopf 1. Biomed Pharmacother Biomed Pharmacothe 97:1243–1249
Dincel AS, JøRGENSEN NR (2023) New emerging biomarkers for bone disease: sclerostin and dickkopf-1 (DKK1) [J]. Calcif Tissue Int 112(2):243–257
Everson TM, Niedzwiecki MM, Toth D et al (2021) Metal biomarker mixtures and blood pressure in the United States: cross-sectional findings from the 1999–2006 national health and nutrition examination survey (NHANES). Environ Health Glob Access Sci Sour 20(1):15
Fang X, Chen C, Wang ZX et al (2023) Serum DKK-1 level in ankylosing spondylitis: insights from meta-analysis and mendelian randomization. Front Immunol 14:1193357
Faul JD, Kim JK, Levine ME et al (2023) Epigenetic-based age acceleration in a representative sample of older Americans: associations with aging-related morbidity and mortality. Proc Natl Acad Sci USA 120(9):e2215840120
Gracey E, Yao Y, Green B et al (2016) Sexual dimorphism in the Th17 signature of ankylosing spondylitis. Arthr Rheumatol 68(3):679–689
Hanson A, Brown MA (2017) Genetics and the causes of ankylosing spondylitis. Rheum Dis Clin North Am 43(3):401–414
Hao J, Liu Y, Xu J et al (2017a) Genome-wide DNA methylation profile analysis identifies differentially methylated loci associated with ankylosis spondylitis. Arthritis Res Ther 19(1):177
Hao X, Li D, Wu D et al (2017b) The Relationship between hematological indices and autoimmune rheumatic diseases (ARDs), a meta-analysis. Sci Rep 7(1):10833
Haynes KR, Pettit AR, Duan R et al (2012) Excessive bone formation in a mouse model of ankylosing spondylitis is associated with decreases in Wnt pathway inhibitors. Arthritis Res Ther 14(6):R253
Henderson J, Pryzborski S, Stratton R et al (2021) Wnt antagonist DKK-1 levels in systemic sclerosis are lower in skin but not in blood and are regulated by microRNA33a-3p. Exp Dermatol 30(1):162–168
Kocemba KA, Groen RW, van Andel H et al (2012) Transcriptional silencing of the Wnt-antagonist DKK1 by promoter methylation is associated with enhanced Wnt signaling in advanced multiple myeloma. PLoS ONE 7(2):e30359
Li X, Wang J, Zhan Z et al (2018) Inflammation intensity-dependent expression of osteoinductive Wnt proteins is critical for ectopic new bone formation in ankylosing spondylitis. Arthr Rheumatol 70(7):1056–1070
Liang T, Chen J, Xu G et al (2021) Platelet-to-lymphocyte ratio as an independent factor was associated with the severity of ankylosing spondylitis. Front Immunol 12:760214
Liao HT, Lin YF, Tsai CY et al (2018) Bone morphogenetic proteins and Dickkopf-1 in ankylosing spondylitis. Scand J Rheumatol 47(1):56–61
Ligthart S, Marzi C, Aslibekyan S et al (2016) DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol 17(1):255
Liu L, Zuo Y, Xu Y et al (2019a) MiR-613 inhibits proliferation and invasion and induces apoptosis of rheumatoid arthritis synovial fibroblasts by direct down-regulation of DKK1. Cell Mol Biol Lett 24:8
Liu R, Zhang X, Jiang G et al (2019b) Gene-gene interaction and association of Wnt/β-catenin signalling pathway gene polymorphisms with ankylosing spondylitis susceptibility in the Chinese Han population. Autoimmunity 52(7–8):281–288
Mazzone R, Zwergel C, Artico M et al (2019) The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics 11(1):34
Meng H, Cao Y, Qin J et al (2015) DNA methylation, its mediators and genome integrity. Int J Biol Sci 11(5):604–617
Mishra N, Aden K, Blase JI et al (2022) Longitudinal multi-omics analysis identifies early blood-based predictors of anti-TNF therapy response in inflammatory bowel disease. Genome Medicine 14(1):110
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38(1):23–38
Muralidharan S, Hanley PJ, Liu E et al (2011) Activation of Wnt signaling arrests effector differentiation in human peripheral and cord blood-derived T lymphocytes. J Immunol 187(10):5221–5232
Ni M, Chen Y, Sun X et al (2022) DNA methylation and transcriptional profiles of IRF5 gene in ankylosing spondylitis: a case-control study. Int Immunopharmacol 110:109033
Peixoto P, Cartron PF, Serandour AA et al (2020) From 1957 to nowadays: a brief history of epigenetics. Int J Mol Sci 21(20):7571
Sheng W, Jiang H, Yuan H et al (2022) miR-148a-3p facilitates osteogenic differentiation of fibroblasts in ankylosing spondylitis by activating the Wnt pathway and targeting DKK1. Exp Ther Med 23(5):365
Sun B, Hu L, Luo ZY et al (2016) DNA methylation perspectives in the pathogenesis of autoimmune diseases. Clin Immunol 164:21–27
Taurog JD, Chhabra A, Colbert RA (2016) Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med 374(26):2563–2574
Uderhardt S, Diarra D, Katzenbeisser J et al (2010) Blockade of dickkopf (DKK)-1 induces fusion of sacroiliac joints. Ann Rheum Dis 69(3):592–597
Vaidya H, Jeong HS, Keith K et al (2023) DNA methylation entropy as a measure of stem cell replication and aging. Genome Biol 24(1):27
van der Linden S, Valkenburg HA, Cats A (1984) Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum 27(4):361–368
Wang X, Bin W, Zhou M et al (2021) Systemic inflammation mediates the association of heavy metal exposures with liver injury: a study in general Chinese urban adults. J Hazard Mater 419:126497
Wu M, Chen M, Ma Y et al (2018) Dickkopf-1 in ankylosing spondylitis: review and meta-analysis clinica chimica acta. , Int J Clin Chem 481:177–183
Wu Y, Chen Y, Sun X et al (2023) DNA methylation and transcriptome signatures of the PDCD1 gene in ankylosing spondylitis. Genes Immun 24(1):46–51
Xie W, Zhou L, Li S et al (2016) Wnt/β-catenin signaling plays a key role in the development of spondyloarthritis. Ann N Y Acad Sci 1364(1):25–31
Xu S, Ma Y, Wu M et al (2020) Neutrophil lymphocyte ratio in patients with ankylosing spondylitis: a systematic review and meta-analysis. Mod Rheumatol 30(1):141–148
Xu S, Gao X, Ma Y et al (2021) Association of methylation level and transcript level in TRAF5 gene with ankylosing spondylitis: a case-control study. Genes Immun 22(2):101–107
Yang H, Chen Y, Xu W et al (2021) Epigenetics of ankylosing spondylitis: recent developments. Int J Rheum Dis 24(4):487–493
Yu T, Liu D, Zhang T et al (2019) Inhibition of Tet1- and Tet2-mediated DNA demethylation promotes immunomodulation of periodontal ligament stem cells. Cell Death Dis 10(10):780
Zeng Y, He R, Liu Y et al (2022) HDAC1 regulates inflammation and osteogenic differentiation of ankylosing spondylitis fibroblasts through the Wnt-smad signaling pathway. J Orthop Surg Res 17(1):343
Zhang L, Ouyang H, Xie Z et al (2016) Serum DKK-1 level in the development of ankylosing spondylitis and rheumatic arthritis: a meta-analysis. Exp Mol Med 48(4):e228
Zhang X, Sun Z, Zhou A et al (2021) Association between infections and risk of ankylosing spondylitis: a systematic review and meta-analysis. Front Immunol 12:768741
Zhao J, Huang C, Huang H et al (2020) Prevalence of ankylosing spondylitis in a Chinese population: a systematic review and meta-analysis. Rheumatol Int 40(6):859–872
Zhu G, Song J, Chen W et al (2021) Expression and role of dickkopf-1 (Dkk1) in tumors: from the cells to the patients. Cancer Manag Res 13:659–675
Zou YC, Yang XW, Yuan SG et al (2016) Downregulation of dickkopf-1 enhances the proliferation and osteogenic potential of fibroblasts isolated from ankylosing spondylitis patients via the Wnt/β-catenin signaling pathway in vitro. Connect Tissue Res 57(3):200–211
Acknowledgements
We thank all the patients and healthy controls who participated in our study.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81773514, 82073655, 82373672) and the scientific research level promotion plan of Anhui Medical University (2020xkjT006).
Author information
Authors and Affiliations
Contributions
XS: study conception and design, data curation, formal analysis, writing—original draft. YD: study conception and design, data curation, investigation. MN: investigation, methodology, resources. TZ and XW: investigation, formal analysis. YW: material preparation and data collection, investigation. ZS: investigation, data analysis. FP: funding acquisition, writing—review & editing.
Corresponding author
Ethics declarations
Conflict of interest
No competing financial interests exist.
Ethical Approval
The study was approved by the Local Ethics Research Committee of Anhui Medical University, and all participants provided their written informed content.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, X., Deng, Y., Ni, M. et al. Aberrant DNA Methylation Profile of Dickkopf-1 in Ankylosing Spondylitis. Biochem Genet (2024). https://doi.org/10.1007/s10528-024-10675-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10528-024-10675-y