
Abstract
The receptor for advanced glycation end-products (RAGE) was initially characterized and named for its ability to bind to advanced glycation end-products (AGEs) that form upon the irreversible and non-enzymatic interaction between nucleophiles, such as lysine, and carbonyl compounds, such as reducing sugars. The concentrations of AGEs are known to increase in conditions such as diabetes, as well as during ageing. However, it is now widely accepted that RAGE binds with numerous ligands, many of which can be defined as pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). The interaction between RAGE and its ligands mainly results in a pro-inflammatory response, and can lead to stress events often favouring mitochondrial dysfunction or cellular senescence. Thus, RAGE should be considered as a pattern recognition receptor (PRR), similar to those that regulate innate immunity. Innate immunity itself plays a central role in inflammaging, the chronic low-grade and sterile inflammation that increases with age and is a potentially important contributory factor in ageing. Consequently, and in addition to the age-related accumulation of PAMPs and DAMPs and increases in pro-inflammatory cytokines from senescent cells and damaged cells, PRRs are therefore important in inflammaging. We suggest here that, through its interconnection with immunity, senescence, mitochondrial dysfunction and inflammasome activation, RAGE is a key contributor to inflammaging and that the pro-longevity effects seen upon blocking RAGE, or upon its deletion, are thus the result of reduced inflammaging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The receptor for advanced glycation end-products (RAGE) expressed in mammals is a 45 kDa transmembrane receptor from the immunoglobulin superfamily (Ig) which also serves as a cell adhesion molecule (Sessa et al. 2014). While it is very strongly expressed in the lung, its basal expression is low in most tissues. While its precise physiological function remains unclear, there is good evidence that it could be involved in immunity due to its pro-inflammatory signalling. Nonetheless, Rage deletion in mice is viable and has shown to be protective against a number of pathologies, including age-related diseases, such as cardiovascular diseases or Alzheimer’s disease (Cai et al. 2016; Park et al. 2011), and even features of physiological ageing (Grossin et al. 2015; Teissier et al. 2019). RAGE interaction with its ligands results primarily in increased inflammation and oxidation. Given the properties of RAGE, its role in age-related diseases and its association with some features of ageing, we here suggest that it could also play a key role in physiological ageing.
Ageing is characterized by a number of events that lead to reduced efficacy of organ functions, to an increased probability of developing so-called age-related diseases and to an increased risk of death with time. Hallmarks or pillars of ageing that cause or result from ageing have recently been described and reviewed (Kennedy et al. 2014a; López-Otín et al. 2013). Chronic inflammation, or rather inflammaging, is considered one of the most important processes of ageing that both explains and is explained by different hallmarks of ageing. We here suggest that RAGE signalling might be an important contributor to inflammaging with respect to its inherent pro-inflammation properties and its suspected role in ageing.
In this review, we summarize the current knowledge of RAGE expression and structure and its interaction with downstream effectors and extracellular ligands. Consequently, we describe inflammaging and its links with immunosenescence, damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) and the senescence-associated secretory phenotype (SASP), and show that RAGE is classifiable as a pattern recognition receptor (PRR) involved in inflammaging and ageing.
RAGE and its ligands
RAGE expression
RAGE is a multiligand protein found in mammals, belonging to the immunoglobulin superfamily and to the family of cell adhesion molecules (Sessa et al. 2014). It is widely expressed across all organs, notably in skeletal muscle, lung, heart, liver and kidney (Brett et al. 1993) and is also expressed in circulating adaptive and innate immune cells (Akirav et al. 2012; Chen et al. 2008; Manfredi et al. 2008; Moser et al. 2007; Narumi et al. 2015; Schmidt 2017) and is involved in their recruitment via vascular cell adhesion molecule 1 (VCAM-1) (Boulanger et al. 2002, 2007). While its expression is relatively low in most tissues under basal conditions, it is more strongly expressed in the lung, notably in alveolar epithelial cells (Cheng et al. 2005; Demling et al. 2006; Shirasawa et al. 2004). At least 20 splice variants have been described so far but only a few of them have a fully reported corresponding protein. While some of these variants are shared between mice and humans, others are exclusive to one or other species and mice potentially express less soluble RAGE (sRAGE) through alternative splicing than humans. Nonetheless, despite these differences, the main reported isoforms of RAGE are conserved across species and the mRNA coding for full-length RAGE is the most prevalent variant across organs (Hudson et al. 2008a; Kalea et al. 2009). There exist some exceptions and sRAGE is dominant in the human hippocampus (Ding and Keller 2005a). With age, RAGE expression is increased, at least in the human heart, and its role might therefore become more important in older people (Simm et al. 2004). Mainly described at the plasma membrane, it is also often found in the cytoplasm. So far, only one study has indicated that it could also be found in the nucleus, having a role in DNA-damage response (DDR) and double-strand break repair, but more work is required to confirm this (Kumar et al. 2017).
RAGE structure
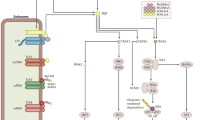
Full-length RAGE is comprised of three Ig domains that form its extracellular part, including one V-type Ig domain (V domain) connected via a short linker to a C-type Ig domain (C1 domain) and then to a second C-type Ig domain (C2 domain). These extracellular domains are then followed by one transmembrane and one intracellular domain which transduces the signalling (Fig. 1) (Bongarzone et al. 2017; Neeper et al. 1992; Xue et al. 2011; Yatime and Andersen 2013). Naturally-occurring isoforms are also found. Two soluble forms (sRAGE) have been widely reported: the endogenous secretory RAGE (esRAGE), which results from alternative splicing of Rage mRNA (Hudson et al. 2008a; Jules et al. 2013), and the cleaved RAGE (cRAGE) which is the result of the extracellular RAGE domain cleaved by metalloproteases such as a disintegrin and metalloproteinase 10 (ADAM10) or matrix metalloproteinase-9 (MMP9) (Raucci et al. 2008; Zhang et al. 2008). Interestingly, sRAGE is a possible biomarker of chronic inflammatory diseases (Maillard-Lefebvre et al. 2009). Two other forms of RAGE have also been reported: the dominant-negative RAGE (DN-RAGE), lacking any intracellular domain, and a form which lacks the V domain (ΔN-RAGE) (Ding and Keller 2005b; Hudson et al. 2008a). Other splice variants have been reported, but the expected proteins are either unknown or untranslated (Hudson et al. 2008a). However, we will focus hereafter upon full-length RAGE.

Adapted from Kierdof et al. (2013)
RAGE structure. Full-length RAGE is comprised of the V, C1 and C2 extracellular domains, of a transmembrane domain and a cytoplasmic domain. While the V and C1 domains are closely bound together, a flexible linker is present between the C1 and C2 domains. The V and C1 domains (in blue) are positively charged with some localized negatively-charged sites, while the C2, transmembrane and cytoplasmic domains (in red) are negatively charged. Most RAGE ligands interact with the VC1 domain. Aβ amyloid β, AOPPs advanced oxidation protein products; CEL carboxyethyllysine; CML carboxymethyllysine; gDNA genomic DNA; HMGB1 high-mobility group box 1; HSP70 heat shock protein 70; LPA lipopolysaccharide lipid component A; LPS lipopolysaccharide; Mac-1 macrophage-1 antigen; MG-H1 methylglyoxal-derived hydroimidazolone-1; mtDNA mitochondrial DNA.
Upon interaction with its ligands, RAGE may be phosphorylated by PKCζ and/or recruit various adaptor proteins or effectors including toll-interleukin 1 receptor domain-containing adaptor protein (TIRAP), MyD88, similar to Toll-like receptors (TLRs) (Sakaguchi et al. 2011), or mammalian diaphanous-1 (mDia1) (Hudson et al. 2008b; Manigrasso et al. 2016; Rai et al. 2012a). In parallel, RAGE can undergo homodimerization, oligomerization (Xie et al. 2008; Yatime and Andersen 2013; Zong et al. 2010) and possibly heterodimerization (Tian et al. 2007; Zong et al. 2010), to engage its signalling pathways. These pathways are diverse and complex, and include the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, c-Jun N-terminal kinase (JNK), the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway, and the mitogen-activated protein kinases (MAPK) pathway with extracellular-signal-regulated kinases (ERK) 1/2 (Huang et al. 2001; Hudson et al. 2008b; Li et al. 2015; Liu et al. 2010; Ray et al. 2016; Wautier et al. 2001; Yeh et al. 2001). These pathways mainly result in the activation of the transcription factors activator protein 1 (AP-1), STATs and more importantly NF-κB, leading to pro-oxidant and pro-inflammatory responses (Bianchi et al. 2010; Bierhaus et al. 2006; Kierdorf and Fritz 2013; Yan et al. 2009b). Interestingly, adaptor proteins such as TIRAP and MyD88, both of which lead to a pro-inflammatory response upon activation of TLRs and RAGE (at least via the NF-κB signalling pathway), suggest similar functions and a potential crosstalk between the two receptor families. Thus, regulating these adaptors might have an important impact on inflammation by modulating the activity of TLRs and RAGE. On the other hand, up- or down-regulation of one of these receptors may alter the activity of other receptors owing to decreased or elevated availability of MyD88 and TIRAP to transduce their signalling. On the contrary, however, RAGE activation can temper NF-κB activation in some cases, as shown in the liver (Zeng et al. 2012). A more detailed description of RAGE downstream signalling pathways is proposed elsewhere (Sorci et al. 2013; Xie et al. 2013).
While RAGE was first described as a receptor for advanced glycation end-products (AGEs), hence its name (Neeper et al. 1992; Schmidt et al. 1992), it is now widely known that RAGE is in fact a multi-ligand receptor and proteins and molecules from various origins interact with RAGE to induce a transduction signal. To better understand how RAGE functions and in what conditions its contribution is involved, it is important to fully describe the ligands that trigger RAGE signalling.
Advanced glycation end-products
Advanced glycation end-products (AGEs) are a family of molecules issuing from the reaction between a nucleophile, such as the amino group of a lysine, and a carbonyl compound such as reducing sugars. Upon this interaction, successive reversible rearrangements produce the early glycation products known as Schiff bases and Amadori products which eventually lead to a final reaction forming AGEs, so far considered as irreversible. This reaction can be influenced by pH and temperature. While it takes months or years to form AGEs in vivo at 37 °C, they can be produced in minutes or seconds in cooking processes utilising high temperatures. Their concentration in vivo is elevated in pathologies such as diabetes (Thornalley et al. 2003), Alzheimer’s Disease (AD) (Shuvaev et al. 2001) and cataracts (Monnier and Cerami 1981) and increases with age (Sell et al. 1996). Tissues composed of proteins with a long half-life are more likely to accumulate AGEs and become altered by this accumulation, as exemplified by crystallin in the lens and collagen in the extracellular matrix (Masters et al. 1977; Monnier and Cerami 1981; Verzijl et al. 2000). Many different AGEs are formed endogenously or are taken in from various exogenous origins and, despite RAGE being named as the receptor for AGEs, owing to their heterogeneity it is far from certain that all AGEs can interact with this receptor. Methylglyoxal-derived hydroimidazolone-1 (MG-H1) has been reported to interact with the V domain of human RAGE with a Kd = 40 nM in a nuclear magnetic resonance (NMR) and fluorescence titration experiment (Xue et al. 2014). Other NMR and fluorescence titration studies have demonstrated that peptide- and protein-bound carboxymethyllysine (CML) and carboxyethyllysine (CEL) were also able to interact with the V domain with a Kd = 100 µM (Xie et al. 2008; Xue et al. 2011). However free CML and CEL, modified lysine not bound to other amino acid residues, do not seem to be able to interact with RAGE (Xie et al. 2008). This was supported by the observation that excess free CML was unable to inhibit CML-bovine serum albumin (CML-BSA) interaction with 125I-sRAGE (Kislinger et al. 1999). In silico docking analysis predicts that pentosidine, argypirimidine, imidazole and pyrraline are able to interact with the C1 domain of RAGE (Fatchiyah et al. 2015).
Calgranulins
Another group of molecules, calgranulins, has also been reported to interact with RAGE. Calgranulins are a family of S100, EF-hand, calcium-binding proteins which are considered as sensors of intracellular Ca2+ and are involved in different cellular processes such as cell migration, cell proliferation or cell differentiation (van Hout et al. 2016). Dysregulated levels of S100 have been associated with cancer, inflammatory response and cardiomyopathies (Bresnick et al. 2015; Most et al. 2006; Oliveira et al. 2018). S100B is increased with age in mouse kidneys and liver and is also associated with increased infiltration of activated macrophages (Son et al. 2017). The majority of the 21 members of the S100 family has been reported to interact with the V, C1 and even C2 domain of RAGE (Bongarzone et al. 2017; Leclerc et al. 2009). Some work suggests that S100B interacts with positively-charged areas of the V domain (Xie et al. 2008), while others suggest an hydrophobic Ca2+-dependent interaction in the C’D loop of the V domain (Park et al. 2010). The reported Kd ranged from µM to nM and were positively affected by S100 oligomerization (Ostendorp et al. 2007; Xie et al. 2007).
HMGB1
High-mobility group box 1 (HMGB1), or amphoterin, is considered to be the most abundant non-histone, DNA-binding protein. It is mainly localized in the nucleus but can transit to the cytoplasm via nuclear pores when acetylated, thus regulating its function. HMGB1 is involved in nucleosome formation and can modulate transcription by interacting with transcription factors and histones (Bianchi and Agresti 2005; Harris and Andersson 2004). Its role in DNA repair remains elusive and the subject of debate (Lange and Vasquez 2009). Its overexpression in breast cancer sensitizes cancer cells to cisplatin, an antitumor drug (He et al. 2000), and its depletion enhances the excision repair mechanism following cisplatin treatment (Li et al. 1997), but it has also been shown to play a role in genomic and mitochondrial DNA repair (Ito et al. 2015; Lange et al. 2008). HMGB1 can also be released in the extracellular medium by active or passive processes. Active release generally involves a severe stress, such as inflammation or ischemia, which can inhibit HMGB1 deacetylation and lead to its sequestration in the cytoplasm until it is finally secreted (Evankovich et al. 2010; Gardella et al. 2002; Tsung et al. 2014; Venereau et al. 2013). Passive release of HMGB1 occurs in conditions such as necrosis (Scaffidi et al. 2002). HMGB1 is increased with age in mouse kidney and liver and is also associated with macrophage infiltration (Son et al. 2017). Circulating HMGB1 is associated with a set of pro-inflammatory processes, senescence and several pathologies (Davalos et al. 2013; Magna and Pisetsky 2014). Among all RAGE ligands, HMGB1 has one of the highest reported affinities with a Kd = 6–10 nM as determined by an in vitro saturation binding assay (Hori et al. 1995).
Amyloid fibrils
Amyloid fibrils have also been shown to interact with RAGE. Amyloid fibrils are characterised by β-sheet-rich, super-coiled, insoluble structures forming fibrils. These fibrils can be formed by the nucleation of soluble proteins to form protofilaments, which in turn give rise to amyloid fibrils. Many proteins such as the amyloid β (Aβ) peptide, serum amyloid A (SAA), Ig light chains or even insulin have been reported to be responsible for different kind of amyloidosis (Iannuzzi et al. 2014; Rambaran and Serpell 2008). While amyloidosis due to proteins such as apolipoprotein A-II (apoA-II) is very rare in humans, it is common in other species like mice mainly because of species-specific differences in methionine content and dimerization, which is also why amyloidosis is specific to some proteins (Gursky 2014; Higuchi et al. 1991; Kitagawa et al. 2003; Morizane et al. 2011). Amyloidosis has been linked to a number of pathologies including AD where Aβ plaques are increased (Glenner and Wong 1984; Masters et al. 1985a, 1985b), scrapie or bovine spongiform encephalopathy caused by prion proteins (PrPc) (Prusiner 1982, 1991), or primary systemic amyloidosis characterized by accumulation of amyloid light (AL) and heavy (AH) chains in multiple organs (Comenzo 2000; Rambaran and Serpell 2008). Among amyloid fibril RAGE ligands, Aβ is the best characterized and interacts with the V domain with a Kd = 70–80 nM. This interaction plays a role in Aβ neurotoxicity and circulating Aβ influx into the brain (Deane et al. 2012; Yan et al. 1996). Other amyloid fibrils of SAA, prion-derived peptides and amylin have also been reported as ligands of RAGE. Notably, these same proteins presented in a random conformation, as well as an all-β sheet protein erabutoxin B, were unable to interact with RAGE, suggesting a specificity of sequence and conformation is important for this interaction (Abedini et al. 2018; Yan et al. 2000). sRAGE was, however, reported to interact with both soluble and fibrillar transthyretin with equal affinity (Kd = 120 nM) (Sousa et al. 2000).
Complement components
The complement system is a major component of immunity and is characterized by multiple cleavage products. Importantly, it participates in pathogen lysis through the formation of a membrane attack complex (MAC) at their surface, allowing free diffusion of molecules through this pore and engendering cell death. It is also involved in a process called “antigen opsonisation”, by which addition of complement molecules to a pathogen's membrane facilitates recognition by immune cells. Chemotaxis of innate immune cells may also be enhanced by complement components. The complement component C3a, resulting from C3 cleavage, is an anaphylatoxin which is possibly involved in systemic lupus erythematosus (Peng et al. 2009; Porcel et al. 1995). C3a was able to bind to RAGE in an ELISA assay and formed a ternary complex with oligonucleotides (Ruan et al. 2010). Another complement component, C1q is able to initiate complement activation and antibody-independent opsonisation and its absence is also associated with systemic lupus erythematosus (Teh et al. 2011). Interaction of monomeric C1q with RAGE exhibited weak affinity (Kd = 5.6 µM) in SPR analysis, but multivalent bonds may enhance this binding. This interaction is suspected to enhance monocyte phagocytosis (Ma et al. 2012), but while few studies have investigated the interaction between components of the complement and RAGE, it plays a key role in immunity and thus merits further characterization in the context of inflammaging.
Other RAGE ligands
Other ligands of RAGE, though relatively poorly studied, have also been reported. Lipopolysaccharide (LPS) and its lipid component A both interact with the V domain, potentially at the KGAPKKPPQRLEWKLN site, with a Kd = 35 nM and Kd = 2 nM, respectively (Yamamoto et al. 2011). RAGE was also able to interact with CpG oligonucleotides, double strand DNA and double strand RNA (Park et al. 2010; Ruan et al. 2010). The leukocyte cell surface molecule Mac-1 (CD11b/CD18) interacts with RAGE with an undefined Kd (Chavakis et al. 2003). Phosphatidylserine binds to sRAGE with a Kd = 0.563 µM (He et al. 2011). Advanced oxidation protein products have been reported to interact with RAGE in an ELISA assay (Guo et al. 2008). Extracellular heat-shock protein 70 was also able to interact with RAGE in an ELISA assay (Somensi et al. 2017). Quinolinic acid, an undesirable and excitotoxic tryptophan metabolite, interacts with RAGE at multiple sites in the VC1 domain with a mean Kd = 43 nM (Serratos et al. 2015). Lysophosphatidic acid, an endogenous phospholipid with roles in homeostasis and diverse pathologies, also binds to the V domain of RAGE with a Kd = 9 nM (Rai et al. 2012b).
As reviewed elsewhere with respect to HMGB1 and S100 proteins binding to RAGE, different RAGE ligands might initiate different signalling pathways (Sparvero et al. 2009). To our knowledge, currently available data are insufficient to clearly depict which ligand activates which pathways and the literature lacks studies designed to exclude non-specific interactions or unequivocally identify the different pathways involved, but some work has provided interesting tools to address issues such as compounds specifically inhibiting interaction between RAGE and adaptor proteins such as mDia1 (Manigrasso et al. 2016). It is therefore necessary to study RAGE's role and signalling in more detail and in a greater variety of contexts in order to fully elucidate its properties.
Unlike ligands that bind to the V domain of RAGE (CML, LPS,…), those binding to the C1 or C2 domain can still bind to the ΔN-RAGE and presumably induce a pro-inflammatory response. Binding affinities should therefore be considered in order to anticipate competition between various ligands—low concentrations of high affinity ligands such as HMGB1 or LPS could compete even with high concentrations of lower affinity ligands such as CML and other AGEs. In events leading to significant release of HMGB1 or LPS into the circulation, these ligands would thus have a proportionally greater effect upon RAGE-mediated inflammation.
The vast heterogeneity of RAGE ligands suggests that these interactions are probably not dependent upon sequence recognition: rather, it is thought that RAGE acts as a PRR whose interaction with ligands is mostly governed by ionic interactions. While it was first described as a PRR in 2003 (Chavakis et al. 2003), its involvement in immunity has been poorly studied in comparison with other PRRs such as toll-like receptors (TLRs) and also, in a more general sense, compared to the work that has been undertaken on AGEs and diabetes. However, as presented below, many studies indicate that RAGE has a key place in inflammaging or its parameters.
RAGE and inflammaging
Inflammaging
Inflammaging was first used as a term in 2000 by Franceschi et al. (Franceschi et al. 2000) following their counterintuitive observation that, upon stimulation by a mitogen, mononuclear cells from healthy elderly donors (~ 80 years old) produced more cytokines such as interleukine-6 (IL-6), IL-1β and tumor necrosis factor-α (TNF-α), than those from younger donors (~ 27 years old) (Fagiolo et al. 1993). Later, it was shown that in plasma from centenarians the overall load of pro-inflammatory cytokines was significantly increased but was not associated with an enhanced immune response (Morrisette-Thomas et al. 2014). Thus, inflammaging was defined as chronic, low-grade and sterile inflammation associated with age, and it is now considered to a "pillar of ageing" since inflammation status is one of the best predictors of longevity and successful ageing (Arai et al. 2015; Franceschi et al. 2000; Kennedy et al. 2014b).
The characterization of inflammaging has evolved to include a number of pro-inflammatory cytokines and other pro-inflammatory molecules such as the already mentioned IL-1β, IL-6 and TNF-α, but also IL-18, C-reactive protein (CRP), monocyte chemoattractant protein-1 (MCP-1), regulated on activation, normal T cell-expressed and secreted (RANTES) protein and macrophage inflammatory protein 1 α (MIP-1α), all of which increase with age in peripheral blood mononuclear cells (PBMCs) or serum (Dinarello 2006; Fagiolo et al. 1993; Ferrucci et al. 2005a, 2005b; Gerli et al. 2000; de Gonzalo-Calvo et al. 2010; Mariani et al. 2006; Morrisette-Thomas et al. 2014; Seidler et al. 2010). Similarly, anti-inflammatory molecules IL-1 receptor antagonist (IL-1ra), soluble TNF-receptor I (sTNF-RI) and sTNF-RII, or markers of anti-inflammation such as soluble CD30 (sCD30), are also increased with age and inflammatory activation (Gerli et al. 2000; Morrisette-Thomas et al. 2014). Overall, there exists a weight of evidence to indicate development of a global imbalance with increasing age favouring secretion of pro-inflammatory molecules and a concurrent increase in circulating anti-inflammatory molecules in response. These markers have been more comprehensively reviewed elsewhere (Minciullo et al. 2016).
Inflammaging is closely linked to immunosenescence, or the ageing of the immune system. With time, both innate and adaptive immunity become dysregulated, the number and efficacy of various cell types are greatly affected in such a way that the immune system becomes less responsive to pathogens (Nguyen et al. 2018), vaccine efficacy is reduced with age (Zhang et al. 2016) and propensity to develop cancers may increase (Pawelec 2017). Among the many changes in innate immunity over time, key changes are that the most pro-inflammatory of the monocytes appear to be increased with age and may contribute to inflammaging (Hearps et al. 2012), while the inflammasome is over-activated by age-related damage-associated molecular patterns (DAMPs), increasing TNF-α levels or mitochondrial dysfunction.
Nonetheless, the relationship between immunosenescence and inflammaging is not straightforward and inflammaging is not simply, nor indeed exclusively, induced by immunosenescence. One recent model proposes an interaction where immunosenescence and inflammaging mutually induce each other: on the one hand, inflammaging can decrease the strength of the adaptive immune response and lead to immunosenescence; on the other hand, a decrease of the adaptive immune response leads to increased innate immunity and inflammaging (Fulop et al. 2018). However current understanding of inflammaging tends to put innate rather than adaptive immunity at the centre of this process, since the former becomes mildly hyperactive with age. PRRs are key element of innate immunity that are able to recognize pattern-associated molecular patterns (PAMPs) and DAMPs and induce inflammation in response, notably through the involvement of NF-κB, AP-1, interferon regulatory factors (IRFs) and of a cytosolic multiprotein complex, the inflammasome (Franchi et al. 2012; Kaufmann and Dorhoi 2016; Man and Kanneganti 2015; Rivera et al. 2016; Wang et al. 2014). With the accumulation of PAMPs and DAMPs with age, such as cytoplasmic or cell-free DNA (Crow and Manel 2015; Dou et al. 2017; Mankan et al. 2014; Shen et al. 2015; Shimada et al. 2012), a “Garb-aging” theory has been proposed where excess production or a lack of clearance of molecular “garbage” might play a central role in the increase of chronic inflammation with age (Franceschi et al. 2017).
Finally, senescence might be another key contributor to inflammaging. Senescent cells are not quiescent cells, they accumulate multiple damages and modifications and have an aberrant secretory phenotype—the SASP. The SASP is mainly characterized by increased secretion of various interleukins (IL-1β, IL-6), chemokins (MCP-1), growth factors and metalloproteinases which can be secreted by a wide range of cell types (Coppé et al. 2010; Malaquin et al. 2016). In addition, cytoplasmic chromatin fragments (CCFs) are a common feature of senescent cells and they are able to induce inflammation through their interaction with the cyclic GMP-AMP synthase (cGAS)/stimulator for interferon genes (STING) pathway (Dou et al. 2017; Ivanov et al. 2013). As such, senescence is able to promote a pro-inflammatory microenvironment which can induce age-related inflammatory diseases such as osteoarthritis (Jeon et al. 2018; Xu et al. 2017). Interestingly, chronic inflammation itself is able to induce telomere dysfunction and engage the DNA damage response involved in senescence and ageing (Jurk et al. 2014). It thus seems sensible to consider senescence and the SASP as contributors to inflammaging. Accordingly, in vitro (Franceschi and Campisi 2014) and in vivo (Xu et al. 2018) studies have shown a causative role between senescent cells and a pro-inflammatory environment, suggesting a strong interconnection and mutual influence between senescence and inflammaging. Of note, senescent cells are normally recognized and removed by the immune system; immunosenescence may impede clearance of senescent cells, however, and accelerate their accumulation with age (Burton and Stolzing 2018), suggesting another interesting link between immunosenescence and inflammaging.
RAGE, mitochondrial dysfunction and inflammasome
Mitochondrial dysfunction is a prominent attribute of ageing and rejuvenation upon removal of mitochondria shows that it is preferable for a cell to rely upon a different source of energy (i.e. glycolysis) than to continue with harmful mitochondria (Correia-Melo et al. 2016). The detrimental effects of RAGE on mitochondria are two-fold, namely mitochondrial damage and the inflammatory responses induced by mitochondrial dysfunction.
Firstly, studies of the AGE-RAGE axis have shown that it is able to disrupt the mitochondria membrane potential by generating ROS, perhaps by activating the NADPH oxidase (Coughlan et al. 2009; Neviere et al. 2016; Tan et al. 2009; Yu et al. 2017). As soon as the membrane potential is perturbed, generation of ROS increases, first because of electron leakage from the respiratory chain (Jastroch et al. 2010; Suski et al. 2012), and then because of accumulating mtDNA mutations. Malondialdehyde, for example, resulting from lipid peroxidation, is highly genotoxic and promotes mutations in the highly accessible mtDNA (Del Rio et al. 2005). Finally, mtDNA mutations have been associated with further increases in ROS generation, mitochondrial dysfunction and chronic inflammatory diseases, probably due to altered mitochondrial protein functions (Kim et al. 2015; Tuppen et al. 2010). RAGE activation by AGEs was responsible for increased mitophagy (i.e. mitochondria renewal) and mitochondrial dysfunction. But it is not clear whether mitophagy is increased upon RAGE activation only or by the resultant dysfunctional mitochondria (Lo et al. 2015). Even though RAGE signalling is able to activate autophagy/mitophagy, it has been associated with mitochondrial-dependent cell death, potentially due to overextended mechanisms (Lo et al. 2015; Mao et al. 2018). Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), an important protein involved in mitochondrial biogenesis and ROS homeostasis, is notably controlled by acetylation/deacetylation processes dependent on sirtuine 1 (SIRT1) activity (Aquilano et al. 2010; Austin and St-Pierre 2012). Interestingly, the AGE/RAGE axis and RAGE expression have been associated with decreased SIRT1 activity and protein levels, suggesting further impaired mitochondrial biogenesis (Grossin et al. 2015; Neviere et al. 2016; Teissier et al. 2019). Accordingly, the presence of AGEs or RAGE overexpression decreased mitochondrial density and aggravated mitochondrial abnormalities and dysfunction (Mao et al. 2018).
Secondly, it has also been demonstrated that both mitochondrial transcription factor A (TFAM) and mtDNA released upon mitochondrial dysfunction induce a RAGE-dependent inflammatory response, notably activation of NF-κB and the inflammasome component ASC, mediated by TLR9 (Ward et al. 2013; Yao et al. 2015). This crosstalk between RAGE and TLR9 might be explained by the TLR9-HMGB1-RAGE complex found elsewhere (Tian et al. 2007). The noncanonical NF-κB pathway has been related to mitochondrial dynamics, and it would be interesting to know whether RAGE is involved here as well (Laforge et al. 2016).
Other relationships between RAGE and inflammasome activation have been investigated which might also be linked to mitochondrial dysfunction. Recent work shows that both mitochondrial damage and the AGE/RAGE axis are able to activate the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome and IL-1β production (Song et al. 2017). Additionally, NLRP3 expression itself is upregulated by AGEs while an antagonist peptide or 4′-Methoxyresveratrol, both targeting RAGE, prevented this upregulation and subsequent inflammation, suggesting that RAGE activation promotes NLRP3 expression (Chen et al. 2017; Shahzad et al. 2015; Yu et al. 2018). In a model of acute pancreatitis, RAGE also actively participates in AIM2 inflammasome activation, notably more so than TLR2 or TLR4, and its knockout significantly reduced the associated damage and systemic inflammation (Kang et al. 2016). Unsurprisingly, another ligand of RAGE, HMGB1, is also responsible for NLRP3 inflammasome activation and RAGE downregulation significantly attenuated this inflammatory response. However, downregulation of TLR2 and TLR4 provided similar results, suggesting common pathways (Kim et al. 2018; Thankam et al. 2018).
Thus, as RAGE ligands accumulate with age (notably AGEs), mitochondrial dysfunction increases over time, and both RAGE activation and inflammasome activation subsequent to mitochondrial dysfunction and/or RAGE signalling are also augmented with age, RAGE represents a potentially important centre of activity in the promulgation of inflammaging.
RAGE and senescence
Interestingly, RAGE expression is increased by its own ligands (Howes et al. 2004) and several important ligands of RAGE increase or accumulate with age, such as HMGB1, S100s or AGEs (Sell et al. 1996; Son et al. 2017). It has been shown that RAGE expression increased with age in diverse organs such as rats’ aorta (Hallam et al. 2010), rat and human hearts (Gao et al. 2008; Simm et al. 2004) and mouse spleen (Cai et al. 2007). We can therefore hypothesize that increasing amounts of RAGE ligands with age contribute to its upregulation and augment its stimulation, thereby enhancing its pro-inflammatory role and favouring increased low-grade, sterile inflammation with age.
HMGB1 itself has not only been described as a promoter of the SASP, but also as a SASP factor which is secreted by senescent cells (Malaquin et al. 2016). In a study focusing on TLR4 rather than RAGE, HMGB1 initiated IL-6 secretion by senescent cells (Davalos et al. 2013). TNF-α, another SASP factor, has also been reported to upregulate RAGE expression through NF-κB signalling and the existence of an NF-κB-binding site on the RAGE promoter (Li and Schmidt 1997; Tanaka et al. 2000). Thus, the factors of the SASP might activate RAGE, producing even more SASP factors which favour both senescence and RAGE expression/activation, thereby forming a vicious circle. Because of the SASP, widespread senescence is one of the main sources of cytokine-sustained inflammaging. While several studies have already linked the RAGE ligands AGEs to senescence and telomere attrition (Navarrete Santos et al. 2017; Sell et al. 1996; Sellier et al. 2015), only a few studies have directly linked senescence to RAGE.
In a study of the murine model of accelerated ageing, senescence-accelerated mouse-prone 8 (SAMP8), and its senescence-resistant counterpart, senescence accelerated mouse-resistant 1 (SAMR1), Kuhla et al. showed that plasma and/or hepatic AGEs, malondialdehyde (a marker of lipid peroxidation and oxidative stress) and RAGE expression were all increased in SAMP8 compared with SAMR1 mice. Acute liver injury induced by a galactosamine/lipopolysaccharide (G/L) challenge induced MAPK signalling associated with elevated apoptosis, necrosis and liver injury markers which were all greater in SAMP8 mice. Blocking the receptor with an anti-RAGE antibody significantly diminished these markers in SAMP8, but paradoxically also increased these markers in SAMR1. The authors suggest this apparent contradiction could be explained by the antibody dimerizing and thereby activating RAGE when RAGE ligands (i.e. AGEs) are at low concentrations (Kuhla et al. 2013). These results nevertheless show that RAGE signalling might be increased during senescence and that inhibiting its activation might prevent its deleterious effects in an ageing context. A study with the same model of inflammatory liver injury, but in young C57Bl/6 mice, showed that mitochondrial dysfunction significantly increased oxidative stress, AGEs and RAGE. Blockage of RAGE significantly reduced both G/L- and mitochondrial dysfunction-associated damage (Kuhla et al. 2010). Mitochondrial dysfunction being an important part of inflammaging, it is interesting to note that the deleterious effects of dysfunctional mitochondria involve RAGE signalling.
In another study in diabetic nephropathy patients, a correlation was found in proximal tubular epithelial cells (PTECs) between senescence-associated β-galactosidase (SA β-gal—a marker of senescence), p21 (a marker of cell cycle arrest), glucose-regulated protein 78 (GRP78—a marker of endoplasmic reticulum (ER) stress) and RAGE. The authors further confirmed in murine PTECs that activation of RAGE by AGEs induced a p21-dependant senescence involving ER stress. Markers of senescence were successfully prevented by blocking the receptor with an anti-RAGE antibody (Liu et al. 2014).
Finally, we have recently shown that RAGE deletion prevents important features of mouse renal ageing such as glomerulosclerosis or apolipoprotein A-II amyloidosis, and that this was associated with significant limitation of the age-related increase in the inflammation markers, IL-6, TNF-α and VCAM-1 (Teissier et al. 2019).
PRRs are important actors in inflammaging. They represent the point of convergence of all DAMPs and PAMPs, to which exposure increases significantly with age, and will lead to a number of pro-inflammatory signalling pathways fuelling inflammaging. As RAGE recognizes multiple ligands (often DAMPs or PAMPs), is expressed on multiple cell types including immunity cells, triggers a pro-inflammatory response similar to other PRRs and has a significant role in ageing as presented above, we here suggest that RAGE is a key PRR in inflammaging (Fig. 2) whose role in this feature of ageing is currently underestimated.

RAGE-induced inflammaging pathways. Three major pathways have been identified leading to inflammaging and induced via RAGE activation by its ligands. The mitochondrial pathway involves NAPDH oxidase activation by RAGE. Subsequent production of ROS impairs mitochondrial function and mitochondria accumulate damage and ROS: the mitochondrial products released can either reactivate RAGE or the inflammasome, thereafter responsible for release of ILs and NF-κB activation. NF-κB can also be activated directly by RAGE or by pro-inflammatory cytokine activation of TNFR or IL-R. NF-κB and other transcription factors, such as AP-1 or STATs, will thereafter promote the transcription of RAGE, NLRP3 and pro-inflammatory cytokines, all of which participate in inflammaging. RAGE activation can also provoke sustained ER stress, inducing a p21-dependant senescence. Subsequently, the SASP will fuel inflammaging and senescent cells will release RAGE ligands such as HMGB1 and S100s. Ultimately, RAGE expression increases with age, as do concentrations of a number of its ligands, thus engaging a vicious circle of RAGE-induced inflammaging with advancing age. The pro-inflammatory components responsible for inflammaging are highlighted in orange. AIM2 absent in melanoma 2; AP-1 activator protein 1; ASC apoptosis-associated speck-like protein containing CARD; ER Endoplasmic reticulum; IL interleukin; IL-R interleukin receptor; mtDNA mitochondrial DNA; NADPH nicotinamide adenine dinucleotide phosphate; NLRP3 NLR pyrin domain-containing 3; RAGE receptor for advanced glycation end-products; ROS reactive oxygen species; SASP senescence associated secretory pathway; STAT signal transducers and activators of transcription; TNF-α tumor necrosis factor-α; TNFR TNF receptor
Collectively, these results show that RAGE is strongly involved in both physiological ageing and inflammaging. As presented below, and in accordance with these data, there is increasing evidence in the literature that RAGE expression and activation are linked to a number of age-related diseases but that, despite the growing literature, its physiological role remains ill-defined.
RAGE and pathologies
Physiological role of RAGE
RAGE possesses all the characteristics of a PRR: its main ligands are almost all DAMPs (HMGB1, S100 s, DNA, RNA) or PAMPs (LPS, DNA, RNA), or else are molecules involved in non-physiological conditions which require resolution (AGEs, amyloid fibrils, complement, Mac-1/CD11b, phosphatidylserine) and engender a pro-inflammatory response. As described earlier, by virtue of being an ubiquitous protein, RAGE is also expressed on innate immunity cells, similar to other PRRs, and its activation mainly triggers PRR-like pro-inflammatory responses, notably through NF-κB signalling (Hofmann et al. 1999; Li et al. 2015; Liu et al. 2010; Yeh et al. 2001).
Similar to TLRs, RAGE has a role in innate immunity and sepsis and Rage−/− mice are protected against lethal septic shock and exhibit significantly reduced NF-κB activity in the lung (Liliensiek et al. 2004). According to its suspected functions and its genomic proximity to other immune system components in the MHC class III locus (Hauptmann and Bahram 2004; Sugaya et al. 1994), RAGE has been labelled a “noncanonical Toll” (Lin 2006). The role of RAGE in innate immunity is not straightforward, however. When infected by S. pneumonia, Rage−/− mice exhibited better survival, a lower pulmonary bacterial load and a decreased dissemination to both blood and the spleen, suggesting a detrimental role of RAGE in host response (van Zoelen et al. 2009). Conversely, M. tuberculosis challenge of Rage−/− mice was associated with increased inflammation, pulmonary bacterial load and loss of body weight, while survival decreased (van Zoelen et al. 2012).
Despite this evidence, the precise role of RAGE remains elusive and other functions of this receptor have been investigated. Since RAGE expression is significantly greater in the lung, we might expect its function to be more important and obvious. However, as reviewed elsewhere, its contribution to lung function remains unclear, under physiological or pathological conditions (Buckley and Ehrhardt 2010). Recent work has shown a diminished elastance and an augmented compliance in the lungs of Rage−/− mice, but these same mice were protected against cigarette smoke-induced lung pathology, probably due to a reduced infiltration of macrophages (Wolf et al. 2017). Other workers have reviewed the negative implications of RAGE in many inflammatory lung diseases (Oczypok et al. 2017). Of note is that, while RAGE is mainly overexpressed in most cancers (Leclerc and Vetter 2015; Shahab et al. 2018; Sparvero et al. 2009), it is on the contrary downregulated in lung cancer (Bartling et al. 2005).
RAGE’s role in lung structure or function remains poorly understood, but its inherent properties and those of its ligands, its signalling pathways and its widespread expression, all seem to suggest it plays a significant role in ageing and inflammaging. A growing body of literature demonstrates RAGE’s involvement in age-related pathologies.
RAGE and age-related diseases
Similar to inflammaging, RAGE is not only involved in a number of pathologies, including retinopathy (Barile and Schmidt 2007), neuropathy (Misur et al. 2004; Sugimoto et al. 2008; Wada and Yagihashi 2005), nephropathies (Forbes et al. 2003; Yamamoto et al. 2007), type 2 diabetes (Kang et al. 2012; Keri et al. 2018) or cancer (Pelucchi et al. 2006; Turner 2015; Yamagishi et al. 2015), but also in more specific age-related diseases such as osteoarthritis (Larkin et al. 2013; Loeser et al. 2005; Sun et al. 2016; Yang et al. 2015), cardiovascular diseases (Barlovic et al. 2010; Fukami et al. 2014; Park et al. 2011; Pettersson-Fernholm et al. 2003) or AD (Batkulwar et al. 2018; Cai et al. 2016; Yan et al. 2009a). With this weight of accumulating evidence, we here suggest that the deleterious effects of RAGE in ageing are closely linked to its pro-inflammatory properties and its contribution to inflammaging.
In a diabetic context, RAGE has been shown to be involved in microvascular and macrovascular complications (Goldin et al. 2006). However, there is also evidence that even in a “healthy” context, the impact of RAGE signalling on vessels is greater than expected, and might even be a driver of arterial ageing. In a study of exposure to CML (an important RAGE ligand), it has been shown that a CML-enriched diet induced accelerated vascular ageing in middle-aged WT mice, characterized by increased vascular stiffening and elastin disruption, reduced aortic relaxation and increased VCAM-1 and RAGE expression, while SIRT1 expression was diminished. However, in Rage−/− mice exposed to the same amount of CML, all of these parameters were completely absent and Rage−/− mice fed a CML-enriched diet exhibited similar vascular ageing to control WT and control Rage−/− mice (Grossin et al. 2015). In another study, aortic relaxation was also measured during physiological ageing and was significantly reduced when old mice were compared with middle-aged mice; strikingly, Rage−/− mice did not exhibit any reduced aortic relaxation at all and were similar to the middle-aged mice in this regard (Teissier et al. 2019). Accordingly, FPS-ZM1, a commercially available RAGE antagonist, was able to efficiently mimic the positive effect of exercise training in old rats by significantly limiting vascular ageing and aorta inflammation (Gu et al. 2014). Finally, RAGE, as much as TLRs, has been proposed as an important target for treatments against atherosclerosis since inflammation is strongly involved in this process (Lin et al. 2009; Olejarz et al. 2018; Senatus and Schmidt 2017).
Dietary CML (dCML) has been shown to preferentially accumulate in kidneys and this accumulation was independent of RAGE (Tessier et al. 2016). However, dCML had little impact on kidney ageing in old mice perhaps due to an increase in competing ligands with age (thus limiting any effect of dCML), an absence of any dCML effect, or too low a dCML concentration being studied. On the other hand, RAGE deletion significantly reduced features of kidney ageing such as fibrosis, tubular atrophy, arteriolar hyalinosis and, more importantly, glomerulosclerosis and glomerular apolipoprotein A-II amyloidosis. Kidney inflammation, characterized by Il-6, Tnf-α and Vcam-1 levels, was also significantly reduced (Teissier et al. 2019). These findings are in line with other results in diabetic nephropathy where RAGE signalling contributes to a pro-inflammatory environment leading to glomerulosclerosis and proteinuria (Daroux et al. 2010; Wendt et al. 2003). Unsurprisingly, chronic inflammation is an important contributor to chronic kidney disease and RAGE could therefore play a role in the occurrence and development of this disease (Akchurin and Kaskel 2015; Silverstein 2009).
Initial studies of RAGE in a murine model of AD showed that RAGE signalling was responsible for inflammation in microglia, critical for the formation of the Aβ amyloid plaques characteristic of the disease (Fang et al. 2010). RAGE activation by AGEs participated in NLRP3 upregulation and in neuroinflammation by activating pro-inflammatory macrophages—this activation was cancelled by the RAGE inhibitor FSP-ZM1 (Chen et al. 2017). Furthermore, RAGE has also been implicated in other features of AD onset independent of inflammation. Its interaction with Aβ peptides is notably thought to have an important role in the transport of Aβ peptides across the blood–brain barrier, favouring the subsequent formation of amyloid plaques (Candela et al. 2010). The use of FPS-ZM1 in a murine model of AD successfully limited Aβ transport as well as associated inflammation, and normalized cognitive functions (Deane et al. 2012). Following these results, a clinical trial was undertaken with another RAGE antagonist, Azeliragon (TTP488); the trial reached phase 3 but was recently terminated owing to a lack of efficacy in the selected cohort. Importantly, however, no significant undesirable effects were reported or were responsible for the cessation of the trial, and it is suggested that the lack of efficacy might be explained by treatment being initiated too late: RAGE blocking remains of interest, therefore, as a therapeutic approach in the early stages of cognitive impairment (Burstein et al. 2018).
Gut ageing is notably associated with changes in microbiota. On the one hand, the diversity of gut microbiota is decreased, while on the other hand pro-inflammatory microbiota such as pathobionts are increased (Biagi et al. 2010; Rampelli et al. 2013) at the expense of anti-inflammatory microbiota such as bifidobacteria, Faecalibacterium prausnitzii or Clostridium cluster IV (Biagi et al. 2016; Hopkins and Macfarlane 2002; Hopkins et al. 2001; Lopetuso et al. 2013; Walton et al. 2012; Zwielehner et al. 2009). It has been demonstrated in mice and non-human primates that such a dysbiosis could lead to increased gut permeability and that gut permeability increases with age (Thevaranjan et al. 2017; Tran and Greenwood-Van Meerveld 2013). As a consequence, altered gut permeability has been linked to leakage to the circulation of bacterial components including the pro-inflammatory LPS (Thevaranjan et al. 2017; Tulkens et al. 2018). While LPS has been described as a ligand of RAGE, little work has been undertaken to clearly describe and understand the importance of this interaction. There is nonetheless a potential role for RAGE where increased LPS leakage from the gut with age could activate RAGE and other LPS receptors to promote inflammaging. Indeed, this hypothesis is strongly supported by a recent study where microbiota from old mice was transferred to young, germ-free mice and promoted several parameters of inflammaging (Fransen et al. 2017). Conversely, older germ free mice had significantly lower plasma IL-6, more functional macrophages and improved survival (Thevaranjan et al. 2017). In addition, in humans, obesity is associated with increased gut permeability, inflammasome activity and RAGE expression (Rainone et al. 2016) and it would be interesting to determine whether this chain of events is also increased in physiological ageing. RAGE signalling is also involved in inflammatory bowel disease in mice and human (Body-Malapel et al. 2019; Wang et al. 2016), suggesting another potential role in gut dysfunction and ageing.
Taken together, then, these findings clearly demonstrate that RAGE is significantly involved in increased chronic inflammation with age, and that this inflammation is closely linked to age-related diseases and perhaps to physiological ageing. Additional, very promising results on the relationship between RAGE, inflammaging and ageing could soon come from a newly generated sRAGEhigh murine model overexpressing circulating sRAGE which is able to block RAGE signalling and/or serves as a decoy for RAGE ligands (Peng et al. 2019).
Conclusion
Historically, RAGE was first isolated from lungs thanks to its binding affinity with AGEs, hence it was first described as the receptor for advanced glycation end-products (Neeper et al. 1992; Schmidt et al. 1992). However, this discovery and the subsequent name and role attributed to RAGE may have diverted attention from its wider physiological role(s) and limited research beyond the effects of RAGE linked to AGEs. Besides, the binding affinity between RAGE and several AGEs is relatively weak in comparison with other ligands such as HMGB1. While an increasingly large body of work has highlighted different functions for RAGE, much remains to be done before the full extent of RAGE’s physiological functions are clearly defined. Its high expression in the lung gives clear hints as to a possible role for RAGE and indeed, as stated above, its expression positively correlates with protection against some pulmonary pathogens (van Zoelen et al. 2012). Thus, close attention should be paid to susceptibility to pathogens when considering the use of RAGE antagonists—however, the recent clinical trial with Azeliragon did not report increased infection rates or mortality associated with this antagonist. Despite the suspected beneficial roles of RAGE, no major defects have been reported upon Rage−/− deletion in mice and, on the contrary, they seem to be protected against a number of physiological and non-physiological conditions.
While full-length RAGE was at the centre of this review, it should be noted that sRAGE, resulting from alternative splicing or RAGE cleavage, is of equivalent importance despite being less expressed in most tissues. Contrary to full-length RAGE, sRAGE is thought to be beneficial by acting as a decoy for RAGE ligands and/or by interacting with RAGE at the membrane, thus preventing further interactions, oligomerization and RAGE signalling. Accordingly, lower levels of circulating sRAGE were found in patients with mild cognitive impairment or AD compared with control subjects (Emanuele et al. 2005; Ghidoni et al. 2008) while sRAGE is the most expressed isoform in the hippocampus (Ding and Keller 2005a), suggesting a protective role of sRAGE against age-related cognitive impairment. Additionally, sRAGE was significantly diminished in precocious patients with acute myocardial infarction compared with young healthy controls while it has been shown to be increased in healthy centenarians (Geroldi et al. 2006). We could surmise that greater sRAGE prevents RAGE signalling and inflammaging and limits the impact of its numerous ligands, particularly if sRAGE remains high with advancing age. This could mirror a paradigm proposed to discriminate successful from unsuccessful ageing: “optimization”, where inflammaging is increased but counteracted by immuno-remodeling and anti-inflammaging as against “deterioration”, where inflammaging is increased but immune-remodeling and anti-inflammaging are insufficient to provide an effective counterbalance (Fulop et al. 2018). Thus, sRAGE could serve as a counterbalance to RAGE and its ligands which increase with age.
Despite its name, RAGE is much more than a receptor for AGEs and attention should not be limited to AGEs or diabetes when studying RAGE and its involvement in different conditions. The variety of its ligands covers a number of molecules involved in many stress events, all leading to a pro-inflammatory response. Thus RAGE should be considered, more than anything else, as a PRR similar to TLRs, NLRs, RLRs or CLRs. As proposed by the literature presented herein, innate immunity is essential in the onset and maintenance of inflammaging and PRRs are important contributors to this innate response. As demonstrated by different studies, the deletion of Rage in mice leads to a pro-longevity phenotype where features of ageing are significantly diminished in vessels or kidneys (Grossin et al. 2015; Teissier et al. 2019), and is associated with a decreased overall inflammatory burden. Evidence is accumulating, therefore, that RAGE plays an important role in inflammaging and that its deletion or blockage has beneficial effects on longevity. In addition, we here suggest that studies on Rage−/− mice or RAGE antagonists provide a proof of concept that PRR blocking is indeed a good target to limit the progression of inflammaging, which increasingly seems to be key component of ageing.
References
Abedini A, Cao P, Plesner A, Zhang J, He M, Derk J, Patil SA, Rosario R, Lonier J, Song F et al (2018) RAGE binds preamyloid IAPP intermediates and mediates pancreatic β cell proteotoxicity. J Clin Invest 128:682–698
Akchurin OM, Kaskel F (2015) Update on inflammation in chronic kidney disease. Blood Purif 39:84–92
Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R, Schmidt AM, Herold KC (2012) RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PLoS ONE 7:e34698
Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR (2010) Peroxisome proliferator-activated receptor γ Co-activator 1α (PGC-1α) and Sirtuin 1 (SIRT1) reside in mitochondria possible direct function in mitochondrial biogenesis. J Biol Chem 285:21590–21599
Arai Y, Martin-Ruiz CM, Takayama M, Abe Y, Takebayashi T, Koyasu S, Suematsu M, Hirose N, von Zglinicki T (2015) Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine 2:1549–1558
Austin S, St-Pierre J (2012) PGC1α and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125:4963–4971
Barile GR, Schmidt AM (2007) RAGE and its ligands in retinal disease. Curr Mol Med 7:758–765
Barlovic DP, Thomas MC, Jandeleit-Dahm K (2010) Cardiovascular disease: what’s all the AGE/RAGE about? Cardiovasc Hematol Disord 10:7–15
Bartling B, Hofmann H-S, Weigle B, Silber R-E, Simm A (2005) Down-regulation of the receptor for advanced glycation end-products (RAGE) supports non-small cell lung carcinoma. Carcinogenesis 26:293–301
Batkulwar K, Godbole R, Banarjee R, Kassaar O, Williams RJ, Kulkarni MJ (2018) Advanced glycation end products modulate amyloidogenic APP processing and tau phosphorylation: a mechanistic link between glycation and the development of Alzheimer’s disease. ACS Chem Neurosci 9:988–1000
Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, Nikkïla J, Monti D, Satokari R, Franceschi C et al (2010) Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 5:e10667
Biagi E, Franceschi C, Rampelli S, Severgnini M, Ostan R, Turroni S, Consolandi C, Quercia S, Scurti M, Monti D et al (2016) Gut microbiota and extreme longevity. Curr Biol CB 26:1480–1485
Bianchi ME, Agresti A (2005) HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15:496–506
Bianchi R, Giambanco I, Donato R (2010) S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging 31:665–677
Bierhaus A, Stern DM, Nawroth PP (2006) RAGE in inflammation: a new therapeutic target? Curr Opin Investig Drugs Lond Engl 2000(7):985–991
Body-Malapel M, Djouina M, Waxin C, Langlois A, Gower-Rousseau C, Zerbib P, Schmidt A-M, Desreumaux P, Boulanger E, Vignal C (2019) The RAGE signaling pathway is involved in intestinal inflammation and represents a promising therapeutic target for Inflammatory Bowel Diseases. Mucosal Immunol 12:468–478
Bongarzone S, Savickas V, Luzi F, Gee AD (2017) Targeting the receptor for advanced glycation endproducts (RAGE): a medicinal chemistry perspective. J Med Chem 60:7213–7232
Boulanger E, Wautier M-P, Wautier J-L, Boval B, Panis Y, Wernert N, Danze P-M, Dequiedt P (2002) AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Int 61:148–156
Boulanger E, Grossin N, Wautier M-P, Taamma R, Wautier J-L (2007) Mesothelial RAGE activation by AGEs enhances VEGF release and potentiates capillary tube formation. Kidney Int 71:126–133
Bresnick AR, Weber DJ, Zimmer DB (2015) S100 proteins in cancer. Nat Rev Cancer 15:96–109
Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A et al (1993) Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143:1699–1712
Buckley ST, Ehrhardt C (2010) The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Corp 3:3. https://doi.org/10.1155/2010/917108
Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L (2018) Development of Azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer’s disease. J Prev Alzheimers Dis 5:149–154
Burton DGA, Stolzing A (2018) Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev 43:17–25
Cai W, He JC, Zhu L, Chen X, Wallenstein S, Striker GE, Vlassara H (2007) Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol 170:1893–1902
Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M, Chang L, Yan L-J, Zhao B (2016) Role of RAGE in Alzheimer’s disease. Cell Mol Neurobiol 36:483–495
Candela P, Gosselet F, Saint-Pol J, Sevin E, Boucau M-C, Boulanger E, Cecchelli R, Fenart L (2010) Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J Alzheimers Dis 22:849–859
Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT et al (2003) The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins. J Exp Med 198:1507–1515
Chen Y, Akirav EM, Chen W, Henegariu O, Moser B, Desai D, Shen JM, Webster JC, Andrews RC, Mjalli AM et al (2008) RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol Baltim Md 1950(181):4272–4278
Chen J, Sun Z, Jin M, Tu Y, Wang S, Yang X, Chen Q, Zhang X, Han Y, Pi R (2017) Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J Neuroimmunol 305:108–114
Cheng C, Tsuneyama K, Kominami R, Shinohara H, Sakurai S, Yonekura H, Watanabe T, Takano Y, Yamamoto H, Yamamoto Y (2005) Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod Pathol 18:1385–1396
Comenzo RL (2000) Primary systemic amyloidosis. Curr Treat Opt Oncol 1:83–89
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118
Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A et al (2016) Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35:724–742
Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan ALY, Fukami K, Thallas-Bonke V, Nawroth PP et al (2009) RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol JASN 20:742–752
Crow YJ, Manel N (2015) Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15:429–440
Daroux M, Prévost G, Maillard-Lefebvre H, Gaxatte C, D’Agati VD, Schmidt AM, Boulanger E (2010) Advanced glycation end-products: implications for diabetic and non-diabetic nephropathies. Diabetes Metab 36:1–10
Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe J-P, Rodier F, Campisi J (2013) p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201:613–629
de Gonzalo-Calvo D, Neitzert K, Fernández M, Vega-Naredo I, Caballero B, García-Macía M, Suárez FM, Rodríguez-Colunga MJ, Solano JJ, Coto-Montes A (2010) Differential inflammatory responses in aging and disease: TNF-alpha and IL-6 as possible biomarkers. Free Radic Biol Med 49:733–737
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ et al (2012) A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122:1377–1392
Del Rio D, Stewart AJ, Pellegrini N (2005) A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis NMCD 15:316–328
Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP (2006) Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res 323:475–488
Dinarello CA (2006) Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr 83:447S–455S
Ding Q, Keller JN (2005a) Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci Lett 373:67–72
Ding Q, Keller JN (2005b) Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim Biophys. Acta BBA 1746:18–27
Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z et al (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550:402–406
Emanuele E, D’Angelo A, Tomaino C, Binetti G, Ghidoni R, Politi P, Bernardi L, Maletta R, Bruni AC, Geroldi D (2005) Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol 62:1734–1736
Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A (2010) High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem 285:39888–39897
Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R (1993) Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol 23:2375–2378
Fang F, Lue L-F, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM et al (2010) RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J 24:1043–1055
Fatchiyah F, Hardiyanti F, Widodo N (2015) Selective inhibition on RAGE-binding AGEs required by bioactive peptide alpha-S2 case in protein from goat Ethawah breed milk: study of biological modeling. Acta Inform Med 23:90–96
Ferrucci L, Guralnik JM, Woodman RC, Bandinelli S, Lauretani F, Corsi AM, Chaves PHM, Ershler WB, Longo DL (2005a) Proinflammatory state and circulating erythropoietin in persons with and without anemia. Am J Med 118:1288
Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub DD, Guralnik JM, Longo DL (2005b) The origins of age-related proinflammatory state. Blood 105:2294–2299
Forbes JM, Cooper ME, Oldfield MD, Thomas MC (2003) Role of advanced glycation end products in diabetic nephropathy. J Am Soc Nephrol JASN 14:S254–S258
Franceschi C, Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol Ser A 69:S4–S9
Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S (2017) Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab 28:199–212
Franchi L, Muñoz-Planillo R, Núñez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332
Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, Savelkoul HFJ, De Jonge MI, Boekschoten MV, Smidt H et al (2017) Aged gut microbiota contributes to systemical inflammaging after transfer to germ-free mice. Front Immunol 8:1385
Fukami K, Yamagishi S-I, Okuda S (2014) Role of AGEs-RAGE system in cardiovascular disease. Curr Pharm Des 20:2395–2402
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C (2018) Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes?. Front, Immunol, p 8
Gao ZQ, Yang C, Wang YY, Wang P, Chen HL, Zhang XD, Liu R, Li WL, Qin XJ, Liang X et al (2008) RAGE upregulation and nuclear factor-kappaB activation associated with ageing rat cardiomyocyte dysfunction. Gen Physiol Biophys 27:152–158
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3:995–1001
Gerli R, Monti D, Bistoni O, Mazzone AM, Peri G, Cossarizza A, Di Gioacchino M, Cesarotti ME, Doni A, Mantovani A et al (2000) Chemokines, sTNF-Rs and sCD30 serum levels in healthy aged people and centenarians. Mech Ageing Dev 121:37–46
Geroldi D, Falcone C, Minoretti P, Emanuele E, Arra M, D’Angelo A (2006) High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J Am Geriatr Soc 54:1149–1150
Ghidoni R, Benussi L, Glionna M, Franzoni M, Geroldi D, Emanuele E, Binetti G (2008) Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment. J Neural Transm 1996(115):1047–1050
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890
Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114:597–605
Grossin N, Auger F, Niquet-Leridon C, Durieux N, Montaigne D, Schmidt AM, Susen S, Jacolot P, Beuscart J-B, Tessier FJ et al (2015) Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol Nutr Food Res 59:927–938
Gu Q, Wang B, Zhang X-F, Ma Y-P, Liu J-D, Wang X-Z (2014) Contribution of receptor for advanced glycation end products to vasculature-protecting effects of exercise training in aged rats. Eur J Pharmacol 741:186–194
Guo ZJ, Niu HX, Hou FF, Zhang L, Fu N, Nagai R, Lu X, Chen BH, Shan YX, Tian JW et al (2008) Advanced oxidation protein products activate vascular endothelial cells via a RAGE-mediated signaling pathway. Antioxid Redox Signal 10:1699–1712
Gursky O (2014) Hot spots in apolipoprotein A-II misfolding and amyloidosis in mice and men. FEBS Lett 588:845–850
Hallam KM, Li Q, Ananthakrishnan R, Kalea A, Zou YS, Vedantham S, Schmidt AM, Yan SF, Ramasamy R (2010) Aldose Reductase and AGE–RAGE pathways: central roles in the pathogenesis of vascular dysfunction in aging rats. Aging Cell 9:776–784
Harris HE, Andersson U (2004) Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol 34:1503–1512
Hauptmann G, Bahram S (2004) Genetics of the central MHC. Curr Opin Immunol 16:668–672
He Q, Liang CH, Lippard SJ (2000) Steroid hormones induce HMG1 overexpression and sensitize breast cancer cells to cisplatin and carboplatin. Proc Natl Acad Sci USA 97:5768–5772
He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y et al (2011) Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep 12:358–364
Hearps AC, Martin GE, Angelovich TA, Cheng W-J, Maisa A, Landay AL, Jaworowski A, Crowe SM (2012) Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11:867–875
Higuchi K, Kitagawa K, Naiki H, Hanada K, Hosokawa M, Takeda T (1991) Polymorphism of apolipoprotein A-II (apoA-II) among inbred strains of mice. Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Biochem J 279(2):427–433
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P et al (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Hopkins MJ, Macfarlane GT (2002) Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol 51:448–454
Hopkins MJ, Sharp R, Macfarlane GT (2001) Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut 48:198–205
Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 270:25752–25761
Howes KA, Liu Y, Dunaief JL, Milam A, Frederick JM, Marks A, Baehr W (2004) Receptor for advanced glycation end products and age-related macular degeneration. Invest Ophthalmol Vis Sci 45:3713–3720
Huang JS, Guh JY, Chen HC, Hung WC, Lai YH, Chuang LY (2001) Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J Cell Biochem 81:102–113
Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM (2008a) Identification, classification, and expression of RAGE gene splice variants. FASEB J 22:1572–1580
Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM (2008b) Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 283:34457–34468
Iannuzzi C, Irace G, Sirangelo I (2014) Differential effects of glycation on protein aggregation and amyloid formation. Front Mol Biosci 1:9
Ito H, Fujita K, Tagawa K, Chen X, Homma H, Sasabe T, Shimizu J, Shimizu S, Tamura T, Muramatsu S et al (2015) HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol Med 7:78–101
Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF et al (2013) Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202:129–143
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD (2010) Mitochondrial proton and electron leaks. Essays Biochem 47:53–67
Jeon OH, David N, Campisi J, Elisseeff JH (2018) Senescent cells and osteoarthritis: a painful connection. J Clin Invest 128:1229–1237
Jules J, Maiguel D, Hudson BI (2013) Alternative splicing of the RAGE cytoplasmic domain regulates cell signaling and function. PLoS ONE 8:e78267
Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R et al (2014) Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun 5:4172
Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI (2009) Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J 23:1766–1774
Kang P, Tian C, Jia C (2012) Association of RAGE gene polymorphisms with type 2 diabetes mellitus, diabetic retinopathy and diabetic nephropathy. Gene 500:1–9
Kang R, Chen R, Xie M, Cao L, Lotze MT, Tang D, Zeh HJ (2016) The receptor for advanced glycation endproducts (RAGE) activates the AIM2 inflammasome in acute pancreatitis. J Immunol Baltim Md 1950(196):4331–4337
Kaufmann SHE, Dorhoi A (2016) Molecular determinants in phagocyte-bacteria interactions. Immunity 44:476–491
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE et al (2014a) Geroscience. Cell 159:709–713
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE et al (2014b) Aging: a common driver of chronic diseases and a target for novel interventions. Cell 159:709–713
Keri KC, Samji NS, Blumenthal S (2018) Diabetic nephropathy: newer therapeutic perspectives. J Community Hosp Intern Med Perspect 8:200–207
Kierdorf K, Fritz G (2013) RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol 94:55–68
Kim H-R, Won SJ, Fabian C, Kang M-G, Szardenings M, Shin M-G (2015) Mitochondrial DNA aberrations and pathophysiological implications in hematopoietic diseases, chronic inflammatory diseases, and cancers. Ann Lab Med 35:1–14
Kim EJ, Park SY, Baek SE, Jang MA, Lee WS, Bae SS, Kim K, Kim CD (2018) HMGB1 increases IL-1β production in vascular smooth muscle cells via NLRP3 inflammasome. Front Physiol 9:313
Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D et al (1999) N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 274:31740–31749
Kitagawa K, Wang J, Mastushita T, Kogishi K, Hosokawa M, Fu X, Guo Z, Mori M, Higuchi K (2003) Polymorphisms of mouse apolipoprotein A-II: seven alleles found among 41 inbred strains of mice. Amyloid 10:207–214
Kuhla A, Hettwer C, Menger MD, Vollmar B (2010) Oxidative stress-associated rise of hepatic protein glycation increases inflammatory liver injury in uncoupling protein-2 deficient mice. Lab. Investig. J. Tech. Methods Pathol. 90:1189–1198
Kuhla A, Hauke M, Sempert K, Vollmar B, Zechner D (2013) Senescence-dependent impact of anti-RAGE antibody on endotoxemic liver failure. Age 35:2153–2163
Kumar V, Fleming T, Terjung S, Gorzelanny C, Gebhardt C, Agrawal R, Mall MA, Ranzinger J, Zeier M, Madhusudhan T et al (2017) Homeostatic nuclear RAGE-ATM interaction is essential for efficient DNA repair. Nucleic Acids Res 45:10595–10613
Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O, Estaquier J (2016) NF-κB pathway controls mitochondrial dynamics. Cell Death Differ 23:89–98
Lange SS, Vasquez KM (2009) HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinog 48:571–580
Lange SS, Mitchell DL, Vasquez KM (2008) High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci USA 105:10320–10325
Larkin DJ, Kartchner JZ, Doxey AS, Hollis WR, Rees JL, Wilhelm SK, Draper CS, Peterson DM, Jackson GG, Ingersoll C et al (2013) Inflammatory markers associated with osteoarthritis after destabilization surgery in young mice with and without receptor for advanced glycation end-products (RAGE). Front Physiol 4:121
Leclerc E, Vetter SW (2015) The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim Biophys Acta 1852:2706–2711
Leclerc E, Fritz G, Vetter SW, Heizmann CW (2009) Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 1793:993–1007
Li J, Schmidt AM (1997) Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 272:16498–16506
Li L, Liu X, Glassman AB, Keating MJ, Stros M, Plunkett W, Yang L-Y (1997) Fludarabine triphosphate inhibits nucleotide excision repair of cisplatin-induced DNA adducts in vitro. Cancer Res 57:1487–1494
Li Y, Wu R, Tian Y, Yu M, Tang Y, Cheng H, Tian Z (2015) RAGE/NF-κB signaling mediates lipopolysaccharide induced acute lung injury in neonate rat model. Int J Clin Exp Med 8:13371–13376
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne H-J, Kurschus FC, Schmidt AM et al (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113:1641–1650
Lin L (2006) RAGE on the Toll Road? Cell Mol Immunol 3:351–358
Lin L, Park S, Lakatta EG (2009) RAGE signaling in inflammation and arterial aging. Front Biosci 14:1403–1413
Liu Y, Liang C, Liu X, Liao B, Pan X, Ren Y, Fan M, Li M, He Z, Wu J et al (2010) AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis 208:34–42
Liu J, Huang K, Cai G-Y, Chen X-M, Yang J-R, Lin L-R, Yang J, Huo B-G, Zhan J, He Y-N (2014) Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal 26:110–121
Lo M-C, Chen M-H, Lee W-S, Lu C-I, Chang C-R, Kao S-H, Lee H-M (2015) Nε-(carboxymethyl) lysine-induced mitochondrial fission and mitophagy cause decreased insulin secretion from β-cells. Am J Physiol Endocrinol Metab 309:E829–E839
Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im H-J, Bursch LS, Yan SD (2005) Articular chondrocytes express the receptor for advanced glycation end products: potential role in osteoarthritis. Arthritis Rheum 52:2376–2385
Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A (2013) Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 5:23
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Ma W, Rai V, Hudson BI, Song F, Schmidt AM, Barile GR (2012) RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell Immunol 274:72–82
Magna M, Pisetsky DS (2014) The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med 20:138–146
Maillard-Lefebvre H, Boulanger E, Daroux M, Gaxatte C, Hudson BI, Lambert M (2009) Soluble receptor for advanced glycation end products: a new biomarker in diagnosis and prognosis of chronic inflammatory diseases. Rheumatol Oxf Engl 48:1190–1196
Malaquin N, Martinez A, Rodier F (2016) Keeping the senescence secretome under control: molecular reins on the senescence-associated secretory phenotype. Exp Gerontol 82:39–49
Man SM, Kanneganti T-D (2015) Regulation of inflammasome activation. Immunol Rev 265:6–21
Manfredi AA, Capobianco A, Esposito A, De Cobelli F, Canu T, Monno A, Raucci A, Sanvito F, Doglioni C, Nawroth PP et al (2008) Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol Baltim Md 1950(180):2270–2275
Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, DeVita RJ, Ramasamy R, Shekhtman A, Schmidt AM (2016) Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci Rep 6:22450
Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, Kubarenko AV, Andreeva L, Hopfner K-P, Hornung V (2014) Cytosolic RNA:DNA hybrids activate the cGAS–STING axis. EMBO J 33:2937–2946
Mao YX, Cai WJ, Sun XY, Dai PP, Li XM, Wang Q, Huang XL, He B, Wang PP, Wu G et al (2018) RAGE-dependent mitochondria pathway: a novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis 9:674
Mariani E, Cattini L, Neri S, Malavolta M, Mocchegiani E, Ravaglia G, Facchini A (2006) Simultaneous evaluation of circulating chemokine and cytokine profiles in elderly subjects by multiplex technology: relationship with zinc status. Biogerontology 7:449–459
Masters PM, Bada JL, Zigler JS (1977) Aspartic acid racemisation in the human lens during ageing and in cataract formation. Nature 268:71–73
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985a) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249
Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K (1985b) Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J 4:2757–2763
Minciullo PL, Catalano A, Mandraffino G, Casciaro M, Crucitti A, Maltese G, Morabito N, Lasco A, Gangemi S, Basile G (2016) Inflammaging and anti-inflammaging: the role of cytokines in extreme longevity. Arch Immunol Ther Exp (Warsz) 64:111–126
Misur I, Zarković K, Barada A, Batelja L, Milicević Z, Turk Z (2004) Advanced glycation endproducts in peripheral nerve in type 2 diabetes with neuropathy. Acta Diabetol 41:158–166
Monnier VM, Cerami A (1981) Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science 211:491–493
Morizane R, Monkawa T, Konishi K, Hashiguchi A, Ueda M, Ando Y, Tokuyama H, Hayashi K, Hayashi M, Itoh H (2011) Renal amyloidosis caused by apolipoprotein A-II without a genetic mutation in the coding sequence. Clin Exp Nephrol 15:774–779
Morrisette-Thomas V, Cohen AA, Fülöp T, Riesco É, Legault V, Li Q, Milot E, Dusseault-Bélanger F, Ferrucci L (2014) Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech Ageing Dev 139:49–57
Moser B, Desai DD, Downie MP, Chen Y, Yan SF, Herold K, Schmidt AM, Clynes R (2007) Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J Immunol Baltim Md 1950(179):8051–8058
Most P, Seifert H, Gao E, Funakoshi H, Völkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Eckhart AD et al (2006) Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation 114:1258–1268
Narumi K, Miyakawa R, Ueda R, Hashimoto H, Yamamoto Y, Yoshida T, Aoki K (2015) Proinflammatory proteins S100A8/S100A9 activate NK cells via interaction with RAGE. J Immunol Baltim Md 1950(194):5539–5548
Navarrete Santos A, Jacobs K, Simm A, Glaubitz N, Horstkorte R, Hofmann B (2017) Dicarbonyls induce senescence of human vascular endothelial cells. Mech Ageing Dev 166:24–32
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004
Neviere R, Yu Y, Wang L, Tessier F, Boulanger E (2016) Implication of advanced glycation end products (Ages) and their receptor (Rage) on myocardial contractile and mitochondrial functions. Glycoconj J 33:607–617
Nguyen THO, Sant S, Bird NL, Grant EJ, Clemens EB, Koutsakos M, Valkenburg SA, Gras S, Lappas M, Jaworowski A et al (2018) Perturbed CD8+ T cell immunity across universal influenza epitopes in the elderly. J Leukoc Biol 103:321–339
Oczypok EA, Perkins TN, Oury TD (2017) All the “RAGE” in lung disease: the receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr Respir Rev 23:40–49
Olejarz W, Łacheta D, Głuszko A, Migacz E, Kukwa W, Szczepański MJ, Tomaszewski P, Nowicka G (2018) RAGE and TLRs as key targets for antiatherosclerotic therapy. Biomed Res Int. https://doi.org/10.1155/2018/7675286
Oliveira JB, Soares AASM, Sposito AC (2018) Inflammatory response during myocardial infarction. Adv Clin Chem 84:39–79
Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B, Heizmann CW, Kroneck PMH, Fritz G (2007) Structural and functional insights into RAGE activation by multimeric S100B. EMBO J 26:3868–3878
Park H, Adsit FG, Boyington JC (2010) The 1.5 Å crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J Biol Chem 285:40762–40770
Park S, Yoon S-J, Tae H-J, Shim CY (2011) RAGE and cardiovascular disease. Front Biosci Landmark Ed 16:486–497
Pawelec G (2017) Immunosenescence and cancer. Biogerontology 18:717–721
Pelucchi C, Galeone C, Levi F, Negri E, Franceschi S, Talamini R, Bosetti C, Giacosa A, La Vecchia C (2006) Dietary acrylamide and human cancer. Int J Cancer 118:467–471
Peng Q, Li K, Sacks SH, Zhou W (2009) The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm Allergy Drug Targets 8:236–246
Peng Y, Park H-S, Tang LA, Horwitz N, Lin L (2019) Generation of sRAGEhigh transgenic mice to study inflammaging. Front Biosci Landmark Ed 24:555–563
Pettersson-Fernholm K, Forsblom C, Hudson BI, Perola M, Grant PJ, Groop P-H (2003) The functional —374 T/A RAGE gene polymorphism is associated with proteinuria and cardiovascular disease in type 1 diabetic patients. Diabetes 52:891–894
Porcel JM, Ordi J, Castro-Salomo A, Vilardell M, Rodrigo MJ, Gene T, Warburton F, Kraus M, Vergani D (1995) The value of complement activation products in the assessment of systemic lupus erythematosus flares. Clin Immunol Immunopathol 74:283–288
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Prusiner SB (1991) Molecular biology of prion diseases. Science 252:1515–1522
Rai V, Maldonado AY, Burz DS, Reverdatto S, Yan SF, Schmidt AM, Shekhtman A (2012a) Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem 287:5133–5144
Rai V, Touré F, Chitayat S, Pei R, Song F, Li Q, Zhang J, Rosario R, Ramasamy R, Chazin WJ et al (2012b) Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J Exp Med 209:2339–2350
Rainone V, Schneider L, Saulle I, Ricci C, Biasin M, Al-Daghri NM, Giani E, Zuccotti GV, Clerici M, Trabattoni D (2016) Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int J Obes 2005(40):1026–1033
Rambaran RN, Serpell LC (2008) Amyloid fibrils. Prion 2:112–117
Rampelli S, Candela M, Turroni S, Biagi E, Collino S, Franceschi C, O’Toole PW, Brigidi P (2013) Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging 5:902–912
Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J Off Publ Fed Am Soc Exp Biol 22:3716–3727
Ray R, Juranek JK, Rai V (2016) RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev 62:48–55
Rivera A, Siracusa MC, Yap GS, Gause WC (2016) Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol 17:356–363
Ruan BH, Li X, Winkler AR, Cunningham KM, Kuai J, Greco RM, Nocka KH, Fitz LJ, Wright JF, Pittman DD et al (2010) Complement C3a, CpG oligos, and DNA/C3a complex stimulate IFN-α production in a receptor for advanced glycation end product-dependent manner. J Immunol Baltim Md 1950(185):4213–4222
Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh N (2011) TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE 6(8):e23132
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Schmidt AM (2017) RAGE and implications for the pathogenesis and treatment of cardiometabolic disorders—spotlight on the macrophage. Arterioscler Thromb Vasc Biol 37:613–621
Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M (1992) Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem 267:14987–14997
Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F (2010) Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol 11:30
Sell DR, Lane MA, Johnson WA, Masoro EJ, Mock OB, Reiser KM, Fogarty JF, Cutler RG, Ingram DK, Roth GS et al (1996) Longevity and the genetic determination of collagen glycoxidation kinetics in mammalian senescence. Proc Natl Acad Sci USA 93:485–490
Sellier C, Boulanger E, Maladry F, Tessier FJ, Lorenzi R, Nevière R, Desreumaux P, Beuscart J-B, Puisieux F, Grossin N (2015) Acrylamide induces accelerated endothelial aging in a human cell model. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc 83:140–145
Senatus LM, Schmidt AM (2017) The AGE-RAGE axis: implications for age-associated arterial diseases. Front, Genet, p 8
Serratos IN, Castellanos P, Pastor N, Millán-Pacheco C, Rembao D, Pérez-Montfort R, Cabrera N, Reyes-Espinosa F, Díaz-Garrido P, López-Macay A et al (2015) Modeling the Interaction between Quinolinate and the Receptor for Advanced Glycation End Products (RAGE): relevance for Early Neuropathological Processes. PLoS ONE 10(3):120221
Sessa L, Gatti E, Zeni F, Antonelli A, Catucci A, Koch M, Pompilio G, Fritz G, Raucci A, Bianchi ME (2014) The receptor for advanced glycation end-products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS ONE 9:e86903
Shahab U, Ahmad MK, Mahdi AA, Waseem M, Arif B, Moinuddin, Ahmad S (2018) The receptor for advanced glycation end products: a fuel to pancreatic cancer. Semin Cancer Biol 49:37–43
Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, Al-Dabet MM, Ranjan S, Wolter J, Wacker C et al (2015) Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int 87:74–84
Shen YJ, Le Bert N, Chitre AA, Koo CX, Nga XH, Ho SSW, Khatoo M, Tan NY, Ishii KJ, Gasser S (2015) Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 11:460–473
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414
Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, Hata Y (2004) Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 9:165–174
Shuvaev VV, Laffont I, Serot JM, Fujii J, Taniguchi N, Siest G (2001) Increased protein glycation in cerebrospinal fluid of Alzheimer’s disease. Neurobiol Aging 22:397–402
Silverstein DM (2009) Inflammation in chronic kidney disease: role in the progression of renal and cardiovascular disease. Pediatr. Nephrol. Berl. Ger. 24:1445–1452
Simm A, Casselmann C, Schubert A, Hofmann S, Reimann A, Silber R-E (2004) Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol 39:407–413
Somensi N, Brum PO, de Miranda Ramos V, Gasparotto J, Zanotto-Filho A, Rostirolla DC, da Silva Morrone M, Moreira JCF, Pens Gelain D (2017) Extracellular HSP70 activates ERK1/2, NF-kB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 42:2507–2522
Son M, Chung W-J, Oh S, Ahn H, Choi CH, Hong S, Park KY, Son KH, Byun K (2017) Age dependent accumulation patterns of advanced glycation end product receptor (RAGE) ligands and binding intensities between RAGE and its ligands differ in the liver, kidney, and skeletal muscle. Immun Ageing A 14:12
Song Y, Wang Y, Zhang Y, Geng W, Liu W, Gao Y, Li S, Wang K, Wu X, Kang L et al (2017) Advanced glycation end products regulate anabolic and catabolic activities via NLRP3-inflammasome activation in human nucleus pulposus cells. J Cell Mol Med 21:1373–1387
Sorci G, Riuzzi F, Giambanco I, Donato R (2013) RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 1833:101–109
Sousa MM, Yan SD, Stern D, Saraiva MJ (2000) Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab Invest 80:1101–1110
Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ et al (2009) RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 7:17
Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, Inoko H, Ikemura T (1994) Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 23:408–419
Sugimoto K, Yasujima M, Yagihashi S (2008) Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des 14:953–961
Sun X-H, Liu Y, Han Y, Wang J (2016) Expression and significance of high-mobility group protein B1 (HMGB1) and the receptor for advanced glycation end-product (RAGE) in knee osteoarthritis. Med Sci Monit Int Med J Exp Clin Res 22:2105–2112
Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR (2012) Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol Clifton NJ 810:183–205
Tan ALY, Sourris KC, Harcourt BE, Thallas-Bonke V, Penfold S, Andrikopoulos S, Thomas MC, O’Brien RC, Bierhaus A, Cooper ME et al (2009) Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am J Physiol Ren Physiol 298:F763–F770
Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H (2000) The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem 275:25781–25790
Teh BK, Yeo JG, Chern LM, Lu J (2011) C1q regulation of dendritic cell development from monocytes with distinct cytokine production and T cell stimulation. Mol Immunol 48:1128–1138
Teissier T, Quersin V, Gnemmi V, Daroux M, Howsam M, Delguste F, Lemoine C, Fradin C, Schmidt A-M, Cauffiez C et al (2019) Knockout of receptor for advanced glycation end-products attenuates age-related renal lesions. Aging Cell 18:e12850
Tessier FJ, Niquet-Léridon C, Jacolot P, Jouquand C, Genin M, Schmidt A-M, Grossin N, Boulanger E (2016) Quantitative assessment of organ distribution of dietary protein-bound (13) C-labeled N(ɛ)-carboxymethyllysine after a chronic oral exposure in mice. Mol Nutr Food Res 60(11):2446–2456
Thankam FG, Roesch ZK, Dilisio MF, Radwan MM, Kovilam A, Gross RM, Agrawal DK (2018) Association of inflammatory responses and ECM disorganization with HMGB1 upregulation and NLRP3 inflammasome activation in the injured rotator cuff tendon. Sci Rep 8:8918
Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP et al (2017) Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21:455.e4–466.e4
Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 375:581–592
Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C et al (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Tran L, Greenwood-Van Meerveld B (2013) Age-associated remodeling of the intestinal epithelial barrier. J Gerontol A 68:1045–1056
Tsung A, Tohme S, Billiar TR (2014) High-mobility group box-1 in sterile inflammation. J Intern Med 276:425–443
Tulkens J, Vergauwen G, Van Deun J, Geeurickx E, Dhondt B, Lippens L, De Scheerder M-A, Miinalainen I, Rappu P, De Geest BG et al (2018) Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut. https://doi.org/10.1136/gutjnl-2018-317726
Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797:113–128
Turner DP (2015) Advanced glycation end-products: a biological consequence of lifestyle contributing to cancer disparity. Cancer Res 75:1925–1929
van Hout GPJ, Arslan F, Pasterkamp G, Hoefer IE (2016) Targeting danger-associated molecular patterns after myocardial infarction. Expert Opin Ther Targets 20:223–239
van Zoelen MAD, Schouten M, de Vos AF, Florquin S, Meijers JCM, Nawroth PP, Bierhaus A, van der Poll T (2009) The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J Immunol Baltim Md 1950(182):4349–4356
van Zoelen MAD, Wieland CW, van der Windt GJW, Florquin S, Nawroth PP, Bierhaus A, van der Poll T (2012) Receptor for advanced glycation end products is protective during murine tuberculosis. Mol Immunol 52:183–189
Venereau E, Schiraldi M, Uguccioni M, Bianchi ME (2013) HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol 55:76–82
Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM (2000) Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem 275:39027–39031
Wada R, Yagihashi S (2005) Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann N Y Acad Sci 1043:598–604
Walton GE, van den Heuvel EGHM, Kosters MHW, Rastall RA, Tuohy KM, Gibson GR (2012) A randomised crossover study investigating the effects of galacto-oligosaccharides on the faecal microbiota in men and women over 50 years of age. Br J Nutr 107:1466–1475
Wang JQ, Jeelall YS, Ferguson LL, Horikawa K (2014) Toll-like receptors and cancer: MYD88 mutation and inflammation. Front, Immunol, p 5
Wang J, Zeng J, Wang H, Ye S, Bi Y, Zhou Y, Li K, Zhou Y (2016) Genetic polymorphisms of RAGE and risk of ulcerative colitis in a Chinese population. Immunol Lett 170:88–94
Ward MS, Fortheringham AK, Cooper ME, Forbes JM (2013) Targeting advanced glycation endproducts and mitochondrial dysfunction in cardiovascular disease. Curr Opin Pharmacol 13:654–661
Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL (2001) Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 280:E685–E694
Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS et al (2003) RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162:1123–1137
Wolf L, Herr C, Niederstraßer J, Beisswenger C, Bals R (2017) Receptor for advanced glycation endproducts (RAGE) maintains pulmonary structure and regulates the response to cigarette smoke. PLoS ONE 12:e0180092
Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A (2007) Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem 282:4218–4231
Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A (2008) Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J Biol Chem 283:27255–27269
Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM (2013) Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 25:2185–2197
Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, Curran GL, Ogrodnik M, Jurk D, Johnson KO et al (2017) Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A 72:780–785
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG et al (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24:1246
Xue J, Rai V, Singer D, Chabierski S, Xie J, Reverdatto S, Burz DS, Schmidt AM, Hoffmann R, Shekhtman A (2011) Advanced glycation end product recognition by the receptor for AGEs. Struct Lond Engl 1993(19):722–732
Xue J, Ray R, Singer D, Böhme D, Burz DS, Rai V, Hoffmann R, Shekhtman A (2014) The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 53:3327–3335
Yamagishi S, Matsui T, Fukami K (2015) Role of receptor for advanced glycation end products (RAGE) and its ligands in cancer risk. Rejuvenation Res 18:48–56
Yamamoto H, Watanabe T, Yamamoto Y, Yonekura H, Munesue S, Harashima A, Ooe K, Hossain S, Saito H, Murakami N (2007) RAGE in diabetic nephropathy. Curr Mol Med 7:752–757
Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S et al (2011) Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol Baltim Md 1950(186):3248–3257
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J et al (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D et al (2000) Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 6:643–651
Yan SD, Bierhaus A, Nawroth PP, Stern DM (2009a) RAGE and Alzheimer’s disease: a progression factor for amyloid-β-induced cellular perturbation? J Alzheimers Dis JAD 16:833–843
Yan SF, Yan SD, Ramasamy R, Schmidt AM (2009b) Tempering the wrath of RAGE: an emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med 41:408–422
Yang HY, Chuang SY, Fang WH, Huang GS, Wang CC, Huang YY, Chu MY, Lin C, Su W, Chen CY et al (2015) Effect of RAGE polymorphisms on susceptibility to and severity of osteoarthritis in a Han Chinese population: a case-control study. Genet Mol Res GMR 14:11362–11370
Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, Zang QS (2015) Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 10:e0139416
Yatime L, Andersen GR (2013) Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J 280:6556–6568
Yeh CH, Sturgis L, Haidacher J, Zhang XN, Sherwood SJ, Bjercke RJ, Juhasz O, Crow MT, Tilton RG, Denner L (2001) Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes 50:1495–1504
Yu Y, Wang L, Delguste F, Durand A, Guilbaud A, Rousselin C, Schmidt AM, Tessier F, Boulanger E, Neviere R (2017) Advanced glycation end products receptor RAGE controls myocardial dysfunction and oxidative stress in high-fat fed mice by sustaining mitochondrial dynamics and autophagy-lysosome pathway. Free Radic Biol Med 112:397–410
Yu W, Tao M, Zhao Y, Hu X, Wang M (2018) 4′-Methoxyresveratrol alleviated AGE-induced inflammation via RAGE-mediated NF-κB and NLRP3 inflammasome pathway. Mol J Synth Chem Nat Prod, Chem, p 23
Zeng S, Zhang QY, Huang J, Vedantham S, Rosario R, Ananthakrishnan R, Yan SF, Ramasamy R, DeMatteo RP, Emond JC et al (2012) Opposing roles of RAGE and Myd88 signaling in extensive liver resection. FASEB J 26:882–893
Zhang L, Bukulin M, Kojro E, Roth A, Metz VV, Fahrenholz F, Nawroth PP, Bierhaus A, Postina R (2008) Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem 283:35507–35516
Zhang H, Puleston DJ, Simon AK (2016) Autophagy and Immune Senescence. Trends Mol Med 22:671–686
Zong H, Madden A, Ward M, Mooney MH, Elliott CT, Stitt AW (2010) Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J Biol Chem 285:23137–23146
Zwielehner J, Liszt K, Handschur M, Lassl C, Lapin A, Haslberger AG (2009) Combined PCR-DGGE fingerprinting and quantitative-PCR indicates shifts in fecal population sizes and diversity of Bacteroides, bifidobacteria and Clostridium cluster IV in institutionalized elderly. Exp Gerontol 44:440–446
Acknowledgements
We are grateful to Mike Howsam for his contribution of editorial assistance and English proofreading.
We thank LES LABORATOIRES SERVIER who have shared, under the creative commons license, some of the graphic elements that were adapted for the figures herein.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Teissier, T., Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 20, 279–301 (2019). https://doi.org/10.1007/s10522-019-09808-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-019-09808-3