
Abstract
Although the majority of renal amyloidosis is caused by either acquired monoclonal immunoglobulin light-chain amyloidosis or reactive systemic amyloid A, some cases are caused by hereditary amyloidosis. Apolipoprotein A-II (apoAII) amyloidosis is a rare form of hereditary amyloidosis and cannot be diagnosed by a routine examination. Thus, the prevalence and etiology of apoAII amyloidosis are uncertain. In humans, a genetic mutation in the stop codon of apoAII is considered to be a cause of amyloid fibril formation. We report on a 68-year-old man who presented with proteinuria by apoAII amyloidosis without family history. His proteinuria gradually increased to 6 g/day within 1 year. A renal biopsy showed amyloid deposition in the glomeruli, however, acquired monoclonal immunoglobulin light-chain amyloidosis and reactive systemic amyloid A were ruled out. Immunohistochemistry revealed apoAII deposition in the glomeruli, but DNA sequencing did not identify any genetic mutation in the coding sequence of apoAII. Here, we report a case of apoAII amyloidosis without a genetic mutation in the coding sequence and discuss the etiology of apoAII amyloidosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyloidosis is a heterogeneous disease characterized by the deposition of amyloid fibrils in an antiparallel beta-pleated sheet configuration. Acquired monoclonal immunoglobulin light-chain (AL) amyloidosis is the most common form of amyloidosis, in which protein derived from immunoglobulin light-chain fragments is deposited. AL amyloidosis is caused by plasma cell dyscrasia that produces monoclonal immunoglobulin. Another common cause of amyloidosis is reactive systemic amyloid A (AA) amyloidosis, which is a complication of chronic inflammatory conditions such as rheumatoid arthritis, spondyloarthropathy, inflammatory bowel disease, or chronic infections.
Although AL and AA amyloidoses are the major causes of amyloidosis, hereditary amyloidosis is also known to occur. Lachmann et al. [1] reported that 10% of all cases diagnosed as AL amyloidosis are actually hereditary amyloidosis. Among the forms of hereditary amyloidosis, transthyretin, fibrinogen A alpha, apolipoprotein A-I (apoAI), apolipoprotein A-II (apoAII), lysozyme, gelsolin and leukocyte chemotactic factor 2 (LECT2) are known to affect the kidney [2, 3].
Thus, hereditary amyloidoses should be considered in all patients with amyloidosis that does not correspond to the AA type and in whom the AL type cannot be confirmed. Here, we report on a patient who presented with renal limited amyloidosis without evidence of AA or AL disease and in whom immunohistochemical findings for kidney biopsy specimen led to a diagnosis of apoAII amyloidosis.
Case report
A 69-year-old Japanese man presented with proteinuria at the age of 67 years. When he was 68 years old, his proteinuria reached a level of 2.3 g/day and we performed a renal biopsy which showed amyloid deposition in the glomeruli with positive congo red staining. Treatment with potassium permanganate dispersed the congo red staining; however, immunostaining against the AA protein was negative. The patient was also negative for serum amyloid A (SAA), and he did not have rheumatoid arthritis, spondyloarthropathy, inflammatory bowel disease, or chronic infections capable of causing AA amyloidosis. Thus, we suspected that his amyloidosis was AL type. However, dysproteinemia was not detected, and we decided to follow him closely without chemotherapy. His proteinuria gradually increased to 6 g/day over the next 12 months, and he was re-admitted to our hospital to undergo chemotherapy for AL amyloidosis.
The patient was 171 cm tall and weighed 74 kg. His blood pressure was 110/70 mmHg. His body temperature was 36.0°C. His conjunctivae were pink, and marked edema was present in the lower extremities. No respiratory or neurologic abnormalities were apparent. He was taking atorvastatin (10 mg) and ezetimibe (10 mg) daily for the treatment of dyslipidemia.
A laboratory evaluation showed a total leukocyte count of 4800/mm3, a hemoglobin level of 14.7 g/dL, and a platelet count of 154000/mm3. Other laboratory parameters were as follows: serum creatinine, 0.8 mg/dL; blood urea nitrogen, 10.4 mg/dL; total protein, 3.9 g/dL; albumin, 1.5 g/dL; total cholesterol, 273 mg/dL; LDL cholesterol, 148 mg/dL; HDL cholesterol, 74 mg/dL; serum sodium, 141.1 mEq/L; serum potassium, 4.1 mEq/L; total serum calcium, 7.4 mg/dL; serum inorganic phosphate, 3.5 mg/dL; immunoglobulin (Ig) G, 335 mg/dL; IgA, 208 mg/dL; IgM, 194 mg/dL; IgD, 2.8 mg/dL; kappa free light chain, 3.11 mg/L (normal value 3.3–19.4 mg/L), lambda free light chain, 12.1 mg/L (normal value 5.71–26.3 mg/L), and kappa/lambda ratio, 0.26 (normal value 0.26–1.65). The complement C3, C4 and CH50 levels were normal. Liver function tests were also normal. Rheumatoid factor was weakly positive (32 IU/mL), but anticyclic citrullinated peptides antibody, SAA and antinuclear antibody were negative. Hepatitis B antigen, hepatitis C antibody and serological test for syphilis were negative, but a QuantiFERON®-TB Gold assay was positive. Urinalysis showed 3+ protein, but no blood or casts. A total of 6.12 g of protein was excreted during a 24-h urine collection, and immunoelectrophoresis of urine and serum samples showed no M protein or Bence-Jones protein.
A chest X-ray showed no abnormal findings in bilateral lungs or the cardiac outline, and the cardiothoracic ratio was 44.4%. A thoracic computed tomography scan showed an old inflammatory change in the apex of bilateral lungs, suggesting a possible latent tuberculosis infection. An abdominal ultrasonography scan revealed no abnormalities of the liver, gallbladder, kidney, spleen or pancreas except for an 11-mm diameter liver cyst and 3-cm diameter renal cysts in both kidneys; the long axes of bilateral kidneys were about 11 cm. A brain magnetic resonance imaging examination showed no abnormal findings except for tiny infarcts of the cerebral white matter, consistent with the patient’s age. An electrocardiogram showed a normal sinus rhythm, and no arrhythmia was found. The transthoracic echocardiography showed no wall motion abnormality or cardiomyopathy and no evidence of cardiac amyloidosis. A gastroscopy disclosed chronic gastritis, and gastric and duodenal biopsies showed no amyloid deposits. A bone marrow biopsy showed no plasma cell dyscrasia and no amyloid deposition. A subcutaneous adipose biopsy was also performed, but no amyloid deposits were found.
The renal biopsy was repeated following admission and provided 21 glomeruli for light microscopic evaluation, and 3 of these glomeruli showed global sclerosis. Periodic acid–Schiff-positive homogenous deposits were found in all of the 18 remaining glomeruli, but the extraglomerular vessels, tubular cells, or interstitium were free of such deposit. The glomerular deposit was congo red-positive with apple-green birefringence under polarized light microscope. Immunofluorescence microscopy revealed no deposition of IgG, IgM, fibrinogen, C4 or C1q, but was positive for IgA (1+) and C3 (2+) immunofluorescence in mesangium. Electron microscopy demonstrated the deposition of nonbranching fibrils in the mesangium and subendothelium, and no other electron dense deposit was found (Fig. 1). The deposition of light chains was also examined using immunofluorescence. Kappa and lambda free light chains were faintly positive, but these findings were interpreted to be nonspecific (Fig. 2a, b).

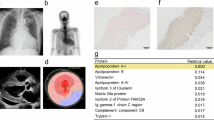
Kidney specimens. a–c Congo-red-positive deposits were observed on the mesangium and intraglomerular vessels, but not on the extraglomerular vessels, tubular cells or interstitium. d, e Immunofluorescence studies showed IgA (1+) and C3 (2+) in mesangium. f Electron microscopy revealed deposition of nonbranching fibrils in the mesangium and subendothelium, and no other electron dense deposit was found

Immunohistochemistry of kidney specimens a, b Free light chains of kappa and lambda were equally weakly positive. c SAA was negative. d Fibrinogen (rabbit polyclonal anti-human fibrinogen, DAKO), e transthyretin (rabbit anti-human prealbumin, DAKO), f lysozyme (rabbit polyclonal anti-lysozyme, DAKO) and g apoAI (goat polyclonal anti-apolipoprotein AI, Abcam) were all negative. h, i The deposition of apoAII (goat polyclonal anti-apolipoprotein AII, Thermo Scientific) on the mesangium and intraglomerular vessels was indicated
We also performed an immunohistochemical study of the renal biopsy specimen using antibodies to SAA, fibrinogen, transthyretin, lysozyme, apoAI and apoAII considering the possibility of various hereditary amyloidoses; the result was positive only with antibody to apoAII which colocalized with the amyloid deposition of the glomeruli (Fig. 2c–i). Thus, we diagnosed the patient as having apoAII amyloidosis. We analyzed the coding sequence of the apoAII in this patient using direct sequencing and 3 pairs of oligonucleotide primers generated to amplify the coding sequence, consisting of exons 2 to 4 (Fig. 3). However, no mutations were found in the coding sequence of apoAII. We also performed a Western blot analysis of the patient’s serum proteins using mouse monoclonal anti-apoAII antibody (WH0000336M1; Sigma) and mass spectrometry of the patient’s serum apoAII [4]. However, no abnormalities were detected using either the Western blot analysis or mass spectrometry (data not shown).

DNA sequencing of apoAII. Genomic DNA was extracted from peripheral blood lymphocytes using a DNA extraction kit according to the manufacturer’s protocol. Three pairs of oligonucleotide primers were originally designed and generated to amplify the coding sequence of apolipoprotein AII (Accession: X02905), consisting of exons 2 to 4. A direct sequencing reaction using the purified PCR product was performed using the dye-terminator method with a BigDye terminator cycle sequencing kit (Applied Biosystems). The resulting sample was then sequenced using an automated DNA sequencer. The following oligonucleotide primers were used: exon 2, 5′-AGAGGCCTGGGCTTCAGTT-3′ and 5′-TGCCAGACCTGGATATAAGAGC-3′; exon 3, 5′-TGGGCAGGAGCTTTGGTTC-3′ and 5′-GAACCCCTTGCCCTGAGA-3′; exon 4, 5′-CTCTAATCCCCTCACCTATCC-3′ and 5′-GGTTGGAAGACAATGGTCTG-3′
We chose to observe his renal amyloidosis closely without performing steroid or chemotherapy and administered isoniazid for the latent tuberculosis infection. At present, 3 years after his initial presentation with proteinuria, his serum creatinine level has gradually increased to 1.5 mg/dL; however, he is still free from other organ involvement, such as cardiac amyloidosis.
Discussion
The possibility of hereditary etiology is not routinely pursued in patients with amyloidosis, and Lachmann et al. [1] reported that hereditary amyloidosis was often misdiagnosed as AL amyloidosis and that amyloidogenic mutations were present in 34 of 350 amyloidosis patients. Thus, genetic causes should be considered in all patients with amyloidosis in whom the diagnosis is not definite for AL or AA amyloidosis.
In our case, we initially suspected AL amyloidosis because the patient did not have any disease that would cause AA amyloidosis and SAA was not detected in the patient’s serum. However, in renal biopsy samples potassium permanganate treatment dispersed the congo red staining of the glomeruli, suggesting that AL amyloidosis was unlikely. In addition, M protein was not detected, and a bone marrow biopsy showed no clonal excess of plasma cells. Thus, neither AL nor AA amyloidosis was diagnosed, and we instead considered the possibility of hereditary amyloidosis, although his family did not have a past history of renal disease.
Among the causes of hereditary amyloidoses, transthyretin, fibrinogen A alpha, apoAI, apoAII, lysozyme, gelsolin and LECT2 are known to cause renal amyloidosis; however, the affected organs differ according to the amyloid protein. ApoAII amyloidosis mainly involves the kidney, compared with other types of amyloid proteins [2]. In our case, amyloid deposits in the subcutaneous fat, bone marrow, stomach and duodenum were not detected in biopsies, and electrocardiogram and echocardiography did not suggest cardiac amyloidosis. Therefore, the amyloid deposits were thought to be limited to the kidneys, consistent with apoAII amyloidosis. LECT2 amyloidosis, which has been recently discovered, also mainly affects the kidney. However, Murphy et al. reported 10 cases of LECT2 amyloidosis where all the cases showed the predominant deposition of amyloid in the renal interstitium. Our case did not show any amyloid deposition in the renal interstitium.
ApoAII amyloidosis is thought to be very rare and the possibility of this condition is not routinely explored in patients with amyloidosis. Thus, the prevalence of apoAII amyloidosis has not been studied in previous large-scale studies [1, 5], and the mechanism of apoAII amyloidosis has not been fully elucidated. In an animal model, apoAII amyloidosis is known to be induced in a senescence-accelerated mouse model of aging [6], and some genetic polymorphisms causing amyloidosis have been identified [7]. In humans, Benson et al. first reported that a mutation in the stop codon of apoAII gene induced a variant apoAII consisting of a 77 amino acid mature apoAII protein plus a carboxyl (C)-terminal peptide elongation, resulting in the formation of amyloid fibrils [8–10]. We analyzed the entire coding sequence of apoAII, but could not detect any genetic mutation, including that of the stop codon. We also performed a Western blot analysis of the patient’s serum protein and a mass spectrometry evaluation of his serum apoAII [4]. Yazaki et al. [9] reported that two bands corresponding to normal apoAII and a larger variant of apoAII were found in an apoAII amyloidosis patient, but we did not find any abnormality in the present patient’s serum apoAII by Western blot and mass spectrometry, suggesting that the mutation of the apoAII gene in the sequence, including introns and exons other than those in the coding sequence, was unlikely.
In our case, the coding sequence of apoAII did not show any mutation, and Western blot and mass spectrometry of the patient’s serum apoAII were normal. Therefore, we concluded that the apoAII amyloid deposition in our case may not have been caused by a genetic mutation, although the possibility of a genetic mutation in the introns or exons other than those in the coding sequence could not be fully excluded. In transthyretin amyloidosis, transthyretin-derived senile systemic amyloidosis without any causative genetic mutation has been previously reported [11]. Therefore, apoAII amyloidosis may also occur without a causative genetic mutation, and some nongenetic mechanisms may exist that cause apoAII to form amyloid fibrils. A prion-like infectious process is another possibility. In a mouse model, apoAII amyloidosis was transmitted by feces and milk excreted by mice with apoAII amyloidosis [12, 13]. A recent study demonstrated the possible transmission of an amyloid fibril conformation through muscle [14]. Although further research is needed to determine the mechanism of apoAII amyloidosis, we report here a case of apoAII amyloidosis without a genetic mutation in the coding sequence of apoAII.
References
Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–91.
Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17:3458–71.
Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56:1100–7.
Ueda M, Misumi Y, Mizuguchi M, Nakamura M, Yamashita T, Sekijima Y, et al. SELDI-TOF mass spectrometry evaluation of variant transthyretins for diagnosis and pathogenesis of familial amyloidotic polyneuropathy. Clin Chem. 2009;55:1223–7.
von Hutten H, Mihatsch M, Lobeck H, Rudolph B, Eriksson M, Röcken C. Prevalence and origin of amyloid in kidney biopsies. Am J Surg Pathol. 2009;33:1198–205.
Higuchi K, Yonezu T, Kogishi K, Matsumura A, Takeshita S, Higuchi K, et al. Purification and characterization of a senile amyloid-related antigenic substance (apoSASSAM) from mouse serum apoSASSAM is an apoA-II apolipoprotein of mouse high density lipoproteins. J Biol Chem. 1986;261:12834–40.
Higuchi K, Kitagawa K, Naiki H, Hanada K, Hosokawa M, Takeda T. Polymorphism of apolipoprotein A-II (apoA-II) among inbred strains of mice. Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Biochem J. 1991;279:427–33.
Benson MD, Liepnieks JJ, Yazaki M, Yamashita T, Hamidi Asl K, Guenther B, et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72:272–7.
Yazaki M, Liepnieks JJ, Barats MS, Cohen AH, Benson MD. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003;64:11–6.
Yazaki M, Liepnieks JJ, Yamashita T, Guenther B, Skinner M, Benson MD. Renal amyloidosis caused by a novel stop-codon mutation in the apolipoprotein A-II gene. Kidney Int. 2001;60:1658–65.
Westermark P, Bergström J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid. 2003;10(Suppl 1):48–54.
Xing Y, Nakamura A, Chiba T, Kogishi K, Matsushita T, Li F, et al. Transmission of mouse senile amyloidosis. Lab Invest. 2001;81:493–9.
Korenaga T, Yan J, Sawashita J, Matsushita T, Naiki H, Hosokawa M, et al. Transmission of amyloidosis in offspring of mice with AApoAII amyloidosis. Am J Pathol. 2006;168:898–906.
Qian J, Yan J, Ge F, Zhang B, Fu X, Tomozawa H et al. Mouse senile amyloid fibrils deposited in skeletal muscle exhibit amyloidosis-enhancing activity. PLoS Pathog. 2010;6:e1000914.
Acknowledgment
We thank Shizuka Fujii for technical assistance.
Conflict of interest
The authors have declared that no conflicts of interest exist.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Morizane, R., Monkawa, T., Konishi, K. et al. Renal amyloidosis caused by apolipoprotein A-II without a genetic mutation in the coding sequence. Clin Exp Nephrol 15, 774–779 (2011). https://doi.org/10.1007/s10157-011-0483-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-011-0483-4