
Abstract
This study aimed to develop and validate a cuproptosis-related gene signature for the prognosis of gastric cancer. The data in TCGA GC TPM format from UCSC were extracted for analysis, and GC samples were randomly divided into training and validation groups. Pearson correlation analysis was used to obtain cuproptosis-related genes co-expressed with 19 Cuproptosis genes. Univariate Cox and Lasso regression analyses were used to obtain cuproptosis-related prognostic genes. Multivariate Cox regression analysis was used to construct the final prognostic risk model. The risk score curve, Kaplan–Meier survival curves, and ROC curve were used to evaluate the predictive ability of Cox risk model. Finally, the functional annotation of the risk model was obtained through enrichment analysis. Then, a six-gene signature was identified in the training cohort and verified among all cohorts using Cox regression analyses and Kaplan–Meier plots, demonstrating its independent prognostic significance for gastric cancer. In addition, ROC analysis confirmed the significant predictive potential of this signature for the prognosis of gastric cancer. Functional enrichment analysis was mainly related to cell–matrix function. Therefore, a new cuproptosis-related six-gene signature (ACLY, FGD6, SERPINE1, SPATA13, RANGAP1, and ADGRE5) was constructed for the prognosis of gastric cancer, allowing for tailored prediction of outcome and the formulation of novel therapeutics for gastric cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is a widespread malignancy ranked 5th in the world’s top ten most common cancers (Ferlay et al. 2021). It remains a significant malignancy globally, with over a million newly diagnosed cases in 2020 (Sung et al. 2021) and although the burden of GC has decreased in China, the number of new cases will reach 509,421 and the deaths will be approximately 400,415 in 2022 (Xia et al. 2022). Despite the rapid improvement of surgical and adjuvant therapeutic technologies, the overall survival (OS) rates for patients with GC continue to be relatively low and vary greatly around the globe. For illustration, the five-year survival rate in the United States is 31%, 26% in Europe, and 19% in the United Kingdom (Allemani et al. 2018). GC is a tremendously heterogeneous malignancy, therefore the conventional TNM classification is difficult to fully cover the indefinite genetic features and further research is required to identify helpful prognostic indicators and potential treatment targets.
Copper is a crucial trace element in the body and a component of numerous compounds such as cytochrome oxidase, catalase, and ceruloprotein (Allemani et al. 2021; Vallet and Ricard-Blum 2019; Fujieda et al. 2020; Katsuyama et al. 2021; Ariöz and Wittung-Stafshede 2018), which promotes the transfer and utilization of iron in tissues (Myint et al. 2018). Moreover, cancer cells have a higher demand for copper (Shanbhag et al. 2021), with higher copper concentrations in the serum and tumor tissues of cancer patients than in healthy individuals (Lelièvre et al. 2020). Copper has been implicated in cancer progression (Luca et al. 2019; Silva et al. 2022) and may trigger cell death, such as autophagy and apoptosis via the production of reactive oxygen species, inhibition of the proteasome, and antiangiogenesis (Jiang et al. 2022) Multiple tightly controlled programmed cell death methods have been identified including pyroptosis, apoptosis, ferroptosis, necroptosis (Bock and Tait 2020), and more recently, cuproptosis. Elesclomol administration did not trigger caspase-3, an apoptosis marker, and inhibiting apoptosis or other defined pathways of programmed cell death could not inhibit copper-induced cell death, demonstrating that there is a difference between copper-induced cell death and other known pathways of cell death (Zheng et al. 2022). Elesclomol has been shown to degrade ATP7A resulting in copper-reliant ferroptosis in cancer cells of the colorectum (Gao et al. 2021) and Xia et al. found that in GC, a novel Schiff base coordinated copper (II) compound promotes apoptosis and prevents tumor development (Xia et al. 2019). Disulfiram/copper also promotes anticancer activity in GC via the NPL4 and ROS/MAPK pathways (Liu et al. 2022). However, the value of copper and copper metabolism-related genes in the diagnosis and prognosis of GC needs to be investigated further.
This study aimed to develop a prognostic gene signature for GC using data from The Cancer Genome Atlas (TCGA) and previously identified cuproptosis-related genes. The properties of the gene signature in the tumor microenvironment were investigated using GO, KEGG, and GSEA, therefore, the developed prognostic assessment model may deepen the intrinsic relationship between GC and cuprotosis.
Materials and Methods
Data Acquisition
The corresponding clinical information and gene expression of GC samples were downloaded from the University of California Santa Cruz (UCSC) Xena browser (https://xenabrowser.net/). The Wilcox test was used to identify differentially expressed genes (DEGs) between the tumor and normal groups, which is suitable for the analysis of large samples and TPM data. The criteria for DEGs were log Fold Change (LogFC) absolute value > 1 and p < 0.05. The UCSC database was accessed on August 1, 2022, to obtain all the data in HTseq-TPM format. The clinical information of all samples was also downloaded including age, sex, TNM stage, AJCC stage, progression-free survival (PFS) time, OS time, and disease-specific survival (DSS) time. In total, the transcriptome data of 450 tissues (36 normal tissues and 414 GC tissues) and 315 patients with complete survival data were retrieved.
Nineteen key genes involved in cancer cuproptosis have been proposed including ATP7B, ATP7A, DBT, CDKN2A, DLD, DLAT, FDX1, DLST, GLS, GCSH, LIPT1, LIAS, MTF1, LIPT2, NLRP3, NFE2L2, PDHB, PDHA1, and SLC31A1 (Tsvetkov et al. 2022; Emami et al. 2022; Deng et al. 2022). Pearson’s correlation analysis was performed between the 19 genes and DEGs to identify cuproptosis-related genes according to the correlation coefficients (Cor Pearson > 0.4) and p values (p < 0.05).
Identification and Validation of the Cuproptosis-Gene Signature
Three hundred fifty-one patients were assigned randomly to the training group (n = 176) with the R package “caret” and validation group (n = 175). Using the chi-square test, the baseline data of participants in the validation and training groups were compared to assess if the data in the two groups were uniform. A Cox regression model of the cuproptosis-related genes was constructed using the training group results. All cuproptosis-related genes underwent overall survival-based univariate Cox regression analysis, then potential prognostic DEGs were screened. The Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis was performed on potential prognostic genes to avoid overfitting the model. When the cross-validation error of LASSO regression is small, the list of prognostic genes corresponding to the best penalty parameter is obtained. Multivariate Cox regression analysis of the LASSO prognostic genes was performed to determine the final prognostic model. Each patient’s risk score was calculated using the formula: risk score = βmRNA1*ExprmRNA1+βmRNA2*ExprmRNA2+…+βmRNAn*ExprmRNAn. Expr represented the expression value of each gene, and β was the estimated coefficient value obtained from the multivariate Cox regression model for this gene.
Prognostic Risk Signature Construction
Based on the median risk score of the training group, subjects in the validation and training groups were grouped into low-risk (below the median risk score) or high-risk (above the median of the risk score). In the entire cohort, validation, and training groups, the risk score was defined as an independent prognostic predictor in GC patients by multivariate and univariate Cox regression analyses. Each subject’s risk score and disease outcome were visualized by plotting the risk score curve, with Kaplan–Meier survival curves used to assess whether there were differences in PFS, DSS, and OS between patients in the high- and low-risk groups. The receiver operating characteristic (ROC) curve and the area under the ROC curve (AUC) were used to assess the risk score and the predictive power of each clinical characteristic for OS. Finally, PCA analysis was used to downscale the three sets of genes (DEGs, cuproptosis genes, cuproptosis-related genes, and risk model genes) in low- and high-risk patients to visualize the discriminative power of the risk scores.
Functional Enrichment Analysis
Differential analysis was performed on the low-risk groups (n = 166) and high-risk groups (n = 167) in the entire cohort, |LogFC|> 0.6 and p < 0.05 were defined as risk-associated DEGs. KEGG and GO enrichment analyses of risk-associated DEGs were performed using the “ClusterProfiler” and “ggplot2” packages in R, with a p value < 0.05. The core genes in DEGs were screened out by constructing a PPI network. GSEA was established to evaluate the regulatory impact of high and low-risk groups on the pathway. GSEA, using the GSEA software (GSEA 4.1.0), was employed for the entire cohort analysis. The h.all.v7.5.symbols.gmt data set in the MsigDB database was used as control data, the random sample permutations were set at 1000, and the nominal p value (NOM p value) < 0. 05, False discovery rates (FDR) < 0. 25 and normalized enrichment score |NES|> 1 were set as the significance threshold.
Statistical Analysis
Statistical analysis was performed by using R (Version 3.6.3). Cox regression was utilized to assess each gene for the prognosis of GC by calculating the hazard ratio (HR) and 95% CI. Log-rank tests were used to evaluate the OS, DSS, and PFS of the three groups (entire, training, and validation). The Pearson correlation test was utilized for correlation analysis. The Ggplot2 R package performed the visualization of the data. Statistical significance was set at p < 0.05.
Results
Data Acquisition
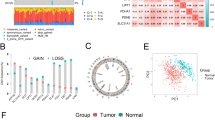
The transcriptome data of 450 tissues were retrieved (36 normal tissues and 414 GC tissues, with a total of 2349 (1980 upregulated genes and 369 downregulated genes, Supplementary Table 1). The DEGs were identified based on the screening criteria of |LogFC|> 1, p < 0.05. All genes are shown in the volcano plot in Fig. 1A and the expression of the risk score genes is shown in Fig. 1B.

Construction process of COX risk model. A Volcano plot of DEGs. B Heatmap about the the expression levels of COX risk model genes in tumor tissues and normal tissues. C The upset plot shows the number and cross-linking of co-expressed genes of the 19 cuproptosis genes. D Distribution of LASSO coefficients of the 70 potential prognostic cuproptosis-related genes in training group. E The cross-validations curve of Laaso regression shows the best penalty parameter value in training group. F The 6 prognostic cuproptosis-related genes signature constructs the COX risk model in training group. G The correlation between 6 genes Signature and 19 cuproptosis genes. FC, fold change, *p < 0:05; **p < 0:01; ***p < 0:001. DEGs: differentially expressed genes
Grouping and Establishment of the Cox Prognostic Model
Three hundred fifty-one patients were assigned randomly to the training group (n = 176), with the R package “caret” and validation group (n = 175). The clinicopathological information of 351 GC patients is shown in Table 1 and there was no significant difference in the clinical characteristics between groups. Correlation analysis was performed between the 19 cuproptosis genes and 2349 DEGs based on the correlation coefficient criterion > 0.4 and p < 0.05 to identify 1314 potential cuproptosis-related genes (Fig. 1C, Supplementary Table 2). Univariate Cox regression analysis of all cuproptosis-related genes identified 70 (Supplementary Table 3) genes with potential prognostic value, which were then analyzed using a LASSO regression model based on OS (Fig. 1D). The result of LASSO regression revealed that the best penalty parameter corresponds to 13 prognostic genes (Fig. 1E, Table 2). The obtained 13 genes were again subjected to multivariate Cox regression analysis showing that six cuproptosis-related genes were independent prognostic factors for GC patients (Fig. 1F). These six genes were therefore the final genes for constructing the prognostic model. The correlation between the six gene signatures and 19 cuproptosis genes is shown in Fig. 1G. The risk score was calculated as follows: (− 0.385*ExpressionSPATA13)+0.515*ExpressionACLY+(− 0.681*ExpressionRANGAP1)+(− 0.419*ExpressionADGRE5)+(0.495*ExpressionFGD6)+(0.225*ExpressionSERPINE1).
Validation of the Cox Risk Model
The risk scores were calculated for each patient based on the prognostic model relative to the training group median risk score (risk score = 1.004) and the participants were classified into high and low-risk groups (risk score higher or no more than the median risk score). The distribution of outcome status, gene profiles, and risk scores for the six-gene signature in the entire, validation and training groups are shown in Fig. 2. As exhibited in Fig. 2A–C, significantly more incidents occurred in the high-risk group compared to the low-risk group.

Risk scores, risk curves and 6 genes signature heatmaps of GC patients in all cohorts. The distribution trend of risk score, OS time, PFS time, DSS time in entire group (A), in training group (B), and in validation group (C). Prognostic signature signal heatmaps in entire cohort (D), in training group (E), and in validation group (F). OS, overall survival; PFS, progression-free survival; DSS, disease-specific survival
The heatmaps show that ACLY, FGD6, and SERPINE1 were overexpressed in high-risk cases, while SPATA13, RANGAP1, and ADGRE5 were downregulated (Fig. 2D–F). In addition, Kaplan–Meier curves showed that high-risk subjects had lower OS, PFS, and DSS (Fig. 3A–C, D–F, and G–I, respectively). Furthermore, the prognostic accuracy of this Cox model was assessed using ROC curve analysis. The AUC in the entire cohort, training group, and validation group was 0.705, 0.719, and 0.715, respectively, higher than other baseline clinical data (Fig. 4A–C). Interestingly, the risk score time-dependent survival ROC curve revealed the good predictive power of 1-, 3-, and 5-years survival compared to other clinical data (Fig. 4D–F).

The differences in survival time of GC patients between high- and low-risk groups. The OS, PFS and DSS time KM curve in entire group (A, D, G), in training group (B, E, H), and in validation group (C, F, I). The differences between the high- and low-risk groups were measured by log-rank. KM, Kaplan–Meier

Validating the prognostic predictive power of the COX risk models. ROC curve of the COX risk model and clinical characteristics in entire group (A), in training group (B), and in validation group (C). ROC curves of risk models predict 1-, 3-, and 5-year OS rates in entire group (D), in training group (E), and in validation group (F). PCA analysis of DEGs (G), cuproptosis genes (H), cuproptosis-related genes (I), and 6 genes Signature (J) in GC patients. ROC, receiver operating characteristic; AUC, area under ROC curve; PCA, principal component analysis; DEGs, differentially expressed genes
PCA analysis was used to investigate the distribution of high- and low-risk groups, showing that compared to DEGs(Fig. 4G) cuproptosis genes (Fig. 4H), and cuproptosis-related genes (Fig. 4I), the six-gene signature can more clearly divide all patients into two risk groups (Fig. 4J).
The Risk Score Independent Prognostic Value
Using multivariate and univariate Cox regression analyses, the risk score was evaluated as an independent prognostic factor compared to several clinicopathological data (age, grade, N stage, T stage, M stage, risk scores, and gender). Univariate Cox analysis demonstrated statistically significant differences between age, risk score, N stage, T stage, and M stage in the entire group (Fig. 5A). In multivariate Cox analysis, the T stage was culled (Fig. 5B). In univariate and multivariate Cox regression analyses, age and the risk score were significantly linked to OS time in the training group (Fig. 5C, D). In the validation group, risk score, T stage, and N stage correlated with OS in univariate Cox regression analyses (Fig. 5E). Multivariate analysis showed that gender was also associated with OS in addition to the univariate inclusions (Fig. 5F). Furthermore, we confirmed that the risk score correlated with OS in the three groups. p < 0.05 was considered a statistically significant difference.

Risk score from COX risk model is an independent predictor of prognosis in GC patients. Univariate and Multivariate COX analysis of risk score and clinical characteristics in entire group (A, B), in training group (C, D), and in validation group (E, F)
KEGG and GO Analyses
DEGs between high- and low-risk groups were used for functional enrichment analysis. In total, 691 genes were upregulated and 107 genes were downregulated (Fig. 6A, B, Supplementary Table 4). Additionally, we completed the protein–protein interaction networks (PPI) analysis of the DEGs based on the STRING database (https://string-db.org/), and Fig. 6C shows the core genes with a degree value greater than 30. According to GO enrichment analysis, the biological processes included extracellular matrix organization, muscle system process, extracellular structure organization, and transmembrane receptor protein serine (cellular component contains cell–cell junction, collagen-containing extracellular matrix, endoplasmic reticulum lumen, and contractile fiber). The molecular functions included glycosaminoglycan binding, heparin-binding, extracellular matrix structural constituent, and sulfur compound binding (Fig. 6D). Analysis of KEGG pathways revealed that the DEGs participated in cancer-related pathways, for instance, focal adhesion, protein digestion and absorption, the PI3K-Akt signaling pathway, vascular smooth muscle contraction, and proteoglycans in cancer (Fig. 6E).

Risk score-related enrichment analysis in GC patients. A, B The volcano plot and heatmap present the difference analysis results between high and low-risk groups. C PPI analysis shows the core genes with a degree value greater than 30 of the DEGs. GO analysis (D) and KEGG analysis (E) results of risk score-related differentially expressed genes. GO, gene ontology, KEGG, Kyoto encyclopedia of genes and genomes
GSEA Analyses
GSEA was utilized to determine the pathway enrichment characteristics in samples from the full cohort of high and low-risk groups, with ten signaling pathways meeting the screening criteria (Fig. 7A–J). Five of these pathways were activated in the high-risk group: focal adhesion, neuroactive ligand-receptor interaction, complement and coagulation cascades, vascular smooth muscle contraction and calcium signaling pathway, and, and five signaling pathways were activated in the low-risk group: base excision repair, DNA replication, proteasome signaling pathway, aminoacyl tRNA biosynthesis and pyrimidine metabolism. It is speculated that the disparity between the low- and high-risk groups is related to the pathogenesis of GC.

Risk score-related GSEA analysis in GC patients. GSEA analysis according to high-risk groups (A–E) and low-risk groups (F–J) of GC patients. GSEA, gene set enrichment analysis
Discussion
A cuproptosis-related six-gene signature for the prognosis of GC was developed using UCSC database mining. First, 1314 cuproptosis-related genes were attained, then the GC patients were randomly assigned to validation and training groups. Using LASSO regression and the Cox regression model, prognostic cuproptosis-related genes were confirmed based on the training group. The risk score was then utilized to classify the patients as low or high risk, with a statistically significant difference in OS, DS, and PFS between the low- and high-risk groups. The prediction ability of the gene signature was indicated by AUC and the Cox analysis indicated that the risk score was an independent predictor of GC. PCA analysis intuitively discriminated between high- and low-risk groups. The six-gene signature was validated in the entire cohort, with GSEA, GO and KEGG analyses revealing the signature-related characteristics and pathways.
Copper is an essential trace element and a transition metal that is a crucial component for all living organisms. The significance of physiological copper was first recognized in 1928 (Jiang et al. 2022) and the disturbance of copper ions can lead to a variety of pathologies (Balsano et al. 2018). Furthermore, copper ions are involved in cancer progression, with serum copper levels in some cancers positively correlating with cancer grade (Lelièvre et al. 2020) and copper can induce cell death (Jiang et al. 2022). Recently, intracellular copper accumulation has been shown to induce the accumulation of lipoylated proteins of the mitochondria as well as the instability of Fe-S cluster proteins, resulting in a unique type of cell death called cuproptosis (Tang et al. 2022). There are three mechanisms involved in cuproptosis. First, Cu-mediated Fenton reaction, oxidative stress, or depletion of antioxidants result in higher ROS levels, thus promoting mitochondrial dysfunction and leading to apoptosis (Aboelella et al. 2021; Hao et al. 2021; Ngamchuea et al. 2016). Second, the inhibition of the proteasome may stimulate the entry of cytochrome c into the cytoplasm as well as activate the caspase cascade, further triggering tumor cell apoptosis (Gałczyńska et al. 2020). Cu (II) ions suppress the proteasome by directly binding and producing partial redox effects in cell-free conditions or HeLa cells (Santoro et al. 2016). Third, copper depletion inhibits the development of new blood vessels that supply the tumor tissue with nutrients. Copper deficiency can shut down the “angiogenic switch,” halt the proliferation of the endothelial cells and arrest the cell cycle in the G0 phase (Narayanan and Natarajan 2018).
However, there are very few studies of cuproptosis in GC. In this paper, a prediction model related to cuproptosis was established and verified through data mining, and the six genes related to cuproptosis were ACLY, FGD6, SERPINE1, SPATA13, RANGAP1, and ADGRE5.
ATP-citrate lyase (ACLY), an enzyme that produces acetyl-CoA from citrate, is the initial rate-regulating enzyme in lipid synthesis (Wen et al. 2019), facilitating metastasis of colon cancer cells by CTNNB1. Studies have shown that the temporal and spatial control of acetyl-CoA production by ACLY is involved in the DNA repair pathway (Sivanand et al. 2017). Copper may, directly and indirectly, interfere with the synthesis of acetyl-CoA and the formation of biological CH4 (Abdel Azim et al. 2019), so ACLY is closely related to copper metabolism. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis (Carrer et al. 2019) and ACLY is a possible independent biomarker for breast cancer recurrence prediction (Chen et al. 2020). SERPINE1 (the Serpin family E member 1), a serine proteinase inhibitor, is a key regulator of extracellular matrix remodeling. Elevated SERPINE1 expression promoted cellular invasiveness in mesenchymal lung cancer cells (Kong et al. 2021). By regulating VEGFA expression, SERPINE1 substantially contributed to the proliferation and apoptosis of TNBC cells (Zhang et al. 2020). Furthermore, SERPINE1 modulates directional movement and cell-substrate adhesion of GBM cells, and its expression is controlled by TGF signaling (Seker et al. 2021). In gastric adenocarcinoma, SERPINE1 promotes tumor cell proliferation, migration, and invasion by controlling EMT (Yang et al. 2019).
ADGRE5/CD97 is a class II TM7 receptor protein belonging to the epidermal growth factor seven-transmembrane (EGF-TM7) family. Numerous studies have implicated CD97 in tumor migration, dedifferentiation, metastasis, and invasiveness. The overexpression of ADGRE5/CD97 in cervical cancer is associated with tumor aggressiveness and it is an indicator of a poor prognosis in cervical cancer (He et al. 2019). ADGRE5 is a crucial regulator of acute myeloid leukemia stem cell function (Martin et al. 2019) and in hepatocellular carcinoma, ADGRE5 increases tumor aggressiveness via G protein-coupled receptor-mediated signaling (Yin et al. 2018).
Faciogenital dysplasia6 (FGD6) is a member of the 7 FGD family of proteins. FGD6 contains two pleckstrin homologous structural domains, the FYVE and the Rho guanine nucleotide exchange factor (GEF) structural domain, which are associated with the PM of the fold and are distributed at the boundaries of the fold (Steenblock et al. 2014). FGD6 can promote tumor epithelial mesenchymal transition by activating CDC42 which belongs to the Rho family of small GTPases (Maldonado and Dharmawardhane 2018). It has been shown that FGD6 promotes proliferation, macrophage increase and tumor growth in pancreatic ductal cell carcinoma (Zhang et al. 2022).
Spermatogenesis-associated protein 13 (SPATA13) is a guanine nucleotide exchange factor (GEF) expressed primarily in discrete brain regions and the extended central amygdala (CeA) in adults (Waseem et al. 2020). This suggests that SPATA13 may play an important role in brain function and output (Bourbia et al. 2019). SPATA13 regulates cell migration and adhesion (Bhattacharya et al. 2022). Khan et al. showed that fusion mutated SPATA3 is associated with highly aggressive meningiomas (Khan et al. 2020). Methylation of SPATA13 may be a biomarker for BRCA1-like breast cancer risk and help prevent and target this cancer subtype.
RAN GTP hydrolase activating protein 1 (RANGAP1), located on the nuclear pore complex (NPC) is closely related to the function of RNA (Lange et al. 2021). RANGAP1 and RANBP1 are overexpressed in melanoma and promote apoptosis through the ERK1/2 pathway (Audia et al. 2023). Other studies suggest that RanGAP1 deletion may lead to chromosome destabilization and promote the development of osteosarcoma. Recent study showed that RanGAP1 deletion may lead to chromosome destabilization and promote the development of osteosarcoma (Gong et al. 2023).
The KEGG pathway enrichment and GO annotation revealed that DEGs are mainly involved in extracellular structure organization, extracellular matrix organization, cell–cell junctions, collagen-containing extracellular matrix, glycosaminoglycan binding, and an extracellular matrix structural constituent. The extracellular matrix and tumor cells interact to determine the biological behavior of the tumor, that is, growth, infiltration, metastasis, etc. (Karamanos et al. 2021). The KEGG pathway analysis showed that cuproptosis-related genes are primarily enriched in focal adhesion, vascular smooth muscle contraction, PI3K-Akt signaling pathway, proteoglycans in cancer, and protein digestion and absorption. The PI3K/Akt signaling pathway is a key cellular signaling pathway involved in cell development, proliferation, metabolism, size, and motility (Alzahrani 2019), and activated PI3K/AKT signaling promotes GC development (Wang et al. 2019). The PI3K pathway is activated in ovarian cancer contributing to enhanced cell chemoresistance and survival (Huang et al. 2020). Focal adhesions are huge macromolecular assemblages implicated in several pathological and cellular processes, including polarization, migration, and the genesis of metastatic cancer (Schumacher et al. 2022). Lin et al. verified a gene signature related to a novel focal adhesion that can be utilized for the prognosis of HCC, providing a possible treatment option (Lin et al. 2021). Activated focal adhesion signaling regulates cell proliferation and tumor growth of CRC (Lin et al. 2022). Proteoglycans (PGs) constitute the majority of ECM macromolecules and interact with numerous growth factors, cytokines, adhesion molecules, cell surface receptors, glycoproteins, and enzymes to affect cell behavior and matrix characteristics, as well as the proliferation, angiogenesis, invasion, and metastasis of cancer cells (Wei et al. 2020). From the above results, the functional enrichment of copper death-related genes mainly affects the ECM. Complex and regular interactions with matrix molecules are spatially and temporally controlled to influence cell behavior and phenotypic via the structure of the ECM. This lack of tissue homeostasis has been associated with several cancer hallmarks, such as metabolic reprogramming (Park et al. 2020). The role of ECM, as a dynamic structure, in the development of cancer is essentially mediated by its constituents, the dysregulated feedback between cellular constituents and their local TEM, and the matrix’s biomechanical and biochemical features (Wei et al. 2020). Inevitably, there are some limitations to our research. To evaluate the efficacy of the signature of cuproptosis-related genes, we employed the UCSC validation group and the complete group. More extra patients can increase the model’s dependability. Additional experimental research is necessary to elucidate the molecular processes of cuproptosis-related genes.
Conclusions
We constructed a scoring model based on six genes associated with cuproptosis. This novel model may offer new research tools for investigating the causes of cuproptosis and provide personalized prognoses for GC patients.
Data availability
The following publicly available datasets were used in this study: https://portal.gdc.cancer.gov/projects/TCGA-STAD; University of California Santa Cruz (UCSC) Xena br er (https://xenabrowser.net/).
References
Abdel Azim A, Rittmann SKR, Fino D, Bochmann G (2019) The physiological effect of heavy metals and volatile fatty acids on Methanococcus maripaludis S2. Biotechnol Biofuels 11(2):301–317
Aboelella NS, Brandle C, Kim T, Ding ZC, Zhou G (2021) Oxidative stress in the tumor microenvironment and its relevance to cancer immunotherapy. Cancers (Basel) 13(5):986–1009
Allemani C, Matsuda T, Di Carlo V et al (2018) Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 391(10125):1023–1075
Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M et al (2021) A genome-wide copper-sensitized screen identifies novel regulators of mitochondrial cytochrome c oxidase activity. J Biol Chem 296:100485–100498
Alzahrani AS (2019) PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol 59:125–132
Ariöz C, Wittung-Stafshede P (2018) Folding of copper proteins: role of the metal? Q Rev Biophys 51:4–15
Audia S, Brescia C, Dattilo V, D’Antona L, Calvano P, Iuliano R et al (2023) RANBP1 (RAN Binding Protein 1): the missing genetic piece in cancer pathophysiology and other complex diseases. Cancers (Basel) 15(2):486–498
Balsano C, Porcu C, Sideri S (2018) Is copper a new target to counteract the progression of chronic diseases? Metallomics 10(12):1712–1722
Bhattacharya A, Freedman AN, Avula V, Harris R, Liu W, Pan C, Lusis AJ et al (2022) Placental genomics mediates genetic associations with complex health traits and disease. Nat Commun 13(1):706–721
Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21(2):85–100
Bourbia N, Chandler P, Codner G, Banks G, Nolan PM (2019) The guanine nucleotide exchange factor, Spata13, influences social behaviour and nocturnal activity. Mamm Genome 30(3–4):54–62
Carrer A, Trefely S, Zhao S, Campbell SL, Norgard RJ, Schultz KC et al (2019) Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov 9(3):416–435
Chen Y, Li K, Gong D, Zhang J, Li Q, Zhao G et al (2020) ACLY: a biomarker of recurrence in breast cancer. Pathol Res Pract 216(9):153076–153096
da Silva DA, De Luca A, Squitti R, Rongioletti M, Rossi L, Machado CML et al (2022) Copper in tumors and the use of copper-based compounds in cancer treatment. J Inorg Biochem 226:111634–111645
De Luca A, Barile A, Arciello M, Rossi L (2019) Copper homeostasis as target of both consolidated and innovative strategies of anti-tumor therapy. J Trace Elem Med Biol 55:204–213
Deng H, Zhu S, Yang H, Cui H, Guo H, Deng J et al (2022) The dysregulation of inflammatory pathways triggered by copper exposure. Biol Trace Elem Res. Epub ahead of print
Emami MH, Sereshki N, Malakoutikhah Z, Dehkordi SAE, Fahim A, Mohammadzadeh S et al (2022) Nrf2 signaling pathway in trace metal carcinogenesis: a cross-talk between oxidative stress and angiogenesis. Comp Biochem Physiol C Toxicol Pharmacol 254:9266–9275
Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M et al (2021) Cancer statistics for the year 2020: An overview. Int J Cancer 149(4):778–789
Fujieda N, Umakoshi K, Ochi Y, Nishikawa Y, Yanagisawa S, Kubo M et al (2020) Copper-oxygen dynamics in the tyrosinase mechanism. Angew Chem Int Ed Engl 59(32):13385–13390
Gałczyńska K, Drulis-Kawa Z, Arabski M (2020) Antitumor activity of Pt(II) Ru(III) and Cu(II) complexes. Molecules 25(15):3492–3506
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C (2021) Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol 15(12):3527–3544
Gong Y, Zou S, Deng D, Wang L, Hu H, Qiu Z et al (2023) Loss of RanGAP1 drives chromosome instability and rapid tumorigenesis of osteosarcoma. Dev Cell 58(3):192–210
Hao YN, Zhang WX, Gao YR, Wei YN, Shu Y, Wang JH (2021) State-of-the-art advances of copper-based nanostructures in the enhancement of chemodynamic therapy. J Mater Chem B 9(2):250–266
He Y, Xu L, Feng M, Wang W (2019) Role of CD97 small isoform in human cervical carcinoma. Int J Exp Pathol 100(1):19–24
Huang TT, Lampert EJ, Coots C, Lee JM (2020) Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat Rev 86:102021–102033
Jiang Y, Huo Z, Qi X, Zuo T, Wu Z (2022) Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine (Lond) 17(5):303–324
Karamanos NK, Piperigkou Z, Passi A, Götte M, Rousselle P, Vlodavsky I (2021) Extracellular matrix-based cancer targeting. Trends Mol Med 27(10):1000–1013
Katsuyama M, Kimura E, Ibi M, Iwata K, Matsumoto M, Asaoka N et al (2021) Clioquinol inhibits dopamine-β-hydroxylase secretion and noradrenaline synthesis by affecting the redox status of ATOX1 and copper transport in human neuroblastoma SH-SY5Y cells. Arch Toxicol 95(1):135–148
Khan AB, Gadot R, Shetty A, Bayley JC V, Hadley CC, Cardenas MF et al (2020) Identification of novel fusion transcripts in meningioma. J Neurooncol 149(2):219–230
Kong HJ, Kwon EJ, Kwon OS, Lee H, Choi JY, Kim YJ et al (2021) Crosstalk between YAP and TGFβ regulates SERPINE1 expression in mesenchymal lung cancer cells. Int J Oncol 58(1):111–121
Lange J, Wood-Kaczmar A, Ali A, Farag S, Ghosh R, Parker J et al (2021) Mislocalization of nucleocytoplasmic transport proteins in human Huntington’s disease PSC-derived striatal neurons. Front Cell Neurosci 15:63–85
Lelièvre P, Sancey L, Coll JL, Deniaud A, Busser B (2020) The multifaceted roles of copper in cancer: a trace metal element with dysregulated metabolism but also a target or a bullet for therapy. Cancers (Basel) 12(12):3594–3604
Lin Z, Miao D, Xu Q, Wang X, Yu F (2021) A novel focal adhesion related gene signature for prognostic prediction in hepatocellular carcinoma. Aging (Albany NY) 13(7):10724–10748
Lin X, Zhuang S, Chen X, Du J, Zhong L, Ding J et al (2022) lncRNA ITGB8-AS1 functions as a ceRNA to promote colorectal cancer growth and migration through integrin-mediated focal adhesion signaling. Mol Ther 30(2):688–702
Liu Y, Guan X, Wang M, Wang N, Chen Y, Li B et al (2022) Disulfiram/copper induces antitumor activity against gastric cancer via the ROS/MAPK and NPL4 pathways. Bioengineered 13(3):6579–6589
Maldonado MDM, Dharmawardhane S (2018) Targeting Rac and Cdc42 GTPases in cancer. Cancer Res 78(12):3101–3111
Martin GH, Roy N, Chakraborty S, Desrichard A, Chung SS, Woolthuis CM et al (2019) CD97 is a critical regulator of acute myeloid leukemia stem cell function. J Exp Med 216(10):2362–2377
Myint ZW, Oo TH, Thein KZ (2018) Copper deficiency anemia: review article. Ann Hematol 97(9):1527–1534
Narayanan IG, Natarajan SK (2018) Peptides derived from histidine and methionine-rich regions of copper transporter 1 exhibit anti-angiogenic property by chelating extracellular Cu. Chem Biol Drug Des 91(3):797–804
Ngamchuea K, Batchelor-McAuley C, Compton RG (2016) The copper(II)-catalyzed oxidation of glutathione. Chemistry 22(44):15937–15944
Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li L et al (2020) Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 578(7796):621–626
Santoro AM, Monaco I, Attanasio F, Lanza V, Pappalardo G, Tomasello MF et al (2016) Copper(II) ions affect the gating dynamics of the 20S proteasome: a molecular and in cell study. Sci Rep 6:33444–33454
Schumacher S, Vazquez Nunez R, Biertümpfel C, Mizuno N (2022) Bottom-up reconstitution of focal adhesion complexes. FEBS J 289(12):3360–3373
Seker F, Cingoz A, Sur-Erdem İ, Erguder N, Erkent A, Uyulur F et al (2021) Identification of SERPINE1 as a regulator of glioblastoma cell dispersal with transcriptome profiling. Cancers (Basel) 11(11):1651–1672
Shanbhag VC, Gudekar N, Jasmer K, Papageorgiou C, Singh K, Petris MJ (2021) Copper metabolism as a unique vulnerability in cancer. Biochim Biophys Acta Mol Cell Res 1868(2):118893–211915
Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J et al (2017) Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol Cell 67(2):252–265
Steenblock C, Heckel T, Czupalla C, Espirito Santo AI, Niehage C, Sztacho M et al (2014) The Cdc42 guanine nucleotide exchange factor FGD6 coordinates cell polarity and endosomal membrane recycling in osteoclasts. J Biol Chem 289(26):18347–18359
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249
Tang DL, Chen X, Kroemerv G (2022) Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res 32(5):417–418
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375(6586):1254–1261
Vallet SD, Ricard-Blum S (2019) Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem 63(3):349–364
Wang Q, He Y, Kan W, Li F, Ji X, Wu X et al (2019) microRNA-32-5p targets KLF2 to promote gastric cancer by activating PI3K/AKT signaling pathway. Am J Transl Res 11(8):4895–4908
Waseem NH, Low S, Shah AZ, Avisetti D, Ostergaard P, Simpson M et al (2020) Mutations in SPATA13/ASEF2 cause primary angle closure glaucoma. PLoS Genet 16(4):1008721–1008733
Wei J, Hu M, Huang K, Lin S, Du H (2020) Roles of proteoglycans and glycosaminoglycans in cancer development and progression. Int J Mol Sci 21(17):5983–6011
Wen J, Min X, Shen M, Hua Q, Han Y, Zhao L et al (2019) ACLY facilitates colon cancer cell metastasis by CTNNB1. J Exp Clin Cancer Res 38(1):401–413
Xia Y, Liu X, Zhang L, Zhang J, Li C, Zhang N et al (2019) A new Schiff base coordinated copper(II) compound induces apoptosis and inhibits tumor growth in gastric cancer. Cancer Cell Int 19:81–87
Xia C, Dong X, Li H, Cao M, Sun D, He S et al (2022) Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl) 135(5):584–590
Yang JD, Ma L, Zhu Z (2019) SERPINE1 as a cancer-promoting gene in gastric adenocarcinoma: facilitates tumour cell proliferation migration and invasion by regulating EMT. J Chemother 31(7–8):408–418
Yin Y, Xu X, Tang J, Zhang W, Zhangyuan G, Ji J et al (2018) CD97 promotes tumor aggressiveness through the traditional G protein-coupled receptor-mediated signaling in hepatocellular carcinoma. Hepatology 68(5):1865–1878
Zhang Q, Lei L, Jing D (2020) Knockdown of SERPINE1 reverses resistance of triple-negative breast cancer to paclitaxel via suppression of VEGFA. Oncol Rep 44(4):1875–1884
Zhang YF, Li Q, Huang PQ, Su T, Jiang SH, Hu LP et al (2022) A low amino acid environment promotes cell macropinocytosis through the YY1-FGD6 axis in Ras-mutant pancreatic ductal adenocarcinoma. Oncogene 41(8):1203–1215
Zheng P, Zhou C, Lu L, Liu B, Ding Y (2022) Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J Exp Clin Cancer Res 41(1):271–280
Funding
This research was funded by the National Natural Science Foundation of China (No. 82160588).
Author information
Authors and Affiliations
Contributions
Mingxu Da proposed the study. Yongli Hu performed the research and wrote the first draft. Yan Du, Zhisheng Qiu, Pengwei Bai, Zhaozhao Bai, Chenglou Zhu, Junhong Wang and Tong Liang participated in data analysis. All authors contributed to interpretation of the study and to further drafts.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no conflict of interest. The funders in the decision to publish the results.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, Y., Du, Y., Qiu, Z. et al. Construction of a Cuproptosis-Related Gene Signature for Predicting Prognosis in Gastric Cancer. Biochem Genet 62, 40–58 (2024). https://doi.org/10.1007/s10528-023-10406-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10406-9