
Abstract
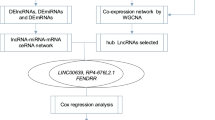
Long non-coding RNAs play crucial role in the tumorigenesis of lung adenocarcinoma (LUAD). However, the function of a large number of lncRNAs in LUAD has not been investigated yet. Weighted gene correlation network analysis (WGCNA) was applied to construct the co-expression module in the TCGA-LUAD cohort. Protein–protein interaction (PPI) network was used to explore the relationship of genes in the key module. The function of the key module on the prognosis in LUAD was analyzed using GO and KEGG analysis. Finally, we constructed the mRNA-lncRNA co-expression network in the key module to identify the hub lncRNAs that play crucial role in the prognosis in LUAD. The most highly expressed 2500 mRNAs and 2500 lncRNAs in the TCGA-LUAD cohort were clustered into 21 modules. After analyzing the correlation between the module and prognostic clinical traits, the Tan module, consisting of 130 genes, was selected as the key module on the prognosis in LUAD. And then, we found that genes in the key module were majorly enriched in ten multiple signaling pathways. Subsequently, we constructed the mRNA-lncRNA co-expression network based on the genes in the key module. Finally, we identified three lncRNAs and nineteen mRNAs that could be the promising prognostic biomarkers for LUAD. We identified three lncRNAs (MIR99AHG, ADAMTS9-AS2, and AC037459.2) and nineteen mRNAs as potential prognostic biomarkers in LUAD, which provided new insight for prognosis monitoring and therapy development in LUAD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer continues to be the leading cause of cancer death, and over 2.2 million patients were diagnosed with lung cancer and approximately 1.8 million patients death caused by lung cancer in 2020 worldwide (Sung et al. 2021). In China, over 87 thousand new cases and approximately 77 thousand new death caused by lung cancer in 2022, which made lung cancer still the cancer with the highest incidence and mortality in China (Xia et al. 2022). Lung adenocarcinoma (LUAD) is a subtype of non-small cell lung cancer, accounting for almost 40% of primary tumors. Owing to decreases in tobacco smoking, increased thoracoscopic surgeries and stereotactic radiation for early-stage disease, and better treatments for advanced-stage disease (Siegel et al. 2019; Boyer et al. 2017), the 5-year survival of lung cancer improved from 15.6% in 2011 to 19.4% in 2019. Even so, the outcome of patients with lung cancer is still very poor and there is an urgent need to identify novel effective prognostic biomarkers for patients with LUAD.
Non-coding RNA (ncRNA) is a kind of RNA with limited or without protein-coding ability. Approximately 97% of RNA transcribed from the human genome is ncRNA, suggesting crucial role of ncRNA in cells (Yan and Bu 2021). According to the length, shape and location, ncRNAs have been majorly divided into three classes, including microRNA (miRNA), long ncRNA (lncRNA), and circular RNA (circRNA). miRNA is a kind of small RNA functioning in posttranscriptional regulation of gene expression (Saliminejad et al. 2019). A large number of studies have demonstrated the powerful regulation of miRNA in cancer (He et al. 2020). Circular RNAs (circRNAs) are characterized by their covalently closed-loop structures without a 5′ cap or a 3′ poly(A) tail (Ju et al. 2022). An increasing number of studies have indicated that circRNA can be the potential target for various diseases including cancer (Najafi 2022). Long non-coding RNA (lncRNA) is a module of non-coding RNA longer than 200nt in length. Although lncRNA has been considered as the noise in the genome for the first time, an increasing number of studies have demonstrated that lncRNA played role in the regulation of multiple biological processes through interaction with other biological substances including sponging target miRNAs, regulation of several signaling pathways, transcription factors, and effector proteins (Sanchez Calle et al. 2018; Palazzo and Koonin 2020; Najafi et al. 2022; Najafi et al. 2021). Besides, many lncRNAs have been investigated as biomarkers in cancer. For example, lncRNA HOTAIR is one of the most important lncRNAs which is highly expressed in lung cancer and correlated with invasion, metastasis, and poor survival in patients with lung cancer (Loewen et al. 2014). Besides, many studies have also indicated that MALAT1 may serve as a diagnostic and prognostic biomarker with sufficient specificity and sensitivity in NSCLC (Li et al. 2018). Long Non-Coding RNA-TMPO-AS1 could predict poor prognosis in LUAD by upregulating the expression of STRIP2 (Wang et al. 2022). Besides, lncRNAs are stable and measurable in body fluids and thereby suitable for measurement via non-invasive procedures, thus, the identification of novel prognostic lncRNAs in LUAD is helpful for patients’ prognosis monitoring. However, there are still a large number of lncRNAs have not been evaluated in LUAD yet and whether there are more effective lncRNAs in the prediction of the prognosis in LUAD arouses our interest.
In this study, we identified the key module that contributed to the prognosis in LUAD using the weighted gene correlation network analysis (WGCNA) and further analyzed the genes in the key module to explore the hub genes that play crucial role in the prognosis in LUAD. Besides, we analyzed the functional mechanism of the key module to reveal how the key module affects the prognosis in LUAD. Finally, we identified the prognostic lncRNAs in LUAD, excepting to provide new clues for the development of clinical biomarkers in LUAD.
Materials and Methods
Data Acquisition
The RNA sequencing data and corresponding clinical information of 585 patients with lung adenocarcinoma were downloaded from the Cancer Genome Atlas database (https://portal.gdc.cancer.gov). Eliminating the sample without complete clinical information and survival data, a total of 514 LUAD samples were enrolled for further analysis. The RNA sequencing data of 514 LUAD samples were normalized by the “Limma” package with the Voom function.
Weighted Correlation Network Analysis (WGCNA)
Weighted correlation network analysis (WGCNA) is a systems biology method that aims to search for coexpressed gene module, and explore the correlation between gene networks and phenotypes of concern, as well as the core genes in the network (hub gene). Correlation networks facilitate network-based gene screening methods that can be used to identify candidate biomarkers or therapeutic targets. WGCNA achieves its goal based on two hypotheses: 1. Genes with similar expression patterns may be co-regulated, functionally related, or in the same pathway. 2. Gene networks conform to scale-free distribution. There are three steps to acquiring the coexpressed gene modules. Firstly, the outlier sample needs to be removed. Secondly, set a soft-threshold to judge whether significant co-expression was found between two genes. Thirdly, clustering genes into different modules and identifying the coexpressed gene modules (Zhang and Horvath 2005). We first analyzed the expression level of mRNA and lncRNA in the LUAD samples. The expression data of 5000 genes, including the top 2500 lncRNA and 2500 mRNA, in 514 LUAD samples were used to develop co-expression modules with the “WGCNA” package (Langfelder and Horvath 2008).
PPI Network
Protein–protein interaction (PPI) and the obtained networks are very important in the majority of biological functions and processes. PPI network analysis investigates the molecular mechanism of diseases and the development of new therapeutic targets through building up a PPI network (Athanasios et al. 2017). The selected module genes were analyzed by inputting them into the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://cn.string-db.org/). A combined score of ≥ 0.5 was considered as significant to construct a PPI network. The PPI network was displayed by Cytoscape software (v3.9.1) (Shannon et al. 2003). The hub genes were chosen based on a higher number of associations with other genes.
mRNA-lncRNA Co-expression Network
Gene co-expression analysis is a data analysis technique that helps identify groups of genes with similar expression patterns across several different conditions (Montenegro 2022). The mRNA‑lncRNA co-expression networks of significant modules were performed to explore the association between mRNA and lncRNA. Significant correlation pairs were applied to build the network in accordance with Pearson correlation coefficients. And the differential co-expression network was visualized using Cytoscape software (Version 3.9.1).
Pathway Enrichment Analysis
Gene ontology (GO) is a controlled vocabulary of terms to represent biology in a structured way. The terms are subdivided in three distinct ontologies that represent different biological aspects: Molecular Function (MF), Biological Process (BP), and Cellular Component (CC). Kyoto encyclopedia of genes and genomes (KEGG) is an encyclopedia of genes and genomes integrating eighteen databases categorized into systems, genomic, chemical and health information (Kanehisa and Goto 2000; Kanehisa et al. 2021). Assigning functional meanings to genes and genomes both at the molecular and higher levels is the primary objective of the KEGG database project (Kanehisa et al. 2017). KEGG pathway enrichment analysis and the GO analysis were performed to explore the possible mechanism of the key module in LUAD. We analyzed the enriched pathways of the genes in the key module using “ClusterProfiler” R package (Yu et al. 2012).
Statistical Analysis
Prognosis analysis was performed by Kaplan-Meier survival analysis using the “Survival” R package. The log-rank test was used to compare the difference between the two groups. A comparison of gene expression levels between the TCGA-LUAD cohort and the GTEX-normal cohort was performed using a t-test. All the statistical analyses were performed using the R (version 4.3.1) and a p-value<0.05 was considered a significant difference.
Results
Co-expression Modules Construction in LUAD
We first compared the expression level of all genes contained in the RNA sequencing data in the TCGA-LUAD cohort. The most highly expressed 2500 mRNAs and 2500 lncRNAs in the TCGA-LUAD cohort were selected for WGCNA analysis. And then, we clustered the LUAD samples to detect the outliner, and no sample was identified as outliner (Supplementary Figure 1). Thus, all 514 LUAD samples were used for WGCNA analysis. The basic clinical characteristics of 514 samples in the TCGA-LUAD cohort are shown in Table 1. The independence and the average connectivity degree of the co-expression modules were decided by power value (β) and scale R2 value. First, a set of soft-thresholding powers were plotted. When the power value was equal to 5, the scale R2 was up to over 0.8 (Fig. 1A). Therefore, we define the adjacency matrix using soft-thresholding with β = 5 to construct and identify distinct co-expression gene modules in LUAD. A cluster dendrogram of all selected genes was constructed based on a TOM-based dissimilarity measure. These identified co-expression modules were distributed in different colors (Fig. 1B). A total of twenty-one co-expression modules were identified and the number of genes in each module is shown in Table 2. Besides, we analyzed the correlation between these twenty-one modules using Pearson’s correlation analysis (Fig. 1C).

Identification of key module by WGCNA. A Analysis of the scale-free index for various soft-threshold powers (β). B Analysis of the mean connectivity for various soft-threshold powers. C Dendrogram of all differentially expressed genes clustered based on the measurement of dissimilarity (1-TOM). The color band shows the results obtained from the automatic single-block analysis. The heatmap shows the correlation between each module
Identification of Key Module in the Prognosis in LUAD
To identified the key module that was associated with the prognosis in LUAD, we selected the clinical traits that had significant prognosis value in LUAD using univariate Cox analysis and three clinical traits were significant factors for the prognosis in LUAD, including TNM stage, T, N, and M. And then, we analyzed the relationship between the co-expression modules and these three prognostic clinical traits. We found that the Yellow module was negatively correlated with the N (Pearson’s r = −0.2, p = 3e−06). The Tan module was positively correlated with both TNM stage (Pearson’s r = 0.19, p = 2e−05) and N (Pearson’s r = 0.2, p = 5e−06). The turquoise module was positively correlated with N (Pearson’s r = 0.2, p = 7e−06) (Fig. 2A). And then, we analyzed the correlation between gene significance (GS) and module membership (MM) in the co-expression module. As displayed in the scatterplots, we found that MM in the Tan module significantly correlated with TNM stage (Fig. 2B, r = 0.52, p = 2.3e−10) and N (Fig. 2C, r = 0.6, p = 4.6e−14), and genes with high module membership often had high gene significance, which suggested that hub genes of the co-expression Tan module tend to be highly correlated with selected clinical characteristics. Considering the correlation coefficient, p-value, and consistency between module-trait relationships plot and the scatterplot, we chose the Tan module for further analysis.

Identification of modules associated with the clinical traits of LUAD. A Heatmap of the correlation between the module and clinical traits of LUAD. B The relationship between the module membership in the Tan module and gene significance in group N. C The relationship between the module membership in the Tan module and gene significance in group tumor stage
Functional Enrichment Analysis
One hundred and thirty genes in the Tan module were used to perform both the KEGG pathway enrichment analysis and GO analysis. For KEGG pathway enrichment analysis, the top ten enriched pathways included Antigen processing and presentation, cell cycle, central carbon metabolism in cancer, Glycolysis/Gluconeogenesis, Glyoxylate and dicarboxylate metabolism, Spliceosome, Protein processing in endoplasmic reticulum, Tight junction, Biosynthesis of amino acids, and Carbon metabolism (Fig. 3A). The complete result of the KEGG pathway enrichment analysis is shown in Supplementary Table 1. For GO analysis, the Tan module genes were enriched in 50 BP pathways, nine CC pathways, and eight MF pathways (Supplementary Table 2). The most ten BP enriched pathways included protein folding, chaperone-mediated protein folding, protein refolding, mRNA catabolic process, ribonucleoprotein complex biogenesis, regulating of DNA biosynthetic process, RNA catabolic process, positive regulation of DNA biosynthetic process, response to unfolded protein, and chaperone cofactor-dependent protein refolding (Fig. 3A). The enriched CC pathways included ficolin-1-rich granule, chaperone complex, region of cytosol, proteasome complex, endopeptidase complex, secretory granule lumen, cytoplasmic vesicle lumen, and vesicle lumen (Fig. 3A). The enriched MF pathways included protein folding chaperone, unfolded protein binding, heat shock protein binding, ubiquitin protein ligase binding, ubiquitin-like protein ligase binding, misfolded protein binding, cadherin binding, and ATPase activity (Fig. 3A).

Analysis of genes in the Tan module. A Functional analysis using GO analysis and KEGG pathway enrichment analysis. B PPI network. C mRNA-lncRNA co-expression network
Association of Genes in the Tan Module
We constructed the PPI network based on the genes in the Tan module to explore the interaction between genes in the module. We found that five genes interacted with other genes in the Tan module strongly, including GAPDH, HSP90AA1, HSPA8, HSPA4, and CCT7 (Fig. 3B). These five genes were the hub genes in the Tan module, which might play crucial role in the prognosis in LUAD. And then, we further constructed the mRNA-lncRNA co-expression network in the tan module using the mRNA-lncRNA pairs with a correlation coefficient larger than 0.5 and a p-value less than 0.01. The result showed that eleven lncRNAs (AC092071.1, TYMSOS, AC037459.2, AL157394.1, MEF2C-AS1, MRI99AHG, AC008669.1, ADAMTS9-AS2, AL356599.1, AC004982.1, and AL031058.1) correlated with 72 mRNAs in the tan module (Fig. 3C).
Identification of Prognostic lncRNAs in LUAD
We identified novel prognostic indicators in LUAD based on the mRNA-lncRNA co-expression network. The lncRNAs and mRNAs in the network with both significantly different expression levels between the TCGA-LUAD cohort and the GTEX-normal cohort, and between the high and low expression groups were defined as potential prognostic biomarkers in LUAD. Finally, we found that three lncRNAs, including MIR99AHG, ADAMTS9-AS2, and AC037459.2, were significantly down-expressed in LUAD compared to normal samples, which were all associated with poor prognosis (Fig. 4). Besides, nineteen mRNAs, including AHSA1, SLC2A1, FKBP4, HMGA1, PSMD2, GAPDH, MCM7, ARPC1A, CCT7, TUBA1C, ACTG1, ATIC, MDH2, TIP1, CCT8, EIF2S2, PA2G4, EIF4A3, and SHMT2, were highly expressed in LUAD and associated with poor prognosis (Figs. 5, 6).

Identification of prognostic lncRNAs in LUAD. ****p-value < 0.0001

Kaplan-Meier survival curve of prognostic mRNAs in LUAD

Comparison of the expression level of prognostic mRNAs in the TCGA-LUAD cohort and the GTEX-normal cohort. **p-value < 0.01; ****p-value < 0.0001
Discussion
Cancer ranks as one of the deadliest diseases worldwide and lung cancer continues to be one of the most prevalent cancers with poor prognosis globally. The outcomes of patients with lung cancer remain poor even many new treatment have been developed clinically. Therefore, searching novel promising biomarker for lung cancer is very important to provide basis for prognosis management and therapy development in lung cancer. Nowadays, many lncRNAs have been claimed as useful biomarkers for the diagnosis and prognosis of cancer patients. However, the function of a large number of lncRNAs in lung cancer still needs to be further investigated. In this study, we identified three lncRNAs as the potential prognostic indicators for patients with LUAD using the WGCNA analysis including MIR99AHG, ADAMTS9-AS2, and AC037459.2. A study has demonstrated that MIR99AHG was significantly correlated with survival in lung squamous cell carcinoma (Ning et al. 2018). Besides, another study further revealed that the axis of MIR99AHG had significant diagnostic and prognosis prediction values in human lung cancer (Sweef et al. 2022) Our findings suggested that MIR99AHG might play role in LUAD by cooperating mRNA HMGA1, which provided new knowledge for a better understanding the ceRNA network in LUAD biology. In addition, some studies have also demonstrated that ADAMTS9-AS2 played inhibitory role in LUAD acting as a prognostic biomarker (Liu et al. 2021; Lin et al. 2021). Our findings were consistent with the results of these previous studies and provided more evidence to support that lncRNA ADAMTS9-AS2 could be a promising prognostic biomarker clinically. Moreover, we also found a new lncRNA AC037459.2 that have not been reported yet. We found that lncRNA AC037459.2 was down-regulated in LUAD and associated with poor prognosis, suggesting lncRNA AC037459.2 might be a promising prognostic indicator for LUAD. We further found that lncRNA AC037459.2 interacted with 28 mRNAs in LUAD, including some mRNAs with crucial role in LUAD. For example, SNRPB has been illuminated to promote tumorigenesis and negatively regulate cisplatin resistance in non-small cell lung cancer (Liu et al. 2019, 2021). TUBA1C and PSMD2 have been found to be closely correlated with tumor-infiltrating immune cells in LUAD (Bian et al. 2021; Zhao and Lu 2022). Many studies have been illuminated that EIF4A3 played essential role in lung cancer acting as a target for various lncRNA or circRNA (Zhang et al. 2022; Takahashi et al. 2021x; Yang et al. 2020; Xu et al. 2023). Therefore, our findings provided a new clue for better understanding the mechanism of tumorigenesis and development in LUAD.
LncRNA has limited or even no ability to code a protein. It is generally speaking that lncRNA regulates the expression of mRNA by sponging miRNA which is small RNA that can bind mRNA resulting in the inhibition of mRNA translation. Therefore, the ceRNA hypothesis has emerged to reveal the mechanism of RNA interaction. The lncRNA-mRNA axis is important to reveal the mechanism of lncRNA in LUAD. In this study, we found that the prognostic lncRNAs were interacting with a large number of mRNA, and nineteen of them were associated with poor prognosis in LUAD which were valuable to investigate further, including AHSA1, SLC2A1, FKBP4, HMGA1, PSMD2, GAPDH, MCM7, ARPC1A, CCT7, TUBA1C, ACTG1, ATIC, MDH2, TIP1, CCT8, EIF2S2, PA2G4, EIF4A3, and SHMT2. Although some of these nineteen mRNAs have been reported as prognostic biomarkers in LUAD, including SLC2A1 (Guo et al. 2020), FKBP4 (Meng et al. 2020), HMGA1 (Saed et al. 2022), PSMD2 (Zhao and Lu 2022), MCM7 (Wang et al. 2017), TUBA1C (Bian et al. 2021), ATIC (Niu et al. 2022), MDH2 (Ma et al. 2021), EIF4A3 (Qiu et al. 2022), and SHMT2 (Luo et al. 2021), the function of the other mRNAs has been reported in some of the other types of cancer, but not in LUAD. For example, overexpression of AHSA1 was associated with poor prognosis in hepatocellular carcinoma, breast cancer, and osteosarcoma (Li and Liu 2022; Shi et al. 2022; Shao et al. 2016). Besides, the prognostic relevance of CCT8 in multiple types of cancer has also been investigated (Liao et al. 2021; Liu et al. 2019). Our findings provided new clues for these mRNAs that might also be promising prognostic biomarkers in LUAD. Notably, we found that GAPDH was both the hub gene and prognostic indicator in LUAD. GAPDH is well-known as the housekeeping gene and widely used as an internal control in experiments on proteins, mRNA, and DNA. However, some studies have illuminated that GAPDH played important role in tumor progression by regulating energy metabolism. Our findings provided new clues that GAPDH might also be the prognostic indicator for patients with LUAD. In conclusion, we identified novel prognostic lncRNAs and mRNAs in LUAD, expecting to help the improvement of LUAD patients’ outcomes.
However, there are still some limitations in this study. For example, our findings were generated using the in-silicon analysis and validating our result with in-house cohort would be advantaged. Besides, the mechanism of the prognostic lncRNAs in LUAD has not been fully investigated. In the future, we will attempt to overcome these shortcomings with in-vivo and in-vitro experiments.
Conclusion
This study identified three lncRNAs (MIR99AHG, ADAMTS9-AS2, and AC037459.2) and nineteen mRNAs as potential prognostic biomarkers in LUAD, which provided new insight for prognosis monitoring and therapy development in LUAD.
Data Availability
The datasets generated and/or analyzed during the current study are available in the TCGA repository, https://portal.gdc.cancer.gov.
References
Athanasios A, Charalampos V, Vasileios T, Ashraf GM (2017) Protein-protein interaction (PPI) network: recent advances in drug discovery. Curr Drug Metab 18(1):5–10
Bian T, Zheng M, Jiang D, Liu J, Sun H, Li X et al (2021) Prognostic biomarker TUBA1C is correlated to immune cell infiltration in the tumor microenvironment of lung adenocarcinoma. Cancer Cell Int 21(1):144
Boyer MJ, Williams CD, Harpole DH, Onaitis MW, Kelley MJ, Salama JK (2017) Improved survival of stage I non-small cell lung cancer: a VA central cancer registry analysis. J Thorac Oncol 12(12):1814–23
Guo W, Sun S, Guo L, Song P, Xue X, Zhang H et al (2020) Elevated SLC2A1 expression correlates with poor prognosis in patients with surgically resected lung adenocarcinoma: a study based on immunohistochemical analysis and bioinformatics. DNA Cell Biol 39(4):631–44
He B, Zhao Z, Cai Q, Zhang Y, Zhang P, Shi S et al (2020) miRNA-based biomarkers, therapies, and resistance in Cancer. Int J Biol Sci 16(14):2628–47
Ju J, Song YN, Chen XZ, Wang T, Liu CY, Wang K (2022) circRNA is a potential target for cardiovascular diseases treatment. Mol Cell Biochem 477(2):417–30
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45(D1):D353–D61
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M (2021) KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 49(D1):D545–D51
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559
Li W, Liu J (2022) The prognostic and immunotherapeutic significance of AHSA1 in pan-cancer, and its relationship with the proliferation and metastasis of hepatocellular carcinoma. Front Immunol 13:845585
Li ZX, Zhu QN, Zhang HB, Hu Y, Wang G, Zhu YS (2018) MALAT1: a potential biomarker in cancer. Cancer Manag Res 10:6757–68
Liao Q, Ren Y, Yang Y, Zhu X, Zhi Y, Zhang Y et al (2021) CCT8 recovers WTp53-suppressed cell cycle evolution and EMT to promote colorectal cancer progression. Oncogenesis 10(12):84
Lin Z, Huang W, Yi Y, Li D, Xie Z, Li Z et al (2021) LncRNA ADAMTS9-AS2 is a prognostic biomarker and correlated with immune infiltrates in lung adenocarcinoma. Int J Gen Med 14:8541–55
Liu N, Wu Z, Chen A, Wang Y, Cai D, Zheng J et al (2019) SNRPB promotes the tumorigenic potential of NSCLC in part by regulating RAB26. Cell Death Dis 10(9):667
Liu P, Kong L, Jin H, Wu Y, Tan X, Song B (2019) Differential secretome of pancreatic cancer cells in serum-containing conditioned medium reveals CCT8 as a new biomarker of pancreatic cancer invasion and metastasis. Cancer Cell Int 19:262
Liu N, Chen A, Feng N, Liu X, Zhang L (2021) SNRPB is a mediator for cellular response to cisplatin in non-small-cell lung cancer. Med Oncol 38(5):57
Liu W, Luo W, Zhou P, Cheng Y, Qian L (2021) Bioinformatics analysis and functional verification of ADAMTS9-AS1/AS2 in lung adenocarcinoma. Front Oncol 11:681777
Loewen G, Jayawickramarajah J, Zhuo Y, Shan B (2014) Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol 7:90
Luo L, Zheng Y, Lin Z, Li X, Li X, Li M et al (2021) Identification of SHMT2 as a potential prognostic biomarker and correlating with immune infiltrates in lung adenocarcinoma. J Immunol Res 2021:6647122
Ma YC, Tian PF, Chen ZP, Yue DS, Liu CC, Li CG et al (2021) Urinary malate dehydrogenase 2 is a new biomarker for early detection of non-small-cell lung cancer. Cancer Sci 112(6):2349–60
Meng W, Meng J, Jiang H, Feng X, Wei D, Ding Q (2020) FKBP4 accelerates malignant progression of non-small-cell lung cancer by activating the Akt/mTOR signaling pathway. Anal Cell Pathol (Amst) 2020:6021602
Montenegro JD (2022) Gene co-expression network analysis. Methods Mol Biol 2443:387–404
Najafi S (2022) Circular RNAs as emerging players in cervical cancer tumorigenesis; a review to roles and biomarker potentials. Int J Biol Macromol 206:939–53
Najafi S, Ghafouri-Fard S, Hussen BM, Jamal HH, Taheri M, Hallajnejad M (2021) Oncogenic roles of small nucleolar RNA host gene 7 (SNHG7) long noncoding RNA in human cancers and potentials. Front Cell Dev Biol 9:809345
Najafi S, Khatami SH, Khorsand M, Jamali Z, Shabaninejad Z, Moazamfard M et al (2022) Long non-coding RNAs (lncRNAs); roles in tumorigenesis and potentials as biomarkers in cancer diagnosis. Exp Cell Res 418(2):113294
Ning P, Wu Z, Hu A, Li X, He J, Gong X et al (2018) Integrated genomic analyses of lung squamous cell carcinoma for identification of a possible competitive endogenous RNA network by means of TCGA datasets. PeerJ 6:e4254
Niu N, Zeng J, Ke X, Zheng W, Fu C, Lv S et al (2022) ATIC facilitates cell growth and migration by upregulating Myc expression in lung adenocarcinoma. Oncol Lett 23(4):131
Palazzo AF, Koonin EV (2020) Functional long non-coding RNAs evolve from junk transcripts. Cell 183(5):1151–61
Qiu M, Chen M, Lan Z, Liu B, Xie J, Li X (2022) Plasmacytoma variant translocation 1 stabilized by EIF4A3 promoted malignant biological behaviors of lung adenocarcinoma by generating circular RNA LMNB2. Bioengineered 13(4):10123–40
Saed L, Jelen A, Mirowski M, Salagacka-Kubiak A (2022) Prognostic significance of HMGA1 expression in lung cancer based on bioinformatics analysis. Int J Mol Sci 23(13):6933
Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, Ghaffari SH (2019) An overview of microRNAs: biology, functions, therapeutics, and analysis methods. J Cell Physiol 234(5):5451–65
Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T (2018) Emerging roles of long non-coding RNA in cancer. Cancer Sci 109(7):2093–100
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–504
Shao J, Wang L, Zhong C, Qi R, Li Y (2016) AHSA1 regulates proliferation, apoptosis, migration, and invasion of osteosarcoma. Biomed Pharmacother 77:45–51
Shi W, Qi L, You XB, Chen YC, Xu YL, Yu WB et al (2022) Identification of AHSA1 as a potential therapeutic target for breast cancer: bioinformatics analysis and in vitro studies. Curr Cancer Drug Targets 22(2):142–52
Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69(1):7–34
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–49
Sweef O, Yang C, Wang Z (2022) The oncogenic and tumor suppressive long non-coding RNA-microRNA-messenger RNA regulatory axes identified by analyzing multiple platform omics data from Cr(VI)-transformed cells and their implications in lung cancer. Biomedicines 10(10):2334
Takahashi S, Noro R, Seike M, Zeng C, Matsumoto M, Yoshikawa A et al (2021) Long non-coding RNA CRNDE Is involved in resistance to EGFR tyrosine kinase inhibitor in EGFR-mutant lung cancer via eIF4A3/MUC1/EGFR signaling. Int J Mol Sci 22(8):4005
Wang H, Zhou C, Su B, Teng G, Zheng Y, Zhou X et al (2017) MCM7 expression is correlated with histological subtypes of lung adenocarcinoma and predictive of poor prognosis. Int J Clin Exp Pathol 10(12):11747–53
Wang J, Yuan Y, Tang L, Zhai H, Zhang D, Duan L et al (2022) Long non-coding RNA-TMPO-AS1 as ceRNA binding to let-7c-5p upregulates STRIP2 expression and predicts poor prognosis in lung adenocarcinoma. Front Oncol 12:921200
Xia C, Dong X, Li H, Cao M, Sun D, He S et al (2022) Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl) 135(5):584–90
Xu Q, Zhao T, Han H, Fan J, Xie W (2023) EIF4A3 stabilizes the expression of lncRNA AGAP2-AS1 to activate cancer-associated fibroblasts via MyD88/NF-kappab signaling. Thorac Cancer 14(5):450–61
Yan H, Bu P (2021) Non-coding RNA in cancer. Essays Biochem 65(4):625–39
Yang H, Yang W, Dai W, Ma Y, Zhang G (2020) LINC00667 promotes the proliferation, migration, and pathological angiogenesis in non-small cell lung cancer through stabilizing VEGFA by EIF4A3. Cell Biol Int 44(8):1671–80
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–7
Zhang X, Lei G, Zhao K, Zhang X, Dang C (2022) CircTADA2A up-regulates MAPK8 by targeting MiR-214-3p and recruiting EIF4A3 to promote the invasion and migration of non-small cell lung cancer cells. Histol Histopathol. https://doi.org/10.14670/HH-18-529
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4:Article17
Zhao H, Lu G (2022) Prognostic implication and immunological role of PSMD2 in lung adenocarcinoma. Front Genet 13:905581
Acknowledgements
We would like to thank the support of Liaoning Health Industry Group Fukuang General Hospital in this work.
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
Can Zhao, Han Xu and Chang Liu designed the study and acquired the data. Can Zhao and Han Xu analyzed and interpreted the data and did the statistical analysis. The manuscript was drafted by Can Zhao and Han Xu. Chang Liu provided administrative, technical or material support. The subject was supervised by Chang Liu. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, C., Xu, H. & Liu, C. Identification of Novel Prognostic Long Non-coding RNAs in Lung Adenocarcinoma Using WGCNA Analysis. Biochem Genet 62, 264–280 (2024). https://doi.org/10.1007/s10528-023-10424-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10424-7