
Abstract
The number of patients with COVID-19 caused by severe acute respiratory syndrome coronavirus 2 is still increasing. In the case of COVID-19 and tuberculosis (TB), the presence of one disease affects the infectious status of the other. Meanwhile, coinfection may result in complications that make treatment more difficult. However, the molecular mechanisms underpinning the interaction between TB and COVID-19 are unclear. Accordingly, transcriptome analysis was used to detect the shared pathways and molecular biomarkers in TB and COVID-19, allowing us to determine the complex relationship between COVID-19 and TB. Two RNA-seq datasets (GSE114192 and GSE163151) from the Gene Expression Omnibus were used to find concerted differentially expressed genes (DEGs) between TB and COVID-19 to identify the common pathogenic mechanisms. A total of 124 common DEGs were detected and used to find shared pathways and drug targets. Several enterprising bioinformatics tools were applied to perform pathway analysis, enrichment analysis and networks analysis. Protein–protein interaction analysis and machine learning was used to identify hub genes (GAS6, OAS3 and PDCD1LG2) and datasets GSE171110, GSE54992 and GSE79362 were used for verification. The mechanism of protein-drug interactions may have reference value in the treatment of coinfection of COVID-19 and TB.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Novel coronavirus pneumonia (COVID-19), caused by SARS-COV-2, is affecting the world on a massive scale (Meo 2021). The spike protein (S) is the site where COVID-19 binds to human ACE2 and where mutations frequently occurs. The primary symptoms were fever, dry cough and fatigue. Some patients had the loss of smell and taste as the first manifestations, and a few had symptoms such as nasal congestion, sore throat, diarrhea myalgia, and conjunctivitis (Walls et al. 2020).
TB is a pulmonary infectious disease caused by Mycobacterium tuberculosis (M. tuberculosis) (Fogel 2015). Its transmission through the respiratory tract affects the lungs, producing symptoms of severe cough, hemoptysis, fever, chest pain or even dyspnea. Before COVID-19, the single infectious disease that caused the highest death rate was TB, surpassing AIDS.
There will be a catastrophe for patients if TB, a chronic infectious disease, and COVID-19, an acute infectious disease, coinfect (Tapela et al. 2020). The 2021 global TB report issued by the WHO also clearly states that the number of global TB deaths decreased until 2019, but it began to increase again in 2020. TB is being forgotten in regard to diagnosis or treatment due to the prevalence of COVID-19. Missing TB diagnosis or improper treatment may cause TB exacerbation, which is an undesirable effect of COVID-19 treatment and its prognosis, and even some complications.
Studies have shown that both active TB and latent TB are at increased risk for COVID-19 and worsening infection (Motta et al. 2020; Mousquer et al. 2021; Stochino, et al. 2020). One reason is that damage to the lungs and effects on local immunity caused by tuberculosis make the body more susceptible to airborne pathogens (Mousquer et al. 2021). Moreover, the relationship between TB and SARS-COV-1, which shares 80% of the genome with SARS-CoV-2, shows that TB delays viral clearance, exacerbates disease and raises the stakes of disease transmission. SARS-CoV-2, in turn, has the potential to cause susceptibility to M. tuberculosis or reactivate latent TB since infection with other agents can disrupt the maintenance of granulomas caused by good immune regulation (Motta et al. 2020; Crisan-Dabija et al. 2020).
The delayed or weakened response to SARS-COV-2 caused by the coinfection of tuberculosis and COVID-19 leads to a systemic depletion of T cells, further bringing about a proliferation of cytokines and a remarkable increase in the number of neutrophils (Crisan-Dabija et al. 2020; Muefong and Sutherland 2020; Diao et al. 2020; Miotto et al. 2001). IFN-γ increases the expression of ACE2 receptors on cells; IL-4 and IL-13 are involved in immunopathological damage, and neutrophilia leads to increased inflammation and tissue damage (Heitmann et al. 2014; Vaz de Paula, et al. 2020).
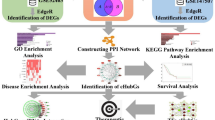
Two datasets, GSE114192 and GSE163151, for TB and COVID-19, respectively, from the GEO database were available further to explore the interaction between TB and COVID-19. First, we identified the DEGs from the two diseases and then obtained the common DEGs. In addition, we conducted pathway analysis and enrichment analysis, constructed a PPI network, constructed DEGs-miRNAs, DEGs- transcription factors (TFs) and protein-drug interactions networks, performed disease ontology analysis to determine the impact of COVID-19 on TB and searched for potential biomarkers and therapeutic targets to facilitate the development of treatment. Here, we have made a flowchart, as shown in Fig. 1.

Schematic illustration of the flow-process diagram of this study
Materials and Methods
Summary of Datasets
Both the microarray and RNA sequencing datasets of SARS-CoV-2 and TB were obtained from the GEO database of the National Center for Biotechnology Information (NCBI) (Barrett, et al. 2013). To ensure the consistency of the test samples and the adequacy of the sample size for the two diseases, we found that the sample source from whole blood was the best choice, and the COVID-19 dataset (GSE163151) and TB dataset (GSE114192) were the best combination. The COVID-19 dataset GSE163151 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163151) was from the GPL24676 Illumina NovaSeq 6000 platform (Homo sapiens), with contributions from Chiu CY et al. We selected samples derived from whole blood and sorted 7 COVID-19 groups and 20 healthy controls. The TB dataset GSE114192 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi) was from the GPL18573 Illumina NextSeq 500 platform (Homo sapiens) provided by Eckold C et al. Its samples are also from whole blood, containing 44 TB groups and 38 healthy controls. Both datasets were obtained by high-throughput sequencing. COVID-19 dataset GSE171110 and TB datasets GSE54992 and GSE79362 were used as validation cohorts. Table 1 shows the basic information for the databases.
DEGs and the Common DEGs of COVID-19 and TB
Screening standards (P value < 0.05 and |log2(FC)|≥ 1) were used to ascertain important DEGs in both datasets. Visualization of sequencing data for DEGs was performed using the DESeq2 and EdgeR packages in NetworkAnalyst (https://www.networkanalyst.ca/), each for COVID-19 and TB. NetworkAnalyst is a platform for visualizing comprehensive gene expression profiling and meta-analysis (Zhou, et al. 2019). The mutual DEGs of GSE114192 and GSE163151 were obtained by the ggplot2 package of the R programming language (v3.6.3), and the corresponding Venn diagrams were drawn.
Mutual Functional Enrichment Analysis
The “clusterProfiler” package of R programming language was applied to conduct gene ontology analysis in three categories of biological process (BP), cellular components (CC), molecular functions (MF) and pathway enrichment. The GO database and KEGG (Kyoto Encyclopedia of Genes and Genomes) were used for analysis. GO is a database that aims to define and describe the functions of genes and proteins for each species (Ashburner et al. 2000). The KEGG pathway is generally regarded as a process that modifies metabolism and has considerable utility in genome analysis and gene annotation (Kanehisa and Goto 2000). The standard metric P value < 0.05 was used to quantify the top paths.
Analysis of Protein Interactions
The protein interaction network shows the participation and association of related genes in the protein interaction network. The STRING tool retrieved the DEGs to obtain information about the protein interaction network. STRING (https://string-db.org/) provides experimental and predicted interaction information to produce protein interaction networks (Szklarczyk et al. 2017).
Extraction and Verification of the Hub Gene
The Cytoscape plugin CytoHubba (Cytoscape App Store—cytoHubba) ranks nodes in network features (Chin et al. 2014). We used Cytohubba, applying degree topological algorithm to obtain the top 10 hub genes, which are the most concentrated nodes. Then, the COVID-19 dataset GSE171110 and TB datasets GSE54992 and GSE79362 were used to verify the hub genes. Then, two machine learning methods were used to further screen out the most critical differentially expressed genes. By using the SVM-REF classifier, which applied the "e1071" R package, we further screened the hub genes most related to COVID-19 from the initially screened hub genes. Random Forest (RF) with the application of the "randomForest" R package was used to screen the most critical genes in TB. Finally, the intersection through a Venn diagram of the two screening results was used to obtain the most critical genes that connect COVID-19 and TB.
Analysis of DEG-TFs and DEG-miRNAs Interactions
Transcription factors (TFs) distinguish the specific DNA sequences that control transcription and are the basis of many different aspects of human physiology, disease, and variation (Lambert et al. 2018). We sought topologically credible TFs from the JASPAR database through the NetworkAnalyst platform. JASPAR is an open-access database for TFs across multiple species in six taxonomic groups (Fornes et al. 2020). Mature miRNAs bind to sites of their complementary mRNAs and regulate gene expression through base pairing. NetworkAnalyst was also used to construct the miRNA-gene interaction network to detect the miRNAs that play an essential regulatory role in target genes. MirTarBase and Tarbase were used for analysis. MirTarBase is the primary experimental database for miRNA-gene interactions, comprising the largest number of validated MTIs in contrast to akin databases (Hsu, et al. 2011). Additionally, we used a degree filter to select the top miRNAs and TFs at a high level and test biological functions and characteristics.
Association Between Gene and Disease
The disease ontology (DO) (http://disease-ontology.org) was used to search for the etiology of human disease (Schriml et al. 2019). The Genetic Association Database (GAD) is a database of genetic association data from inherited, acquired and human developmental diseases. R software was used to perform DO enrichment with the “DOSE” package. We also used DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/) to perform disease enrichment, which uses GAD as the secondary source (Huang et al. 2009a, b). We combined the data from the two approaches and sorted out the top 15 gene-enriched diseases.
Estimate the Correlative Drugs
We used DrugBank via NetworkAnalyst based on the DEGs of TB and COVID-19 to recognize drug molecules by identifying protein-drug interactions. DrugBank (http://www.drugbank.ca), a drug database, possesses well-rounded molecular information about drugs (Wishart, et al. 2018).
Results
Identification of the Shared DEGs Between COVID-19 and TB
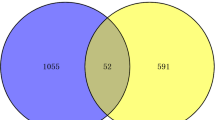
The GSE163151 dataset was used for the identification of COVID-19 differentially expressed genes. A total of 5894 differentially expressed genes were identified, of which 5212 were up-regulated and 682 were down-regulated. Similarly, 411 TB genes were differentially expressed in the GSE114192 dataset, including 295 upregulated genes and 116 downregulated genes. The 124 common DEGs are visualized by a Wayne diagram (Fig. 2).

Common DEGs of COVID-19 and TB representation through a venn diagram. This integrated analysis revealed 124 common DEGs shared between COVID-19 and TB
Analysis of Gene set Enrichment
Through basic molecular or biological processes, pathway analysis illustrates the interactions with diverse diseases. The study illustrated the top 10 GO terms based on the log of the P value and z score for each of the subsections (BP, MF and CC) (Table 2) (Fig. 3), and the top 6 terms of pathway enrichment analysis were also based on the P value. The top three GO terms of BP included response to molecule of bacterial origin, response to lipopolysaccharide, and regulation of inflammatory response. Blood microparticle, collagen-containing extracellular matrix and high-density lipoprotein particle were the top three of CC. Meanwhile, MF included serine-type peptidase activity, serine hydrolase activity, endopeptidase activity, etc. The top 6 KEGG human pathways included tryptophan metabolism, Staphylococcus aureus infection, African trypanosomiasis, cGMP-PKG signaling pathway, type II diabetes mellitus, complement, and coagulation cascades (Table 3) (Fig. 4).

Gene ontology analysis of DEGs in both the GSE163151 and GSE114192 datasets. A GO enrichment of DEGs in biological process. B GO enrichment of DEGs in molecular function. C GO enrichment of DEGs in cellular components

KEGG pathway enrichment analysis of DEGs in both GSE163151 and GSE114192
Screening and Validation of Hub Genes and Module Analysis
The PPI network made by the STRING online tool was applied to identify the interaction among proteins containing 107 nodes and 66 edges (Fig. 5). The top ten identified hub genes selected by the Cytohubba plugin in Cytoscape were ATF3, IL27, CLEC11A, PDCD1LG2, GAS6, HPR, MMP1, C2, OAS3 and CETP(Fig. 6).After validation of datasets GSE171110, GSE54992 and GSE79362, 8 hub genes were identified: IL27, CLEC11A, PDCD1LG2, GAS6, HPR, C2, OAS3 and CETP (Fig. 7). Through machine learning, four of the most critical genes for COVID-19 were obtained by the SVM-REF algorithm, and five of the most critical genes for TB were obtained by the RF algorithm. After the intersection of the two, three common hub genes (GAS6, OAS3 and PDCD1LG2) were obtained, which were taken as the final key genes in COVID-19 and TB (Fig. 8).

PPI network of common DEGs between COVID-19 and TB. In the figure, the circle nodes represent DEGs, and the line thickness indicates the strength of data support

Determination of hub genes between COVID-19 and TB from the PPI network

Hub gene verification results. A Validation of hub genes in the COVID-19 dataset GSE171110. B–C Validation of hub genes in the TB datasets GSE54992 and GSE79362

Machine learning screening for hub genes. A SVM-RFE algorithm to screen candidate genes based on the COVID-19 dataset GSE163151. The point highlighted indicates the optimal accuracy, and the corresponding genes at this point are the best signature selected by SVM-RFE. B–C The random forest algorithm to screen candidate genes based on the TB dataset GSE114192. The random forest algorithm shows the error in TB, ranking of the relative importance of genes. D Venn diagram showing the hub genes shared by SVM-RFE algorithms and random forest algorithm
DEGs–miRNA and TF–DEGs Interactions
To identify transcription factors and regulatory molecules that work together in both diseases, we applied a network-based approach through the NetworkAnalyst online tool to demonstrate. The DEGs–miRNAs interactions network contains 154 nodes and 365 edges. (Fig. 9) The TFs-DEGs network comprised 111 nodes and 604 edges(Fig. 10). Degree represents the number of connections between two nodes. We use degree as the screening standard for network hubs. Therefore, we screened out the top ten TFs (FOXC1, GATA2, YY1, USF2, FOXL1, JUN, RELA, TFAP2A, E2F1 and HINFP) as hubs and screened out ten hub miRNAs. They are hsa-mir-335-5p, hsa-mir-26b-5p, hsa-mir-124-3p, hsa-mir-665, hsa-mir-6840-3p, hsa-mir-4695-5p, hsa-mir-98-5p hsa-mir-1273e, hsa-mir-17-5p, and hsa-mir-30a-5p. Node color reflects the degree of connection and node size reaction interaction intensity as reference standards.

The interconnected regulatory network of DEGs-miRNAs. The square nodes and the circle nodes are miRNAs and gene symbols, respectively

The cohesive regulatory interaction of DEG-TFs. The circle nodes and the square nodes are gene symbols and TFs, respectively
Identification of Disease Association
DO analysis was used to predict the possible complications of TB and COVID-19 coinfection and to design therapeutic strategies for diseases. Bacterial infectious disease, lymphoma, non-Hodgkin’s lymphoma, lymphadenitis, type 2 diabetes, and hepatitis C are shown with the highest correlation according to the P value. (Fig. 11).

Bar graph of the top 10 diseases sorted by p value ranking
Screening of Candidate Drugs
Figure 12 shows the interaction between drugs and the six most essential genes in the common DEGs of COVID-19 and TB, including MAOB, C1QC, IL10, EDNR8, CDO1 and CCL2. Candidate drugs representing common drugs for COVID-19 and TB were selected.

Protein-drug interaction analysis using the common DEGs between COVID-19 and TB. A Interaction between drugs and MAOB. B Interaction between drugs, ITGA2B and C1QC. C Interaction between drugs and IL10. D Interaction between drugs and EDNRB. E Interaction between drugs and CDO1. F Interaction between drugs and CCL2
Discussion
Both TB and COVID-19 have similar symptoms, such as fever, cough and dyspnea. Under the current situation in which medical resources are focused on the treatment of COVID-19 when TB patients are coinfected with SARS-COV-2, the diagnosis of TB is easily overlooked. Meanwhile, the pathogenesis of coinfection is complex, posing a challenge to treatment. Transcriptome analysis of SARS-CoV-2 and TB revealed that a total of 124 DEGs showed shared expression in both. These 124 common DEGs were used to find common pathways and molecular markers between TB and COVID-19 to identify the relationship between TB and COVID-19 and seek drugs with significant therapeutic effects on the coinfection of TB and COVID-19 and its comorbidities. GO analysis, pathway analysis, PPIs, TF-gene interactions, miRNA-gene coregulatory networks, DO enrichment analysis and candidate drug detection were performed to complete this study.
In biological processes, the response to molecules of bacterial origin and the response to lipopolysaccharide (LPS) are the two top GO pathways. Studies have shown that LPS can be used as a biomarker in TB (Larrouy-Maumus 2019). The S protein of SARS-COV-2 specifically binds with LPS to produce a synergistic effect, promoting the response of proinflammatory cells in vivo and in vitro and leading to excessive inflammation (Petruk et al. 2020; Tumpara, et al. 2021). Pulmonary infection with M. tuberculosis or SARS-COV-2 leads to the activation of alveolar macrophages and pulmonary epithelial cells and further the release of proinflammatory cytokines, which increases the permeability of the pulmonary endothelium, allowing the transmission of bacteria or viruses (Polidoro et al. 2020). For the cellular component, the top GO terms were blood microparticles and collagen-containing extracellular matrix. Blood microparticles are critical cellular components in the inflammatory response and are mainly released by platelets and megakaryocytes. Microparticles can send signals to surface receptors and affect the inflammatory response and function of receptor cells, which can also be used as biomarkers for the clinical diagnosis of inflammation (Sahler et al. 2014). Collagen plays an essential role in cell growth and development or healing and regeneration (Fu et al. 2018). According to the molecular function, serine-type endopeptidase activity and oxidoreductase activity were among the top GO terms. Serine endopeptidase catalyzes the hydrolysis of α-peptide bonds in polypeptide chains and makes a difference in coagulation and complement systems. Among them, type II transmembrane serine endopeptidase can cleave the spike protein of coronavirus, making it a potential therapeutic target of coronavirus (Iwata-Yoshikawa, et al. 2019).
Pathway analysis is used to reflect the response of an organism through internal changes. The top 6 KEGG human pathways included tryptophan metabolism, Staphylococcus aureus infection, African trypanosomiasis, cGMP-PKG signaling pathway, type II diabetes mellitus, complement, and coagulation cascades. Here, tryptophan metabolism regulates different physiological processes; thus, changes in its content can help predict diseases. The changes in tryptophan and its metabolites are closely related to the critical pathophysiological processes of both SARS-COV-2 and TB (Anderson et al. 2021; Cho et al. 2020). The increased AHR ligands and the activation of rate-limiting enzymes ( Indoleamine 2,3-dioxygenase and tryptophan-2,3-dioxygenase) that convert tryptophan (the precursor of serotonin synthesis) to kynurenine are both caused by differential regulation of tryptophan metabolites. They are associated with cytokine storms induced by SARS-COV-2 infection and affect serotonin synthesis levels (Anderson et al. 2021, 2020). Studies have shown that l-tryptophan (L-TRP) is decreased in patients with active TB and that its metabolites [including l-Kynurenine(KYN)] are increased, thus affecting immune function (Cho et al. 2020; Feng et al. 2015; Isa et al. 2018; Vrieling et al. 2019; Weiner et al. 2012). Determination of the L-TRP/KYN ratio or IDO activity is conducive to the diagnosis and prognosis of TB, and patients with low IDO activity have a better prognosis (Crowther and Qualls 2020).
Three hub proteins (PDCD1LG2, GAS6, and OAS3) were identified to be involved in these diseases and can be considered potential biomarkers or novel drug targets. The remaining five genes that were not selected for the final hub gene also played an important role. Activated platelets in COVID-19 and TB stimulate the release of large amounts of IL-27, while polymorphisms of IL27 play a protective role in susceptibility to TB (Diao et al. 2020; Taus et al. 2020). CLEC11A is a growth factor of primitive hematopoietic progenitor cells. PDCD1LG2 is a biomarker of tuberculosis and engages in the negative regulation of activated T-cell proliferation, interferon-γ production and interleukin-10 production (Huang et al. 2020). GAS6 is involved in stimulating cell proliferation and is often overexpressed in many cancers. It is also a key regulator of inflammation and vascular injury responses, participates in coagulation-related pathology, and acts importantly on SARS-COV-2 infection and progressive complications. Its elevated expression level is also associated with various diseases, such as venous thromboembolic disease and systemic lupus erythematosus (Tutusaus, et al. 2020). HPR can efficiently bind to hemoglobin and play a specific role in innate immune defense (Yang et al. 2018). The C2 component is a serum glycoprotein, a part of the classical pathway of the complement system (Urban et al. 2021). The OAS3 enzyme is induced by interferon and functions to inhibit cellular protein synthesis and resistance to viral infection (Xiao et al. 2019). CETPP participates in reverse cholesterol transport from high-density lipoprotein to others. It is associated with type 2 diabetes, chronic kidney disease and cardiovascular disease (Schmidt et al. 2021; Srirojnopkun et al. 2018). Potential biomarkers in physiopathological processes were sought from the regulatory biomolecules. Here, the transcriptional and posttranscriptional regulators of the mutual DEGs were detected by TF-gene and miRNA-gene interaction analyses. We found that some miRNAs engage in the regulation of inflammation (miR-124-3p, miR-26b-5p) (Zhang et al. 2020; Zhu et al. 2021), cancer (miR-335-5p, miR-665, miR-4695-5p, miR-98-5p, miR-17-5p) (Fan 2018; Shi et al. 2020; Zhao et al. 2019), cardiovascular disease (miR-124-3p, miR-26b-5p, miR-335-5p, miR-665) (Fan 2018; Lv et al. 2021; Sun et al. 2021), and neurotrophic protection (miR-26b-5p, miR-124-3p) (Berg et al. 2020; Geng et al. 2017). Mechanistically, miR-665 promotes macrophage apoptosis and autophagy, worsening TB (Jiang et al. 2021). MiR-335-5p inhibits SLC2A4 expression, which aggravates type 2 diabetes (Li and Zhang 2021). From the network, CDCP1 interacts with other genes and miRNAs at a high rate. The top five miRNAs with high degree values were miR-335-5p, miR-26b-5p, miR-124-3p, miR-665, and miR-6840-3p. Most miRNAs are involved in cancer and cardiovascular disease.
From the TF-gene network, FOXC1, GATA2, YY1, USF2, FOXL1, JUN, RELA, TFAP2A, E2F1 and HINFP were most associated with the common DEGs. FOXC1 and YY1 mediate immunosuppression. E2F1 together with FOXC1 and YY1 facilitates tumor progression (Hays and Bonavida 2019; Shen et al. 2020). GATA2 functions notably in the regulation of gene transcription during the development and proliferation of hematopoietic and endocrine cell lines. Its deficiency is the basis of mononucleosis and Mycobacterium infection (Spinner et al. 2014). FOXL1 regulates the function of lung fibroblasts, which is related to pulmonary fibrosis and has a tumor-suppressive effect (Miyashita et al. 2020). HINFP is highly expressed in both type 2 diabetes mellitus and neurological diseases (Rahman, et al. 2020).
Disease ontology enrichment analysis was available for predicting possible complications of TB and COVID-19 coinfection. The common DEGs-related diseases of COVID-19 and TB mainly included infectious diseases, autoimmune diseases, metabolic syndrome, and cardiovascular diseases. These results will help us predict the complications of coinfection with COVID-19 and TB.
Statistics indicate that the majority of hospitalized COVID-19 patients acquire secondary bacterial infections. This is mainly due to the decline in immunity and immune system disorders caused by viral infection (Mirzaei et al. 2020). Similarly, research showing slight impairment of innate and cellular immunity in TB patients and statistical data imply that TB patients possess a higher probability of having other bacterial infections (Antas et al. 2006; Attia et al. 2019). Lymphadenitis is a common symptom of both TB and SARS-COV-2 infection (Franco-Paredes 2018; Wang et al. 2021). CD147 is operative in hepatitis and is upregulated in both COVID-19 and TB, facilitating the entry of viruses into cells and regulating ACE2 expression levels (Fenizia, et al. 2021; Feruglio et al. 2015). Both COVID-19 and TB can disrupt the gut microbiome by releasing cytokines and toxins, leading to a loss of immune regulation of the intestinal mucosa and further inflammation (Chai et al. 2018; Vodnar et al. 2020). Autoimmune diseases are often accompanied by COVID-19 and TB. Both TB and COVID-19 can cause macrophage activation syndrome (MAS), which is also connected with autoimmune diseases such as connective tissue disease (McGonagle et al. 2021). For example, the increased production of cytokines such as IFN-γ and TNF-α in TB induces the expansion of autoreactive T cells. The activation of TLR-mediated signaling pathways induced by TB is associated with autoimmune disease. The cytokine storm caused by MAS can also lead to other diseases, such as acute respiratory distress, cardiac dysfunction, and Alzheimer's disease (AD). Mechanisms of injury include increased capillary permeability, which leads to plasma deposition in tissues and further perfusion depletion.
Alzheimer's disease is commonly seen as a central nervous system complication of COVID-19. Systemic inflammation affects cognitive function and promotes the progression of neurodegenerative diseases. The MAS triggers various inflammatory pathways that eventually weaken the blood‒brain barrier, allowing infections to spread to the brain. Both diseases increase the likelihood of severe cytokine storms due to the involvement of microglia and neurons. The cytokine storms can lead to cognitive impairment and neurodegeneration; for example, increased IL-1β leads to impairment of long-term potentiation and cognitive performance (Mandal et al. 2020; Xia et al. 2021). M. tuberculosis manipulates foamy macrophages in atherosclerotic plaques and alters M1/M2 polarization of macrophages, together with cellular immune-mediated immune responses, promoting atherosclerosis (Mandal et al. 2020; Xia et al. 2021). High expression of ACE2, toxins and infected necrotic cells in cardiomyocytes and vascular endothelium can also lead to circulatory disease (Chang et al. 2021). Studies have shown a higher prevalence of diabetes mellitus among TB patients and COVID-19 patients (Chai et al. 2018; Vodnar et al. 2020). Metabolic reprogramming usually occurs when either innate or adaptive immune cells are activated, promoting metabolic syndromes such as type 2 diabetes (Chai et al. 2018; Vodnar et al. 2020).
By learning more about some candidate drugs from the gene-drug networks, we found that the drug mechanisms of etanercept, rituximab, bevacizumab, AV411, bosentan, sitaxentan, ambrisentan, macitentana, acetyl-l-cysteine and NADH may have important reference value in the treatment of COVID-19 combined with TB.
Immunosuppressants commonly used in COVID-19 treatment, such as TNF-α blockers, mostly increase susceptibility to M. tuberculosis and damage the structural integrity of granuloma. Etanercept, one of the candidate drugs, has the lowest M. tuberculosis infection rate among all TNF-α blockers (Wong et al. 2008). Rituximab, also a cytokine blocker that acts on B cells through a different mechanism, is a T-cell costimulatory modulator and has little effect on susceptibility to M. tuberculosis (Prieto-Peña and Dasgupta 2021; Morrison 2014). Bevacizumab is presently being studied for the treatment of COVID-19 complications (Samudrala et al. 2020). It is a monoclonal antibody against vascular endothelial growth factor (VEGF), which is indicated for COVID-19-induced elevated VEGF levels associated with pulmonary edema, dyspnea, acute respiratory distress and acute lung injury. It inhibits the immune response and helps anti-TB antibiotics infiltrate granulation tissue by normalizing the vascular system of TB granulomas, shortening the treatment cycle of TB (Zheng et al. 2016). AV411 has anti-inflammatory and neuroprotective effects through, for example, inhibition of nitric oxide synthesis and reduction of reactive oxygen species (Yagi, et al. 2010). Bosentan is a treatment for pulmonary hypertension and is known to relieve symptoms and improve respiratory function in acute respiratory distress syndrome, the most severe complication of COVID-19. It is considered a promising drug in COVID-19 treatment (Puk et al. 2022). Sitaxentan, ambrisentan and macitentana are all used for hypertension, pulmonary hypertension, congestive heart failure, and connective tissue disease. Among these, sitaxentan blocks the binding of endothelin (a vasoconstrictor that is highly expressed in lung infections and pulmonary hypertension) to its receptors, thereby eliminating the harmful effects of endothelin (Barst et al. 2002; Sidharta et al. 2015; Venitz et al. 2012). Reduced levels of acetyl-l-cysteine (NAC) (a precursor of reduced glutathione (GSH)) result from lung infection, affecting a variety of disease states and immune dysfunction, increasing susceptibility to viral infections, and exacerbating oxidative damage to the lungs. NAC, widely used to restore or prevent glutathione depletion, has antioxidant and anti-inflammatory mechanisms. Meanwhile, studies showed that the binding affinity of ACE2 and SARS-COV-2 was significantly reduced when disulfide bonds of the S proteins were reduced to sulfhydryl groups. Additionally, NAC can protect against the harmful effects of angiotensin II (Flora et al. 2020). NADH exhibits the best multitarget activity in some effective interactions with SARS-CoV-2 structural components and critical residues of host proteins (Artese et al. 2020). It has been considered a promising drug for COVID-19 and is also for treating cardiovascular disease, Alzheimer’s disease, chronic fatigue syndrome and Parkinson’s disease (Belenky et al. 2007). Drugs interacting with ITGA2B mostly have antiplatelet and anticoagulation effects, applying to the common symptoms of COVID-19 and TB (the increase in platelet and blood coagulation) (Tcheng et al. 2003).
Conclusions
Our study revolved around the relationship between TB and COVID-19 in the context of transcriptomic analysis. We performed DEGs and common gene identification in two databases to figure out the influence of COVID-19 on TB. A total of 124 common DEGs were identified. We used them to perform the bioinformatics analysis and pathways analysis. A PPI network was constructed to identify the top 15 hub genes and further identify potential biomarkers or novel drug targets. We also constructed DEGs–miRNAs and TFs–genes interactions networks to identify transcriptional and post transcriptional regulators in both diseases. Gene–disease (GD) analysis shows the common related diseases of the two diseases as a reference to predict the complications caused by their coinfection. The results of candidate drugs are suitable for the treatment of both TB and COVID-19, which to some extent can address the adverse effects of current COVID-19 drugs on TB and are of vital significance in the treatment of the coinfection and complications of TB and COVID-19.
Data Availability
Data sharing not applicable—no new data generated.
References
Anderson G, Carbone A, Mazzoccoli G (2020) Aryl hydrocarbon receptor role in co-ordinating SARS-CoV-2 entry and symptomatology: linking cytotoxicity changes in COVID-19 and cancers modulation by racial discrimination stress. Biology. https://doi.org/10.3390/biology9090249
Anderson G, Carbone A, Mazzoccoli G (2021) Tryptophan metabolites and aryl hydrocarbon receptor in severe acute respiratory syndrome, coronavirus-2 (SARS-CoV-2) pathophysiology. Int J Mol Sci. https://doi.org/10.3390/ijms22041597
Antas PR et al (2006) Decreased CD4+ lymphocytes and innate immune responses in adults with previous extrapulmonary tuberculosis. J Allergy Clin Immunol 117(4):916–923. https://doi.org/10.1016/j.jaci.2006.01.042
Artese A et al (2020) Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist Updat 53:100721. https://doi.org/10.1016/j.drup.2020.100721
Ashburner M et al (2000) Gene ontology: tool for the unification of biology. Gene Ontol Consort Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Attia EF et al (2019) Tuberculosis and other bacterial co-infection in Cambodia: a single center retrospective cross-sectional study. BMC Pulm Med 19(1):60. https://doi.org/10.1186/s12890-019-0828-4
Barrett T et al (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. https://doi.org/10.1093/nar/gks1193
Barst RJ et al (2002) Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: open-label pilot study. Chest 121(6):1860–1868. https://doi.org/10.1378/chest.121.6.1860
Belenky P, Bogan KL, Brenner C (2007) NAD+ metabolism in health and disease. Trends Biochem Sci 32(1):12–19. https://doi.org/10.1016/j.tibs.2006.11.006
Chai Q, Zhang Y, Liu CH (2018) Mycobacterium tuberculosis: an adaptable pathogen associated with multiple human diseases. Front Cell Infect Microbiol 8:158. https://doi.org/10.3389/fcimb.2018.00158
Chang WT et al (2021) Cardiac involvement of COVID-19: a comprehensive review. Am J Med Sci 361(1):14–22. https://doi.org/10.1016/j.amjms.2020.10.002
Chin C-H et al (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 8(S4):S11. https://doi.org/10.1186/1752-0509-8-s4-s11
Cho Y et al (2020) Identification of serum biomarkers for active pulmonary tuberculosis using a targeted metabolomics approach. Sci Rep 10(1):3825. https://doi.org/10.1038/s41598-020-60669-0
Crisan-Dabija R et al (2020) Tuberculosis and COVID-19: lessons from the past viral outbreaks and possible future outcomes. Can Respir J 2020:1401053. https://doi.org/10.1155/2020/1401053
Crowther RR, Qualls JE (2020) Metabolic regulation of immune responses to mycobacterium tuberculosis: A spotlight on l-arginine and l-tryptophan metabolism. Front Immunol 11:628432. https://doi.org/10.3389/fimmu.2020.628432
da Huang W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13. https://doi.org/10.1093/nar/gkn923
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. https://doi.org/10.1038/nprot.2008.211
De Flora S, Balansky R, La Maestra S (2020) Rationale for the use of N-acetylcysteine in both prevention and adjuvant therapy of COVID-19. Faseb j 34(10):13185–13193. https://doi.org/10.1096/fj.202001807
Diao B et al (2020) Reduction and functional exhaustion of t cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11:827. https://doi.org/10.3389/fimmu.2020.00827
Fan J et al (2018) MiR-665 aggravates heart failure via suppressing CD34-mediated coronary microvessel angiogenesis. Aging 10(9):2459–2479. https://doi.org/10.18632/aging.101562
Feng S et al (2015) Analysis of serum metabolic profile by ultra-performance liquid chromatography-mass spectrometry for biomarkers discovery: application in a pilot study to discriminate patients with tuberculosis. Chin Med J 128(2):159–168. https://doi.org/10.4103/0366-6999.149188
Fenizia C et al (2021) SARS-CoV-2 Entry: At the Crossroads of CD147 and ACE2. Cells. https://doi.org/10.3390/cells10061434
Feruglio SL et al (2015) Early dynamics of T helper cell cytokines and T regulatory cells in response to treatment of active Mycobacterium tuberculosis infection. Clin Exp Immunol 179(3):454–465. https://doi.org/10.1111/cei.12468
Fogel N (2015) Tuberculosis: a disease without boundaries. Tuberculosis 95(5):527–531. https://doi.org/10.1016/j.tube.2015.05.017
Fornes O et al (2020) JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48(D1):D87-d92. https://doi.org/10.1093/nar/gkz1001
Franco-Paredes C et al (2018) Cutaneous mycobacterial infections. Clin Microbiol Rev. https://doi.org/10.1128/cmr.00069-18
Fu X et al (2018) Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128(5):2127–2143. https://doi.org/10.1172/jci98215
Geng L, Liu W, Chen Y (2017) miR-124-3p attenuates MPP(+)-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp Biol Med (maywood) 242(18):1757–1764. https://doi.org/10.1177/1535370217734492
Hays E, Bonavida B (2019) YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist Updat 43:10–28. https://doi.org/10.1016/j.drup.2019.04.001
Heitmann L et al (2014) The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J Pathol 234(3):338–350. https://doi.org/10.1002/path.4399
Hsu SD et al (2011) miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. https://doi.org/10.1093/nar/gkq1107
Huang KC et al (2020) Prognostic relevance of programmed cell death 1 ligand 2 (PDCD1LG2/PD-L2) in patients with advanced stage colon carcinoma treated with chemotherapy. Sci Rep 10(1):22330. https://doi.org/10.1038/s41598-020-79419-3
Isa F et al (2018) Mass spectrometric identification of urinary biomarkers of pulmonary tuberculosis. EBioMedicine 31:157–165. https://doi.org/10.1016/j.ebiom.2018.04.014
Iwata-Yoshikawa N et al (2019) TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. https://doi.org/10.1128/jvi.01815-18
Jiang F et al (2021) LncRNA MIAT regulates autophagy and apoptosis of macrophage infected by Mycobacterium tuberculosis through the miR-665/ULK1 signaling axis. Mol Immunol 139:42–49. https://doi.org/10.1016/j.molimm.2021.07.023
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Lambert SA et al (2018) The human transcription factors. Cell 172(4):650–665. https://doi.org/10.1016/j.cell.2018.01.029
Larrouy-Maumus G (2019) Lipids as biomarkers of cancer and bacterial infections. Curr Med Chem 26(11):1924–1932. https://doi.org/10.2174/0929867325666180904120029
Li G, Zhang L (2021) miR-335-5p aggravates type 2 diabetes by inhibiting SLC2A4 expression. Biochem Biophys Res Commun 558:71–78. https://doi.org/10.1016/j.bbrc.2021.04.011
Lv L et al (2021) MiR-124-3p reduces angiotensin II-dependent hypertension by down-regulating EGR1. J Hum Hypertens 35(8):696–708. https://doi.org/10.1038/s41371-020-0381-x
Mandal N et al (2020) Correlation between CNS tuberculosis and the COVID-19 pandemic: the neurological and therapeutic insights. ACS Chem Neurosci 11(18):2789–2792. https://doi.org/10.1021/acschemneuro.0c00546
McGonagle D, Ramanan AV, Bridgewood C (2021) Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat Rev Rheumatol 17(3):145–157. https://doi.org/10.1038/s41584-020-00571-1
Meo SA et al (2021) Omicron SARS-CoV-2 new variant: global prevalence and biological and clinical characteristics. Eur Rev Med Pharmacol Sci 25(24):8012–8018. https://doi.org/10.26355/eurrev_202112_27652
Miotto D et al (2001) Expression of IFN-gamma-inducible protein; monocyte chemotactic proteins 1, 3, and 4; and eotaxin in TH1- and TH2-mediated lung diseases. J Allergy Clin Immunol 107(4):664–670. https://doi.org/10.1067/mai.2001.113524
Mirzaei R et al (2020) Bacterial co-infections with SARS-CoV-2. IUBMB Life 72(10):2097–2111. https://doi.org/10.1002/iub.2356
Miyashita N et al (2020) FOXL1 regulates lung fibroblast function via multiple mechanisms. Am J Respir Cell Mol Biol 63(6):831–842. https://doi.org/10.1165/rcmb.2019-0396OC
Morrison VA (2014) Immunosuppression associated with novel chemotherapy agents and monoclonal antibodies. Clin Infect Dis 59(Suppl 5):S360–S364. https://doi.org/10.1093/cid/ciu592
Motta I et al (2020) Tuberculosis, COVID-19 and migrants: preliminary analysis of deaths occurring in 69 patients from two cohorts. Pulmonology 26(4):233–240. https://doi.org/10.1016/j.pulmoe.2020.05.002
Mousquer GT, Peres A, Fiegenbaum M (2021) Pathology of TB/COVID-19 co-infection: the phantom menace. Tuberculosis 126:102020. https://doi.org/10.1016/j.tube.2020.102020
Muefong CN, Sutherland JS (2020) Neutrophils in tuberculosis-associated inflammation and lung pathology. Front Immunol 11:962. https://doi.org/10.3389/fimmu.2020.00962
Petruk G et al (2020) SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol 12(12):916–932. https://doi.org/10.1093/jmcb/mjaa067
Polidoro RB et al (2020) Overview: systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front Immunol 11:1626. https://doi.org/10.3389/fimmu.2020.01626
Prieto-Peña D, Dasgupta B (2021) Biologic agents and small-molecule inhibitors in systemic autoimmune conditions: an update. Pol Arch Intern Med 131(2):171–181. https://doi.org/10.20452/pamw.15438
Puk O et al (2022) Pulmonary artery targeted therapy in treatment of COVID-19 related ARDS. Lit Rev Biomed Pharmacother 146:112592. https://doi.org/10.1016/j.biopha.2021.112592
Rahman MH et al (2020) A Network-Based Bioinformatics Approach to Identify Molecular Biomarkers for Type 2 Diabetes that Are Linked to the Progression of Neurological Diseases. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph17031035
Sahler J, Woeller CF, Phipps RP (2014) Microparticles engineered to highly express peroxisome proliferator-activated receptor-γ decreased inflammatory mediator production and increased adhesion of recipient monocytes. PLoS ONE 9(11):e113189. https://doi.org/10.1371/journal.pone.0113189
Samudrala PK et al (2020) Virology, pathogenesis, diagnosis and in-line treatment of COVID-19. Eur J Pharmacol 883:173375. https://doi.org/10.1016/j.ejphar.2020.173375
Schmidt AF et al (2021) Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat Commun 12(1):5640. https://doi.org/10.1038/s41467-021-25703-3
Schriml LM et al (2019) Human disease ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Res 47(D1):D955-d962. https://doi.org/10.1093/nar/gky1032
Shen C et al (2020) Advancement of E2F1 in common tumors. Zhongguo Fei Ai Za Zhi 23(10):921–926. https://doi.org/10.3779/j.issn.1009-3419.2020.101.32
Shi YP et al (2020) miR-17-5p knockdown inhibits proliferation, autophagy and promotes apoptosis in thyroid cancer via targeting PTEN. Neoplasma 67(2):249–258. https://doi.org/10.4149/neo_2019_190110N29
Sidharta PN, Treiber A, Dingemanse J (2015) Clinical pharmacokinetics and pharmacodynamics of the endothelin receptor antagonist macitentan. Clin Pharmacokinet 54(5):457–471. https://doi.org/10.1007/s40262-015-0255-5
Spinner MA et al (2014) GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123(6):809–821. https://doi.org/10.1182/blood-2013-07-515528
Srirojnopkun C et al (2018) Association of APOE and CETP TaqIB polymorphisms with type 2 diabetes mellitus. Arch Med Res 49(7):479–485. https://doi.org/10.1016/j.arcmed.2019.02.005
Stochino C et al (2020) Clinical characteristics of COVID-19 and active tuberculosis co-infection in an Italian reference hospital. Eur Respir J. https://doi.org/10.1183/13993003.01708-2020
Sun D et al (2021) Overexpressed miR-335-5p reduces atherosclerotic vulnerable plaque formation in acute coronary syndrome. J Clin Lab Anal 35(2):e23608. https://doi.org/10.1002/jcla.23608
Szklarczyk D et al (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45(D1):D362–D368. https://doi.org/10.1093/nar/gkw937
Tapela K, Ochieng’ Olwal C, Quaye O (2020) Parallels in the pathogenesis of SARS-CoV-2 and M. tuberculosis: a synergistic or antagonistic alliance? Future Microbiol 15:1691–1695. https://doi.org/10.2217/fmb-2020-0179
Taus F et al (2020) Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol 40(12):2975–2989. https://doi.org/10.1161/atvbaha.120.315175
Tcheng JE et al (2003) Benefits and risks of abciximab use in primary angioplasty for acute myocardial infarction: the controlled abciximab and device investigation to lower late angioplasty complications (CADILLAC) trial. Circulation 108(11):1316–1323. https://doi.org/10.1161/01.Cir.0000087601.45803.86
Tumpara S et al (2021) Boosted pro-inflammatory activity in human PBMCs by lipopolysaccharide and SARS-CoV-2 spike protein is regulated by α-1 antitrypsin. Int J Mol Sci. https://doi.org/10.3390/ijms22157941
Tutusaus A et al (2020) Role of vitamin K-dependent factors protein S and GAS6 and TAM receptors in SARS-CoV-2 infection and COVID-19-associated immunothrombosis. Cells. https://doi.org/10.3390/cells9102186
Urban A et al (2021) Gain-of-function mutations R249C and S250C in complement C2 protein increase C3 deposition in the presence of C-reactive protein. Front Immunol 12:724361. https://doi.org/10.3389/fimmu.2021.724361
van den Berg MMJ et al (2020) Circulating microRNAs as potential biomarkers for psychiatric and neurodegenerative disorders. Prog Neurobiol 185:101732. https://doi.org/10.1016/j.pneurobio.2019.101732
Vaz de Paula CB et al (2020) IL-4/IL-13 remodeling pathway of COVID-19 lung injury. Sci Rep 10(1):18689. https://doi.org/10.1038/s41598-020-75659-5
Venitz J et al (2012) Clinical pharmacokinetics and drug-drug interactions of endothelin receptor antagonists in pulmonary arterial hypertension. J Clin Pharmacol 52(12):1784–1805. https://doi.org/10.1177/0091270011423662
Vodnar DC et al (2020) Coronavirus disease (COVID-19) caused by (SARS-CoV-2) infections: a real challenge for human gut microbiota. Front Cell Infect Microbiol 10:575559. https://doi.org/10.3389/fcimb.2020.575559
Vrieling F et al (2019) Plasma metabolomics in tuberculosis patients with and without concurrent type 2 diabetes at diagnosis and during antibiotic treatment. Sci Rep 9(1):18669. https://doi.org/10.1038/s41598-019-54983-5
Walls AC et al (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281-292.e6. https://doi.org/10.1016/j.cell.2020.02.058
Wang XX et al (2021) Histopathological features of multiorgan percutaneous tissue core biopsy in patients with COVID-19. J Clin Pathol 74(8):522–527. https://doi.org/10.1136/jclinpath-2020-206623
Weiner J 3rd et al (2012) Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS ONE 7(7):e40221. https://doi.org/10.1371/journal.pone.0040221
Wishart DS et al (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46(D1):D1074–D1082. https://doi.org/10.1093/nar/gkx1037
Wong M et al (2008) TNFalpha blockade in human diseases: mechanisms and future directions. Clin Immunol 126(2):121–136. https://doi.org/10.1016/j.clim.2007.08.013
Xia X, Wang Y, Zheng J (2021) COVID-19 and Alzheimer’s disease: how one crisis worsens the other. Transl Neurodegener 10(1):15. https://doi.org/10.1186/s40035-021-00237-2
Xiao X et al (2019) Exome sequencing reveals a heterozygous OAS3 mutation in a Chinese family with juvenile-onset open-angle glaucoma. Invest Ophthalmol vis Sci 60(13):4277–4284. https://doi.org/10.1167/iovs.19-27545
Yagi K et al (2010) Ibudilast inhibits cerebral aneurysms by down-regulating inflammation-related molecules in the vascular wall of rats. Neurosurgery 66(3):551–559. https://doi.org/10.1227/01.Neu.0000365771.89576.77
Yang L et al (2018) Lectin microarray combined with mass spectrometry identifies haptoglobin-related protein (HPR) as a potential serologic biomarker for separating nonbacterial pneumonia from bacterial pneumonia in childhood. Proteomics Clin Appl 12(6):e1800030. https://doi.org/10.1002/prca.201800030
Zhang Z et al (2020) Gene correlation network analysis to identify regulatory factors in sepsis. J Transl Med 18(1):381. https://doi.org/10.1186/s12967-020-02561-z
Zhao XG et al (2019) miR-665 expression predicts poor survival and promotes tumor metastasis by targeting NR4A3 in breast cancer. Cell Death Dis 10(7):479. https://doi.org/10.1038/s41419-019-1705-z
Zheng XW et al (2016) Subcutaneous tuberculosis formation during FOLFIRI and bevacizumab treatment: a case report. Int J Colorectal Dis 31(4):943–944. https://doi.org/10.1007/s00384-015-2368-6
Zhou G et al (2019) NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 47(W1):W234-w241. https://doi.org/10.1093/nar/gkz240
Zhu Y et al (2021) Blocking SNHG14 antagonizes lipopolysaccharides-induced acute lung injury via SNHG14/miR-124-3p axis. J Surg Res 263:140–150. https://doi.org/10.1016/j.jss.2020.10.034
Acknowledgements
We sincerely acknowledge the GEO database for offering their platform and their contributions for uploading their valuable dataset.
Funding
Not applicable for that section.
Author information
Authors and Affiliations
Contributions
ZH, JK, JXL, and PC designed the study, acquired all datasets, analyzed the data, prepared the figure and table and wrote the main manuscript. JL and YH analyzed the data, collected the specimens and interpreted the data. QL, HC, NH, and TL supervised the project and revised the manuscript. XG: evaluated and guided the full text and granted final approval of the version to be submitted. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
No potential conflict of interest was reported by the author(s).
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, ZM., Kang, JQ., Chen, PZ. et al. Identifying the Interaction Between Tuberculosis and SARS-CoV-2 Infections via Bioinformatics Analysis and Machine Learning. Biochem Genet 62, 2606–2630 (2024). https://doi.org/10.1007/s10528-023-10563-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10563-x