
Abstract
One of the most prevalent sensorineural disorders, autosomal recessive non-syndromic hearing loss (ARNSHL) which can affect all age groups, from the newborn (congenital) to the elderly (presbycusis). Important etiologic, phenotypic, and genotypic factors can cause deafness. So far, the high genetic variability that explains deafness makes molecular diagnosis challenging. In Morocco, the GJB2 gene is the primary cause of non-syndromic hereditary deafness, while the existence of a variant in the LRTOMT gene is the second cause of this condition. After excluding these two frequently occurring GJB2 and LRTOMT variants, whole-exome sequencing was carried out in two Moroccan consanguineous families with hearing loss. As a result, two novel variants in the TMPRSS3 (c.1078G>A, p. Ala 360Thr) and FOXI1 (c.6C>G, p. Ser 2Arg) genes have been discovered in deaf patients and the pathogenic effect has been anticipated by several bioinformatics and molecular modeling systems. For the first time, these variants are identified in the Moroccan population, showing the population heterogeneity and demonstrating the value of the WES in hearing loss diagnosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the entire world, hearing loss is one of the most common sensory problems (Bakhchane et al. 2016). The majority of congenital hearing loss cases have a genetic cause, while it can also be caused by environmental factors (Salime et al. 2017). Hearing loss can result from a single gene pathogenic variant or from a combination of variants in various genes (Pan et al. 2022). Hereditary deafness can be syndromic and non-syndromic which 173 loci have been reported in syndromic presentations, and 131 genes have been discovered for non-syndromic deafness (https://hereditaryhearingloss.org/). Hereditary deafness study in the Moroccan population has been undertaken for a long time whose GJB2 and LRTOMT genes are the primary genetic causes of hereditary hearing loss in Morocco (Bakhchane et al. 2015).
FOXI1 (MIM* 601,093) (also known as FKHL10) belongs to the forkhead-box (FOX) transcription factors family, which is characterized by the FOX ~ 100 amino acid monomeric DNA-binding region (Moreno-Estrada et al. 2010). It encodes a transcription factor that binds to the SLC26A4 promoter region and controls the upstream regulation of the gene (Lin et al. 2019). Research done in Sweden in 1998 found that FOXI1 should be included as a candidate gene for deafness in humans since it was thought to be an early regulator required for the development of the cochlea and vestibule (Hulander et al. 1998). Additionally, FOXI1 has been linked to the control of vascular H+-ATPase proton pumps in the kidney, epididymis, and inner ear (Landa et al. 2013).
Transmembrane protease serine 3 (TMPRSS3) (MIM * 605511) an enzyme present in the stria vascularis of the cochlear duct, spiral ganglion neurons, and inner hair cells is involved in the growth and maintenance of the perilymph and endolymph in the inner ear (Fan et al. 2014). Whereas TMPRSS3 gene's role in the auditory system is unclear, its modification has been associated with non-syndromic hereditary hearing loss (Moon et al. 2021). TMPRSS3 variants are linked to two types of hereditary non-syndromic recessive sensorineural deafness (DFNB8 and DFNB10) and appear to alter the proteolytic activity of TMPRSS3 (Holder et al. 2021). In several populations from Palestine, Pakistan, Tunisia, Japan, China, Korea, and Turkey, TMPRSS3 pathogenic variants have been identified (Moon et al. 2021). However, less than 1% of White people with non-syndromic genetic deafness have nonetheless been shown to carry this gene (Moon et al. 2021).
In this study, using whole exome sequencing (WES), we present the identification of two homozygous variants in FOXI1 and TMPRSS3 of two Moroccan families affected by hearing loss.
Patients and Methods
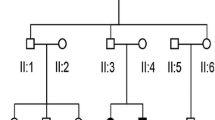
In this study, we enrolled two families with several deaf patients, family 1 and 2 (Fig. 1). All of the patients had severe non syndromic bilateral congenital hearing loss and came from consanguineous families. The study was approved by the Pasteur Institute of Morocco's committee on research ethics and was conducted in line with the Helsinki Declaration's procedure.

A Pedigree and electropherograms of the family 1 presenting the variant in FOXI1. B Pedigree and electropherograms of the family 2 harboring TMPRSS3 variant
From the patient's peripheral blood, genomic DNA was isolated by QIAamp DNA Blood Mini Kit Qiagen. WES was done at IntegraGen (Evry, France) on a single index case of each family (SF14 and SF144). IntegraGen configures a library, capture, sequence and perform a sequence variant detection and annotation. The libraries preparation is achieved by the Agilent Human exome V5 (50 Mb) capture kit, accompanied by paired end sequencing on an Illumina Hiseq2000. The capturing of the sequence was carried out in accordance with the manufacturer's instructions. An image analysis and base calling may be produced using the Illumina Real-Time Analysis Pipeline version 1.14 with the default settings.
The human genome reference sequence hg19 is used for the alignment of paired-end short reads (GRCh37). Based on the Illumina pipeline (CASAVA 1.8), bioinformatic analysis of sequencing data was performed. The selection of pathogenic candidate variants was conducted using succession of filters as known genes causing hereditary deafness were prioritized first. As the pedigree of the families suggested an autosomal recessive inheritance pathway, we screened homozygous and heterozygous compound missence, nonsense, frameshift and splice-site variants with allele frequencies < 0.01 using gnomAD, the 1000 genome project and dbSNP (build 132) databases. The pathogenicity of the remaining variants was examined using PolyPhen-2, Mutation Taster, and SIFT. The conservation analysis was done by Consurf (http://consurf.tau.ac.il/), a web server that analyzes the level of conservation to determine the functional sections of a protein. Sanger sequencing was used to confirm the variation of the candidate gene to validate the familial segregation with the disease phenotype.
To determine amino acid change effect on protein structure, a molecular modeling strategy was used. First, the UniProt database was used to download the FOXI1 (Q12951) and TMPRSS3 (P57727) amino acid sequences in FASTA format. Next, we needed the three-dimensional (3D) structure of the native protein. This was done by homology modeling using the ITasser server and Chimera software to obtain the two proteins mutated structure (Yang et al. 2015; Rodríguez-Guerra Pedregal and Maréchal 2018). After obtaining the PDB files, the energy minimization for all these 3D structures was achieved with the YASARA Energy Minimization Server (Krieger and Vriend 2014).
Results
The whole exome sequencing of the family 1 patient’s DNA was obtained with an average depth on target of 164.25 and a coverage of 99.4%.
WES results revealed a homozygous missense variant in FOXI1 (NM 012188.5: c.6C>G; p. Ser 2Arg), present in both the patients of family 1. Sanger sequencing confirmed that this variant segregated with the disease and the healthy parents were heterozygous (Fig. 1A, B). The variation was absent in all data base and it’s predicted to be disease causing by Mutation Taster (0.999), damaging by SIFT (0.002) and possibly damaging by Polyphen2 (0.909). The American College of Medical Genetics and Genomics (ACMG) has classified this variation as pathogen by the criteria PM1, PM2, PP2, PP3, and PP1 added after confirming segregation. On the report of the conservation scale findings, we can see that the p. Ser 2Arg substitution is located in a conserved location (8 on the conservation scale), and according to the neural-network algorithm, represents an exposed residue (Fig. 2A).

Conservation results of FOXI1 and TMPRSS3 proteins variant
For the family 2, the WES of its proband's DNA was performed with an average depth on target of 197.42 and a coverage of 99.2%. After the analysis of the WES results, a homozygous variant in the TMPRSS3 gene (NM_024022.3: c.1078 G>A) leading to a substitution of alanine in position 360 for threonine (p. Ala 360Thr) and referenced in gnomAD Exomes with a frequency of 3.98 × e−6 was selected as candidate variant. This variant was confirmed in homozygous state in the index patient and his sick brother, in heterozygous state in both unaffected parents and one brother and in wild type sate in two other brothers (Fig. 1B). Prediction tools distributed the variant as disease causing by Mutation Taster (0.999), damaging by SIFT (0.003) and probably damaging by Polyphen2 (0.979). This variant was classified as likely pathogenic according to the ACMG criteria PM1, PM2, PP3 and PP1 added after confirming the segregation. The color-coded conservation scale indicates that the site of the TMPRSS3 protein is in a conserved region (8 on the conservation scale) (Fig. 2B).
Using YASARA, we visualized the structures of wild-type and mutant proteins and discovered differences in the hydrophobic interactions and hydrogen bonds between amino acids. For the p. Ser2Arg substitution, serine had with the amino acid Glu 24, one hydrogen bond and one hydrophobic interaction, the change from serine to arginine lost the previous two bonds and built two hydrogen bonds with Ile 21 and hydrophobic interaction with Glu 24 and Phe4. The RMSD value between mutated and wild-type structures was 1.0259 A (Fig. 3).

Potential structural impact of the p.Ser2Arg FOXI1 amino acid change by molecular modeling. A Wild-type (Ser2). B Muted form (Arg2)
Interactions between amino acids showed that p. Ala360Thr amino acid change affected neither the hydrogen bonds nor the hydrophobic bonds, but the RMSD value between mutated and wild type structures was 0.8296 A (Fig. 4).

Potential structural impact of the p.Ala360Thr TMPRSS3 amino acid change by molecular modeling. A Wild-type (Ala360). B Muted form (Thr360)
Discussion
Whole exome sequencing is the most effective method for detecting causing disease variant. Two variations of the FOXI1 and TMPRSS3 genes were identified in this study in two consanguineous Moroccan families with non-syndromic hearing loss.
FOXI1, a member of the Forkhead family of transcription factors, is able to interact with the ATP6V0A4 promoter and directly regulate the expression of the ATP6V0A4 gene in the kidneys, inner ear, and epididymis (Klarov et al. 2022). Additionally, FOXI1 has been linked to the structuring of the distal nephron epithelium and adequate acid–base homeostasis in the kidney, which leads to distal renal tubular acidosis (dRTA). FOXI1’s variants have been associated to sensorineural deafness and dRTA (Table 1) (Enerbäck et al. 2018). In this study, we identified for the first time a homozygous FOXI1 variant (c.6C>G; p.Ser2Arg) in two patients with isolated hearing loss at the time of enrollment but no follow-up was possible to assess any evolution of a dRTA. This variant is located in a conserved residue of the FOXI1 protein and exposed according to the neural network algorithm According to the ACMG, this variant's classification is uncertain significance. However, the structural modeling analysis conducted to examine the effects of this causative variant on protein structure has able to demonstrate that the p.Ser2Arg variant affected the protein structure as it gained and lost interactions and bonds with surrounding amino acids. These changes may have affected the protein's integrity and stability evaluated by the high RMSD score between wild type and mutated structure. Most of FOXI1 variants were heterozygous causing a syndromic hearing loss, and located in a functional region between 121 and 211 named as Fork-head Domain, a conserved DNA-binding domain of about 100 amino acid residues, also known as a (winged helix) (Weigel et al. 1989). Although this domain is present in a variety of transcription factors, they all play a role in the early developmental decisions about cell fate during embryogenesis (Blomqvist et al. 2004). Enerbäck et al. reported two homozygous FOXI1 missense variants (p.Leu146Phe and p.Arg213Pro) that do not directly affect membrane transport proteins but may affect the highly preserved DNA binding domain. This significantly lowers the activation of several target genes, especially membrane transport proteins, which appropriate expression depends on FOXI1 connections causing a severe acidosis and deafness syndrome two novel loss-of-function variants (Enerbäck et al. 2018). The heterozygous variant c.214C>A (p.Pro72Thr) in exon1, was discovered in the Chinese population, according to (Yalan Liu), who claims that the transcription factor FOXI1 is a key player in deafness associated with EVA (Liu et al. 2020). Another variant, c.565G>A (p.Asp189Asn), was reported in this gene in the Chinese population as well. This pathogenic variant compromised the binding affinity of FOXI1 with the promoter region of SLC26A4 and was situated in the conserved fork-head DNA-binding domain of FOXI1 (Lin et al. 2019).
The type II transmembrane serine protease encoded by TMPRSS3 is essential for the morphological and functional development of the inner ear as well as the preservation of the perilymph and endolymph’s contents (Lee et al. 2023). TMPRSS3’s variants are associated with two different phenotypes which are a hearing loss linked to the prelingual DFNB10 and another linked to the post-lingual DFNB8 (Liang et al. 2022). To our knowledge, this is the first report of a TMPRSS3 missense variant, c.1078G>A; p.Ala360Thr, causing hearing loss in the Moroccan population. Analysis using Polyphen-2 software predicted him to be probably damaging, and it was also identified as damaging by analysis using SIFT and the site of the TMPRSS3 protein is in a conserved region. Moon and al summarized and examined all TMPRSS3 variants related to hearing loss between May 2000 and August 2021 (Moon et al. 2021). The p.Ala360Thr variant found in this study is located in exon 11 which is in a region between two pathogenic variants p.Glu347X and p.Tyr376Tyr in a serine protease domain (Ben-Yosef et al. 2001; Song et al. 2020). According to the data on variants overviews by (Gao et al.), missense mutations especially those that are present in the serine protease domain or adjacent to the active site, have severe effects (Gao et al. 2017). In the Chinese population the c.535G>A missense variant in exon 6 was found (c.535G>A) in a homozygous state, predicted as a disease-causing variant affecting the protein function by prediction algorithms (Fan et al. 2014). Following Next Generation Sequencing, Battelino et al. discovered a frame shift variation of TMPRSS3 c.208delC (p.His70Thrfs * 19) linked to hereditary non-syndromic deafness in Slovenia as it was carried in a homozygous state by a patient and his mother and as a digenic compound heterozygote by his father (Battelino et al. 2016). Furthermore, to evaluate the structural impact of variations, a molecular modeling analysis for FOXI1 and TMPRSS3 was carried out. Due to changes in hydrophobic and hydrogen interactions, the FOXI1 protein’s 3D structure has changed between its native and mutant states, while the TMPRSS3 protein has not changed between its native and mutated states.
Conclusion
Whole exome sequencing of two deaf Moroccan consanguineous families discovered novel variations on TMPRSS3 and FOXI1 in hearing-impaired patients. This study confirmed the population heterogeneity for this disease and demonstrates the value of the WES on molecular analysis.
Data Availability
Data will be provided by the authors upon request.
References
Bakhchane A, Charoute H, Nahili H et al (2015) A novel mutation in the TMC1 gene causes non-syndromic hearing loss in a Moroccan family. Gene 574:28–33. https://doi.org/10.1016/j.gene.2015.07.075
Bakhchane A, Bousfiha A, Charoute H et al (2016) Update of the spectrum of GJB2 gene mutations in 152 Moroccan families with autosomal recessive nonsyndromic hearing loss. Eur J Med Genet 59:325–329. https://doi.org/10.1016/j.ejmg.2016.05.002
Battelino S, Klancar G, Kovac J et al (2016) TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss. Eur Arch Otorhinolaryngol 273:1151–1154. https://doi.org/10.1007/s00405-015-3671-0
Ben-Yosef T, Wattenhofer M, Riazuddin S et al (2001) Novel mutations of TMPRSS3 in four DFNB8/B10 families segregating congenital autosomal recessive deafness. J Med Genet 38:396–400. https://doi.org/10.1136/jmg.38.6.396
Blomqvist SR, Vidarsson H, Fitzgerald S et al (2004) Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Investig 113:1560–1570. https://doi.org/10.1172/JCI20665
Cirello V, Bazzini C, Vezzoli V et al (2012) Molecular and functional studies of 4 candidate loci in Pendred syndrome and nonsyndromic hearing loss. Mol Cell Endocrinol 351:342–350. https://doi.org/10.1016/j.mce.2012.01.013
Enerbäck S, Nilsson D, Edwards N et al (2018) Acidosis and deafness in patients with recessive mutations in FOXI1. J Am Soc Nephrol 29:1041–1048. https://doi.org/10.1681/ASN.2017080840
Fan D, Zhu W, Li D et al (2014) Identification of a novel homozygous mutation, TMPRSS3: c.535G>A, in a Tibetan family with autosomal recessive non-syndromic hearing loss. PLoS ONE 9:e114136. https://doi.org/10.1371/journal.pone.0114136
Gao X, Huang S-S, Yuan Y-Y et al (2017) Identification of TMPRSS3 as a significant contributor to autosomal recessive hearing loss in the Chinese population. Neural Plast 2017:3192090. https://doi.org/10.1155/2017/3192090
Holder JT, Morrel W, Rivas A et al (2021) Cochlear implantation and electric acoustic stimulation in children with TMPRSS3 genetic mutation. Otol Neurotol 42:396–401. https://doi.org/10.1097/MAO.0000000000002943
Hulander M, Wurst W, Carlsson P, Enerbäck S (1998) The winged helix transcription factor Fkh10 is required for normal development of the inner ear. Nat Genet 20:374–376. https://doi.org/10.1038/3850
Klarov LA, Pshennikova VG, Romanov GP et al (2022) Analysis of SLC26A4, FOXI1, and KCNJ10 gene variants in patients with incomplete partition of the cochlea and enlarged vestibular aqueduct (EVA) anomalies. Int J Mol Sci 23:15372. https://doi.org/10.3390/ijms232315372
Krieger E, Vriend G (2014) YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30:2981–2982. https://doi.org/10.1093/bioinformatics/btu426
Landa P, Differ A-M, Rajput K et al (2013) Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med Genet 14:85. https://doi.org/10.1186/1471-2350-14-85
Lee SJ, Lee S, Han JH et al (2023) Structural analysis of pathogenic TMPRSS3 variants and their cochlear implantation outcomes of sensorineural hearing loss. Gene 865:147335. https://doi.org/10.1016/j.gene.2023.147335
Liang J, Yu Z, Wang Z et al (2022) A frameshift mutation of TMPRSS3 in a Chinese family with non-syndromic hearing loss. Front Pediatr 10:1032659. https://doi.org/10.3389/fped.2022.1032659
Lin Y-H, Wu C-C, Lin Y-H et al (2019) Targeted next-generation sequencing facilitates genetic diagnosis and provides novel pathogenetic insights into deafness with enlarged vestibular aqueduct. J Mol Diagn 21:138–148. https://doi.org/10.1016/j.jmoldx.2018.08.007
Liu Y, Wang L, Feng Y et al (2016) A new genetic diagnostic for enlarged vestibular aqueduct based on next-generation sequencing. PLoS ONE 11:e0168508. https://doi.org/10.1371/journal.pone.0168508
Liu Y, Wen J, Sang S et al (2020) Next-generation sequencing-based mutation analysis of genes associated with enlarged vestibular aqueduct in Chinese families. Eur Arch Otorhinolaryngol 277:3331–3339. https://doi.org/10.1007/s00405-020-06050-3
Moon IS, Grant AR, Sagi V et al (2021) TMPRSS3 gene variants with implications for auditory treatment and counseling. Front Genet 12:780874. https://doi.org/10.3389/fgene.2021.780874
Moreno-Estrada A, Aparicio-Prat E, Sikora M et al (2010) African signatures of recent positive selection in human FOXI1. BMC Evol Biol 10:267. https://doi.org/10.1186/1471-2148-10-267
Pan J, Ma S, Teng Y et al (2022) Whole-exome sequencing identifies genetic variants of hearing loss in 113 Chinese families. Clin Chim Acta 532:53–60. https://doi.org/10.1016/j.cca.2022.05.020
Pique LM, Brennan M-L, Davidson CJ et al (2014) Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ 2:e384. https://doi.org/10.7717/peerj.384
Rodríguez-Guerra Pedregal J, Maréchal J-D (2018) PyChimera: use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 34:1784–1785. https://doi.org/10.1093/bioinformatics/bty021
Salime S, Charif M, Bousfiha A et al (2017) Homozygous mutations in PJVK and MYO15A genes associated with non-syndromic hearing loss in Moroccan families. Int J Pediatr Otorhinolaryngol 101:25–29. https://doi.org/10.1016/j.ijporl.2017.07.024
Song MH, Jung J, Rim JH et al (2020) Genetic inheritance of late-onset, down-sloping hearing loss and its implications for auditory rehabilitation. Ear Hear 41:114–124. https://doi.org/10.1097/AUD.0000000000000734
Weigel D, Jürgens G, Küttner F et al (1989) The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 57:645–658. https://doi.org/10.1016/0092-8674(89)90133-5
Yang T, Vidarsson H, Rodrigo-Blomqvist S et al (2007) Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet 80:1055–1063. https://doi.org/10.1086/518314
Yang J, Yan R, Roy A et al (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. https://doi.org/10.1038/nmeth.3213
Acknowledgements
The authors are indebted to the families who contributed to this study. This Project was supported by the Institut Pasteur du Maroc (IPM), we also thanks Dr. Detsouli Mostafa for the clinical auditory investigation.
Funding
No funds, grants, or other support was received.
Author information
Authors and Affiliations
Contributions
Conceptualization: [Houria Abdelghaffar], [Abdelhamid Barakat] ; Formal analysis: [Imane Ait Raise], [Ghita Amalou] and [Amale Bousfiha]; Funding acquisition: [Abdelhamid Barakat]; Investigation: [Imane Ait Raise], [Ghita Amalou]; Methodology: [Imane Ait Raise], [Ghita Amalou], [Crystel Bonnet] and [Amina bakhchane] ; Resources: [Christine Petit] and [Abdelhamid Barakat]; Software: [Imane Ait Raise]; Writing – original draft: [Imane Ait Raise], [Majida charif]; Writing – review & editing: [Houria Abdelghaffar], [Crystel Bonnet], and [Abdelhamid Barakat].
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The genetic study was approved by the ethics committee for the biomedical research of Rabat.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
AitRaise, I., Amalou, G., Bakhchane, A. et al. Homozygous Missense Variants in FOXI1 and TMPRSS3 Genes Associated with Non-syndromic Deafness in Moroccan Families. Biochem Genet 62, 1914–1924 (2024). https://doi.org/10.1007/s10528-023-10515-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10515-5