
Abstract
Hypoxia is a common stressor in shrimp, but the molecular mechanisms underlying the adaptation of the ridgetail white prawn, Exopalaemon carinicauda, to hypoxia and reoxygenation remain poorly understood. In the present study, ridgetail white prawns were exposed to gradual changes in hypoxia for 6 h via oxygen consumption in a static respiration chamber and then rapidly aerated to allow reoxygenation for 8 h. The dissolved oxygen concentration after 6 h of hypoxia was 1.04 mg/L, and the survival rate of the shrimp was 47%. A transcriptomic analysis of hepatopancreas tissues after 0 (control), 3 (hypoxic starting point), and 6 (hypoxic lethal half point) h of exposure to hypoxia and after 1 and 8 h of reoxygenation was conducted using the Illumina HiSeqTM 4000 high-throughput sequencing platform. A total of 93,227 genes were obtained, and 4315 of these genes were identified as differentially expressed genes (DEGs) that respond to hypoxia and reoxygenation. A KEGG pathway enrichment analysis suggested that all identified DEGs were mostly enriched in ribosome biogenesis in eukaryotes, apoptosis, the longevity regulating pathway, the MAPK signaling pathway, and protein processing in the endoplasmic reticulum. In addition, 20,203 genes with RPKM values ≥1 were parsed into 20 modules through a weighted gene coexpression network (WGCNA), and three of these modules were related to hypoxia and reoxygenation. GO and KEGG enrichment analyses of these three modules were then performed, and three hub genes were identified based on their connectivity: RREB1 (Ras-responsive element-binding protein 1-like), UBE1 (ubiquitin-activating enzyme E1), and an unknown gene. This study might help further elucidate the hypoxia tolerance mechanism of E. carinicauda.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dissolved oxygen (DO) is an important factor for the survival of aquatic animals, and hypoxia directly affects the growth, development, and reproduction of aquatic animals (Miller Neilan and Rose 2014; Hou et al. 2020). In general, hypoxia is defined as a DO concentration less than 2 mg/L (Galic et al. 2019) and can be accelerated by many factors, such as high temperature, high-density farming, and poor water quality (Stephen Hopkins et al. 1994). During the shrimp aquaculture process, an extension of the farming time is associated with increased deterioration of the water quality of shrimp ponds (Jiang et al. 2009). Moreover, the bottom layer of pond waters might become hypoxic or even anoxic due to the respiration of the shrimp and the decomposition of organic matter, particularly at night, and these hypoxic conditions can certainly threaten the life of shrimp (Cheng et al. 2003). Typically, shrimp respond to aquatic hypoxia via a series of behavioral, physiological, and biochemical and molecular changes. The mysid shrimp Tenagomysis novae-zealandiae reduces its activity to tolerate moderate hypoxia but shows an escape response at a DO level of 0.5 mg/L (Larkin et al. 2008). To avoid oxidative damage caused by the reactive oxygen species (ROS) and free radicals induced under hypoxia, Litopenaeus vannamei can activate its antioxidant enzyme system to eliminate these molecules (Kniffin et al. 2014; Li et al. 2016). In Litopenaeus vannamei, Palaemonetes sinensis, and Pandalus borealis, hypoxia also induces a change in respiratory metabolism by regulating the expression of key enzymes of aerobic or anaerobic metabolism to ensure an adequate energy supply (Keni et al. 2015; Simón et al. 2018; Bao et al. 2018; Pillet et al. 2016). In addition, many studies have shown that different types of metabolism genes, antioxidants, autophagy, and the cell cycle play important roles in the responses of shrimp to hypoxia stress (Camacho-Jiménez et al. 2018; Trasviña-Arenas et al. 2013; Sun et al. 2019; Nuñez-Hernandez et al. 2019). Hypoxia-inducible factor (HIF), a master regulator of many physiological responses to hypoxia, can affect the physiological state of the body by regulating the transcription of genes involved in glucose metabolism, erythropoiesis, cell growth, and cell proliferation (Kodama et al. 2012). Cota-Ruiz et al. (2016) found that HIF-1 participates in the upregulation of phosphofructokinase transcripts under short-term hypoxia. Hypoxia can also activate apoptosis signaling pathways, and Nuñez-Hernandez et al. (2018) found that caspase-3 expression is significantly increased in the hepatopancreas of Litopenaeus vannamei after 48 h of hypoxia, which indicates that caspase-3 is involved in the molecular process of hypoxia-induced apoptosis.
The ridgetail white prawn (Exopalaemon carinicauda) is one of the most important economic shrimp for marine fisheries and pond culture in China (Wang et al. 2010; Ma et al. 2020). In its natural environment, E. carinicauda can live in seawater or brackish waters in the Indo-West Pacific, Korea, and China (Gao et al. 2017). Due to its various advantages, including rapid growth, strong environmental adaptation, and reproductive performance, the scale of the cultivation of ridgetail white prawns has expanded rapidly and currently contributes to at least 1/3 of the overall output of pond polyculture in eastern China (Li et al. 2019). With the expansion of farming scales, high-density breeds have become the primary mode of pond culture. However, as stocking densities increase, the deterioration of water quality and eutrophication observed in recent years has increased the likelihood of hypoxia, which inevitably leads to hypoxic stress (Sun et al. 2020a, 2020b). Therefore, understanding the mechanisms of the adaptation of ridgetail white shrimp to hypoxia can help its farming industry and the breeding of hypoxia tolerance.
Weighted gene coexpression network analysis (WGCNA) is a transcriptional data analysis method based on large samples that can be used for the rapid identification of core relevant pathways and genes involved in the regulation of transcription and makes it easy to infer the regulatory relationships of unknown genes based on known genes in the gene network (Zhou et al. 2019).
In this study, we performed RNA-seq of hepatopancreas tissue from ridgetail white shrimp subjected to hypoxia and reoxygenation for different time points. Transcriptome and WGCNA analyses were performed to determine the mechanisms and key regulatory genes involved in the response of E. carinicauda to of hypoxia and reoxygenation.
Materials and methods
Experimental shrimp collection and rearing
Healthy E. carinicauda (weight: 2.57 ± 0.21 g, body length: 5.3 ± 0.1 cm) were collected from the Rudong base of Jiangsu Institute of Marine Fisheries, Nantong, China. Before the experiment, all the shrimp were placed in two 800-L constant-aeration cylindrical tanks (each tank contained approximately 500 shrimp and 400 L of marine water) and allowed to acclimate for 1 week. The acclimation conditions were 20~21 °C and 25 ppt, and the tank was continuously aerated to maintain a DO level above 7.5 mg/L. During the acclimation period, the shrimp were regularly fed a commercial diet obtained from Nantong Haid Bio-tech Co., LTD., which has a protein content of 42%, once a day at 3–5% of their wet body weight and were fasted for 24 h before the experiment. Any uneaten food, feces, and dead shrimp were removed in a timely manner through a siphon tube, and one-third of the marine water in each tank was replaced once daily with new marine water (20~21 °C, 25 ppt, DO above 7.5 mg/L).
Experimental design and sample collection
A schematic diagram of the experimental design is shown in Fig. 1. Before the experiments, the terminal time (6 h) of hypoxia stress was determined in a preliminary experiment. After 1 week of acclimatization, the shrimps were randomly transferred into 21 conical flasks (10 shrimps in each bottle) with 5 L of constant-aeration seawater (temperature: 20~21°C, salinity: 25 ppt) and divided into groups (each group was placed in three conical flasks): hypoxia for 0 h (0 h) (as the control group), 3 h (3 h), 4.5 h (4.5 h), and 6 h (6 h) and recovery under constant aeration for 1 h (7 h), 4 h (10 h), and 8 h (14 h). The shrimps were allowed to acclimate in the conical flasks for 30 min, and the conical flasks of the experimental groups were then immediately plugged with rubber plugs; after 6 h of hypoxia, the reoxygenation groups were immediately aerated. At the corresponding time points, water samples were collected in a water bottle and fixed with a solution of manganese chloride and alkaline potassium iodide to determinate the DO level. Hepatopancreas tissue of six/five living shrimp from one treatment bottle was then pooled together to obtain a single replicate, and three biological replicates of each group were obtained; the samples were named as follows: jg-0-1 to 3, jg-3-1 to 3, jg-4.5-1 to 3, jg-6-1 to 3, jg-7-1 to 3, jg-10-1 to 3, and jg-14-1 to 3. All hepatopancreas tissue samples were immediately frozen in liquid nitrogen and stored at −80 °C for further analysis.

Schematic diagram of experimental design. a. inflatable tube. b. conical flasks with 5L seawater. c. air stone. d. clip. e. air inlet. f. rubber plug. g. exhalent siphon
Determination of the DO level in the water samples
As described in “Marine Monitoring Specification——Part 4: Seawater Analysis (GB17378.4–2007),” the water samples were fixed for 1 h (complete precipitation), and the DO level was then measured using the iodometric method.
RNA extraction, library construction, and sequencing
According to the results obtained in “Determination of the DO level in the water samples,” total RNA was extracted from the hepatopancreas tissue obtained after 0 h (control), 3 h (hypoxic starting point), and 6 h (hypoxic lethal half point) of gradual changes in hypoxia and after 1 and 8 h of reoxygenation using a TRIzol reagent kit (Invitrogen) according to the manufacturer’s protocol. The integrity value, purity, and integrity of the extracted RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent), NanoDrop microspectrophotometer, and RNase-free agarose gel electrophoresis, respectively. After quality control, the mRNA was enriched with oligo (dT) beads, and the enriched mRNA was cleaved into short fragments using fragmentation buffer and reverse transcribed into cDNA with random primers. Second-strand cDNA was synthesized using DNA polymerase I, RNase H, dNTPs, and buffer. The cDNA fragments were then purified using a QIAquick PCR extraction kit (Qiagen), subjected to end repair and poly(A) addition, and ligated to Illumina sequencing adapters. The ligation products were size selected by agarose gel electrophoresis, amplified by PCR, and sequenced using an Illumina HiSeqTM 4000 instrument by Gene Denovo Biotechnology Co. (Guangzhou, China).
Data analysis
De novo assembly and gene function annotation
The raw reads were further filtered by fastp (version 0.18.0) (Chen et al. 2018) to remove the adaptor sequences, low-quality reads containing more than 50% low-quality (Q-value≤20) bases, and reads with >10% poly-N to obtain high-quality clean reads. The Trinity package was used for the de novo assembly of the remaining high-quality reads (Grabherr et al. 2011). All unigenes were subjected to a BLASTX similarity search using an E-value threshold of 1e−5 to the NCBI nonredundant protein (NR) database (http://www.ncbi.nlm.nih.gov), the Swiss-Prot protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg), and the COG/KOG database (http://www.ncbi.nlm.nih.gov/COG).
Differential expression analysis
The expression of the unigenes was calculated and normalized to RPKM (reads per kb per million reads) (Mortazavi et al. 2008). RNA differential expression analysis between two groups was performed using DESeq2 (Love et al. 2014). Genes with a false discovery rate (FDR) less than 0.05 and an absolute fold change ≥ 2 were considered differentially expressed genes (DEGs).
Coexpression network construction and module and hub gene selection
Coexpression networks were constructed using Omicsmart (www.omicsmart.com/) from Gene Denovo Biotechnology Co. Genes with RPKM values < 1 were removed from the samples, and the remaining genes were used for WGCNA. As shown in Fig. 2, a soft threshold power of 5 (R2 = 0.9050) was used in this study. Subsequently, a hierarchical clustering dendrogram was constructed based on similar gene expression profiles. We then set the minimum number of genes in each module to 50 and used a threshold of 0.7 to merge similar modules. The correlation coefficients for the samples were calculated using the module eigengenes to identify modules with biological or clinical significance. The intracellular connectivity was calculated for each gene, and genes with high connectivity were often found to be hub genes, which might have important functions. The network of each module was visualized directly in Omicsmart.

Analysis of network topology for different soft threshold power. The left panel displays the effect of soft threshold power on scale free topology model fit index. The right panel shows the effect of soft threshold power on the mean connectivity
Functional analysis of module genes
The biological functions of modules highly associated with hypoxia and reoxygenation were analyzed by gene ontology (GO) (Ashburner et al. 2000) and KEGG (Kanehisa and Goto 2000) pathway enrichment analysis. GO terms and KEGG pathways with P ≤ 0.05 were considered to be significantly enriched.
Experimental validation of DEGs
To further validate the reliability of the transcriptome sequencing, 10 DEGs were randomly selected and analyzed by qRT-PCR. All primers were designed as shown in Table 1. Total RNA was extracted from the samples that were also used for sequencing, and cDNA was synthesized using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Dalian, China). qRT-PCR was performed using TB Green® Premix Ex Taq™ (Tli RNaseH Plus) (TaKaRa, Dalian, China) in 20-μl reactions, which contained 10 μl of TB Green Premix Ex Taq (Tli RNaseH Plus) (2X), 0.8 μl of the reverse primer (10 μM), 0.4 μl of ROX Reference Dye (50X), 2 μl of cDNA, and 6.8 μl of ddH2O. The PCR amplification procedure was performed in a StepOnePlus Real Time PCR System (Applied Biosystems, USA) with the following temperature program: 95 °C for 30 s; 40 cycles of 95 °C for 5 s and 60 °C for 30 s; melt curve detection at 95 °C for 15 s; and 60 °C for 60 s to 95 °C for 15 s at increments of 0.3 °C. All reactions were performed in triplicate. The expression of each gene was normalized using β-actin as the reference gene and calculated using the comparative threshold cycle (2−ΔΔCt) method.
Results
DO concentration at corresponding time points
The results of the DO analysis are shown in Fig. 3. The DO level at 3 h, 4.5 h, and 6 h was significantly (P < 0.05) lower than that in the control group. At 3 h, the DO level was 2.12 mg/L, which was approximately equal to the hypoxia level; thus, 3 h was selected as the start time of hypoxia stress. At 6 h, the survival rate of the shrimp was 47%; thus, 6 h was selected as the longest hypoxia exposure duration, and the DO level at this time point was 1.04 mg/L. In contrast, the DO level in the reoxygenation groups returned to that in the control group level rapidly after the start of reoxygenation, and the levels at 7 h, 10 h, and 14 h were not significantly different from that in the control group (P > 0.05).

Results of DO concentration. Different lowercases indicate significance difference (P<0.05) in different time points
Sequencing, assembly, and gene functional annotation
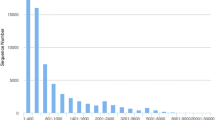
From 15 samples, a total of 864,905,274 raw reads were generated. After filtering, the clean reads from each sample ranged from 44,659,922 to 90,874,932. The average rate of clean reads was 99.91%. The detailed statistics of each sample, including the numbers of raw and clean bases, the Q20 and Q30 values, and the GC percentage, are listed in Table 2. Trinity software was used to assemble and cluster the clean reads into unigenes. A total of 93,227 unigenes, with an average length of 834 bp and an N50 length of 1352 bp, were obtained (Table 3). The assembled unigenes were annotated using four public databases, namely, COG/KOG, NR, KEGG, and Swiss-Prot (Fig. 4). A total of 40,719 unigenes (43.68% of the unigenes) were annotated. The numbers of unigenes annotated with the NR, KEGG, COG/KOG, and Swiss-Prot databases were 37346 (40.01%), 33955 (36.42%), 24238 (26.00%), and 29042 (31.15%), respectively (Table 4). The NR species annotation demonstrated that the majority of unigenes were found in Hyalella azteca, Branchiostoma belcheri, Saccoglossus kowalevskii, Tetrahymena thermophila SB210, Daphnia magna, and Lingula anatine (Fig. 5). The unigenes of E. carinicauda were classified into 25 KOG clusters according to the COG/KOG functional classification, and the cluster with the highest number of unigenes (4949) was the general function prediction cluster, followed by the signal transduction mechanism cluster and the posttranslational modification, protein turnover, and chaperone cluster (4332 and 3020 unigenes, respectively; Fig. 6). In the KEGG classification, 33,955 (36.42%) unigenes were mapped to the five categories of KEGG metabolic pathways, including metabolism, genetic information processing, environmental information processing, cellular processes, and organismal systems, and most of the genes belonged to global and overview maps (1870 unigenes), followed by translation (1283 unigenes), transport and catabolism (1019 unigenes), signal transduction (843 unigenes), and folding, sorting, and degradation (652 unigenes) (Fig. 7). In addition, we also classified the unigenes into three functional categories based on GO classification. In the biological process category, the unigenes were clustered into 24 classifications, and the highest number of genes belonged to metabolic process, followed by cellular process and single-organism process. The unigenes were also classified into 20 classifications in the cellular component category, and cell and cell parts were the most representative. In addition, among the 12 molecular function classifications, catalytic activity and binding were the most representative (Fig. 8).

The Venn diagram of unigenes annotated in COG/KOG, KEGG, NR and Swissprot databases. The number in each color block indicated the number of unigenes that was annotated by single or multiple databases

The top-hits species distribution of unigenes by the NR database

Functional classification of unigenes in COG/KOG

KEGG annotation analysis of all unigenes

GO annotation analysis of all unigenes
Identification and analysis of DEGs
Pairwise comparisons of the different sample groups analyzed (jg-0-vs-jg-3; jg-0-vs-jg-6; jg-0-vs-jg-7; jg-0-vs-jg-14; jg-3-vs-jg-6; jg-3-vs-jg-7; jg-3-vs-jg-14; jg-6-vs-jg-7; jg-6-vs-jg-14; jg-7-vs-jg-14) detected a total of 4315 DEGs (3374 upregulated and 1569 downregulated genes) in the hepatopancreas of E. carinicauda (Fig. 9). Compared with the control group, a total of 274 DEGs (166 upregulated and 108 downregulated genes) were identified in the hypoxia experimental groups (3 h and 6 h), and 1704 DEGs (1470 upregulated and 239 downregulated genes) were identified in the reoxygenation experimental groups (7 h and 14 h). Remarkably, the number of DEGs was higher after hypoxia and reoxygenation. A GO functional enrichment analysis showed that the biological processes of the DEGs were mostly related to metabolic processes, cellular processes, and single-organism processes. Cell and cell part were most enriched GO terms in the cellular component category, and catalytic activity and binding were the most highly enriched molecular function terms (Fig. 10). We also mapped all DEGs to KEGG pathways, and the results showed that the DEGs were significantly enriched (q < 0.05) in ribosome biogenesis in eukaryotes, apoptosis-multiple species, apoptosis-fly, longevity regulating pathway-multiple species, MAPK signaling pathway, MAPK signaling pathway-fly, and protein processing in the endoplasmic reticulum (ER) (Fig. 11).

The number of differential expression genes in fifteen pairwise comparisons. UP: Up-regulated genes, DOWN: Down-regulated genes

Gene ontology annotated analysis of all DEGs

Top 20 of KEGG enrichment pathway of all DEGs
Coexpression network
A weighted gene coexpression network was constructed for the hepatopancreas of E. carinicauda after the hypoxia and reoxygenation treatments. The network contained 20,203 genes and was divided into 20 modules (Fig. 12). In the figure, the blue module had the largest number of genes (11968), whereas the dark magenta module had the smallest number of genes (115) (Fig. 13). Three of the identified modules were found to be specifically related to hypoxia and reoxygenation: the dark gray module with 234 genes, the green module with 1126 genes, and the blue module with 11968 genes were significantly positively correlated with jg-3-1, 2, 3, jg-7-1, 2, 3, and jg-14-1, 2, 3, respectively. Notably, no modules exhibited significant negative correlations with the hypoxia and reoxygenation treatments (Fig. 14).

Clustering dendrograms of 20203 genes. Dissimilarity was based on topological overlap, together with assigned module colors. The 20 co-expression modules are shown in different colors

The number of genes of each module

Correlation heatmap of module-sample association. The colors, ranging from green through white to red, indicate low to high correlations
GO and KEGG enrichment analyses of modules
To further investigate the potential functions of the dark gray, green, and blue modules, GO and KEGG enrichment analyses were performed. The GO enrichment analysis was divided into three categories, biological process, molecular function, and cellular component, and revealed that most of the genes belonging to the three abovementioned modules were enriched in metabolic process, cellular process, single-organism process, cell, cell part, catalytic activity, and binding at level 2 (Fig. 15). A KEGG pathway analysis was performed with the same modules, and the results revealed that 12 pathways, including glycosphingolipid biosynthesis-ganglio series, glycosphingolipid biosynthesis-globo and isoglobo series, and amino sugar and nucleotide sugar metabolism, were significantly (P < 0.05) enriched with the genes in the dark gray module. In addition, 19 pathways, such as the MAPK signaling pathway-fly, MAPK signaling pathway, apoptosis-fly, and Wnt signaling pathway, were significantly (P < 0.05) enriched with the genes in the green module, and the blue module was significantly (P < 0.05) enriched in 21 pathways, such as spliceosome, RNA transport, and N-glycan biosynthesis (Table 5).

Gene ontology annotated analysis of dark grey, blue, and green module. a Dark grey module, b blue module, c green module
Identification of hub genes and network construction
To identify hub genes from the significant modules based on weight values, the top 100 pairs between two genes in each module were selected to construct a gene correlation network. The gene with the highest connectivity was considered a hub gene. In the dark gray module with 35 unigenes, the green module with 38 unigenes, and the blue module with 89 unigenes, unigene0018514 (unknown gene), unigene0018395 (Ras-responsive element-binding protein 1-like, RREB1), and unigene0073542 (ubiquitin-activating enzyme E1, UBE1) were in core positions. RREB1 is a Krüppel-type zinc finger protein that can bind to Ras-responsive elements in promoters and might exert its control via the Ras/Raf pathway (Jiang et al. 2010). UBE1 belongs to the ubiquitin-activating E1 family of enzymes and plays a crucial role in ubiquitylation to activate ubiquitin (Lambert-Smith et al. 2020) (Fig. 16).

Network relationship among the modules. a 35 unigenes with highest weight are in dark grey module, b 38 unigenes with highest weight are in green module, c 89 unigenes with highest weight are in blue module. Note: the direction of the arrow indicates a positive relationship between two genes; the color of lines, ranging from blue through cyan to red, indicate low to high weight values; the size of the circle represents the level of genetic connectivity
Validation of gene expression patterns by qRT-PCR
To verify the reliability of the RNA-seq results, ten candidate DEGs were randomly selected for detection by qRT-PCR. As expected, the qPCR results were generally consistent with the RNA-seq results, confirming the reliability of the RNA-seq results (Fig. 17).

qRT-PCR validation of ten DEGs. The y-axis on the left shows the normalized expression level (RPKM) of RNA-seq (blue bar), and the y-axis on the right shows relative expression level of qRT-PCR (green line). The relative expressions of ten genes were calculated using the 2−△△Ct method, and β-actin was used as a reference gene. The results were expressed as mean ± SD
Discussion
Differential expression of genes during hypoxia and reoxygenation
Hypoxia is an important stressor for shrimp in aquatic environments that can cause respiratory abnormalities and metabolic disturbances, which result in reduced efficiency of bait conversion and retarded growth and development (Bao et al., 2018), and these effects need to be regulated by the underlying genes. The hepatopancreas is an important organ for digestion, absorption, storage, and metabolism in decapods and plays a key role in the initiation of defense responses in shrimp (Jiang et al. 2009). In this study, a total of 4315 DEGs were identified in the hepatopancreas of E. carinicauda after hypoxia and reoxygenation. KEGG analyses showed that the pathways involving these genes were significantly enriched in ribosome biogenesis in eukaryotes, apoptosis, the longevity regulating pathway, the MAPK signaling pathway, and protein processing in the ER.
Hypoxia can induce the production of ROS, and the sudden increase in O2 (reoxygenation) after hypoxia can produce higher levels of ROS (Parrilla-Taylor and Zenteno-Savín 2011). Increased production of ROS mediates oxidative damage, which might affect ER homeostasis, interfere with improper protein folding, and induce apoptosis (Yang et al. 2016). In addition, oxidative stress leads to RNA damage and the incorporation of oxidized nucleotides during RNA synthesis, which impairs protein synthesis and other RNA functions (Magagnin et al. 2007). The ribosome is essential for protein synthesis, and the ER is the site at which newly synthesized, secreted, and transmembrane proteins are folded and plays a key role in the molecular life of each cell (Dominik et al. 2019; Thomson et al. 2013; Magagnin et al. 2007). The biogenesis of ribosomes is a tightly regulated activity that is inevitably linked to other fundamental cellular processes, including growth and cell division (Thomson et al. 2013). However, eukaryotic ribosome biogenesis is a complex process that requires numerous ribosomal proteins to facilitate the modification, folding, and cleavage of rRNA and the binding of ribosomal proteins to pre-rRNA (Horn et al. 2011). The protein synthesis requirements in cells are coordinated with ribosomal biosynthetic activity, and upregulation of the levels of certain key ribosomal proteins can affect cell growth and metabolism by regulating the expression of other proteins (Antonio et al. 2015). We also found that the nucleolar proteins NOP56 and NOP1, which are important components of the Box C/D snoRNAs involved in the early processing of pre-rRNA, were differentially expressed (Andersen et al. 2018; David et al. 1991). A previous study showed that cells exposed to severe hypoxia exhibit an increased synthesis rate of specific proteins (oxygen-regulated proteins, ORPs) (Wilson and Sutherland 1989). However, protein synthesis is an energy-expensive process that consumes a large proportion of the total ATP turnover of all cells and organs. The pelagic red crab, Pleuroncodes planipes, achieves metabolic suppression in zones with minimal oxygen by reducing protein synthesis (Seibel et al. 2018). After 4 h of hypoxia treatment, the Sillago sihama liver inhibits translation and protein synthesis by regulating ribosome activity and tRNA synthesis (Tian et al. 2020). These findings might indicate that ridgetail white prawns also enhance or inhibit protein synthesis under hypoxia and reoxygenation. As a result of hypoxia, the ATP levels decrease, and cell function cannot be maintained; if the stress is not relieved, the cell will eventually die (Sun et al. 2020a, 2020b). Moreover, when energy is depleted, ribosome synthesis is promptly repressed, and nucleolar proteins are degraded by nucleophagy (autophagic degradation of the nucleolus) (Morshed et al. 2019). In addition, oxidative stress can not only alter the function of many cells, which leads to cell death, but also cause ER stress-induced apoptosis (Iurlaro & Munoz-Pinedo., 2016; Sun et al. 2020a, 2020b). The DEG ATF4 is the main transcriptional regulator of the cellular hypoxic response to the unfolded protein response (UPR) and activates genes that promote restoration of normal ER function and survival under hypoxia (Rzymski et al. 2009), and the caspase-7 gene plays primary roles as the executioner of apoptosis (Thomsen et al. 2013). These results are similar to previous findings that hypoxia can initiate apoptosis (Nuñez-Hernandez et al. 2018). Furthermore, stress resistance genes that belong to the longevity regulating pathway (such as superoxide dismutase, SOD, catalase, and CAT) play an important role in resistance to oxidative damage. For example, CAT expression in the gills of white shrimp (Litopenaeus vannamei) is 3.2- and 3-fold higher under hypoxia and reoxygenation conditions (6 and 24 h of hypoxia followed by 1 h of reoxygenation) than under normoxia conditions (Trasviña-Arenas et al. 2013). We also found similar results in our study: this gene was differentially expressed in hepatopancreas tissue under hypoxia and reoxygenation. The MAPK signaling pathway plays a critical role in responding to cellular stress and promoting cell growth and survival (Conrad et al. 2000). ROS can stimulate MAPKs to promote a variety of cellular processes, including apoptosis and the inflammatory response (Lin et al. 2018). In humans, the MAPK signaling pathways are activated during the hypoxia and reoxygenation of villous explants in vitro, and the downstream consequences of this activation is the enhancement of apoptosis (Cindrova-Davies et al. 2007). In response to hypoxia, many genes involved in the MAPK signaling pathway are expressed at higher levels in channel catfish, Ictalurus punctatus (Yang et al. 2018). In this study, abundant MAPK signaling pathway genes, including MEK1, MNK1/2, and MKK3, which directly or indirectly mediate cell apoptosis, were identified as DEGs, which suggests that the hepatopancreas of E. carinicauda might be an important tissue in the response to hypoxia and reoxygenation.
WGCNA
WGCNA is a computational method for constructing a weighted gene coexpression network based on gene expression, and this network can be further divided into coexpression modules. Genes in the same coexpression module are similarly expressed and might have closely related functions (Jia et al. 2020). WGCNA has been widely used to identify genes and modules associated with stress in fish (Huang et al. 2020; Ning and Sun 2020; Kim et al. 2020). However, few studies have investigated the complex coexpression networks in shrimp under hypoxia reoxygenation stress. In this study, 20 modules were identified by WGCNA. Interestingly, three module genes (dark grey, green, and blue) were significantly positively correlated with hypoxia (jg-3-1, 2, 3) and reoxygenation (jg-7-1, 2, and 3 and jg-14-1, 2, and 3). The results of the GO analysis revealed that the genes in the dark gray, green, and blue modules were enriched in metabolic processes. Notably, metabolic processes reportedly play a pivotal role in the hypoxia and reoxygenation responses of shrimp (Li & Brouwer., 2013). The KEGG enrichment analysis showed that the genes in the dark gray module were most significantly enriched in glycosphingolipid biosynthesis. It is worth noting that all the genes in this pathway were annotated as hexosaminidase. Hexosaminidases are exoglycosidases that can cleave N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) residues from the nonreducing terminals of glycoconjugates (Venugopal et al. 2020). In insects and marine invertebrates, hexosaminidases degrade chitin along with chitinases (Venugopal et al. 2020). The role of hexosaminidase in the regulation of the GlcNAc levels is directly related to the cytoprotection of mammalian cardiac tissues against hypoxia and ischemic injury (Jayasundara et al. 2015). We hypothesized that the high expression of hexosaminidase in shrimp might play a role in degradation during exposure to hypoxia for 3 h. The KEGG analysis revealed that genes in the green module were most significantly enriched in the MAPK signaling pathway. The functions of the MAPK signaling pathway have been described previously, and the results suggest that a sudden increase in ROS under reoxygenation for 1 h might stimulate this pathway. The KEGG enrichment analysis showed that the genes in the blue module were mostly enriched in spliceosome and RNA transport. Each pre-mRNA splicing event is performed by an exceptionally dynamic ribonucleoprotein machinery known as the spliceosome (Chen and Moore 2015), and mature spliced mRNAs are then transported to their sites of function throughout the cell (Nakielny et al. 1997). We found a significantly positive correlation between the blue module and reoxygenation for 8 h, which appeared to indicate that the transcription process of shrimp was active.
The identification of hub genes is key to exploring the mechanism of hypoxia and reoxygenation tolerance. In this study, the hub genes of the dark gray, green, and blue modules were unigene0018514 (unknown gene), unigene0018395 (RREB1), and unigene0073542 (UBE1), respectively. Although the hub gene of the dark gray module is unknown, we found that both unigene0000372 (trypsin, TRY) and unigene0002163 (suppressor of tumorigenicity 14 protein, ST14), which showed high correlations, have proteolytic enzyme activity (Baird 2013; Silva et al. 2017), which suggests that these hub genes might also have proteolytic functions and provide energy under hypoxia. RREB1, which is also named HNT, FINB, and LZ321, is a zinc finger transcription factor involved in a variety of biological processes, including DNA damage repair, cell growth and proliferation, cell differentiation, fat development, fasting glucose balance, zinc transport, and transcriptional regulation (Deng et al. 2020). RREB1 has been confirmed to be a downstream effector of the MAPK signaling pathway, which can activate or repress the expression of target genes to respond to Ras signaling (Deng et al. 2020; Kent et al. 2013). This finding is consistent with our results from the KEGG enrichment analysis of the green module. Ubiquitination is often used as a signal of protein degradation, defines the fate and function of proteins, and plays multiple roles in cell survival, differentiation, and development (Carlos et al., 2012). UBE1 catalyzes the first step of the ubiquitin-dependent proteolytic pathway and plays a role in the ubiquitination process (Tokumoto et al. 2000). In addition, the degradation of hypoxia inducible factor-1 alpha (HIF-1α) protein is dependent on ubiquitination (Wang et al. 2016). We thus speculated that UBE1 expression plays a role in the degradation of HIF-1α after reoxygenation for 8 h. In summary, the three hub genes identified from the three related modules might play a key role in the response of E. carinicauda to hypoxia and reoxygenation.
References
Andersen SL, Ardal BK, Hollensen AK, Damgaard CK, Jensen TH (2018) Box C/D snoRNP autoregulation by a cis-acting snoRNA in the NOP56 Pre-mRNA (Article). Mol Cell 72(1):99–111(e5). https://doi.org/10.1016/j.molcel.2018.08.017
Antonio G, Sara CK, George T (2015) A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta 1849(7):812–820. https://doi.org/10.1016/j.bbagrm.2015.02.005
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25(1):25. https://doi.org/10.1038/75556
Baird TT (2013) Trypsin. Columbia Electronic Encyclopedia 1:216–219. https://doi.org/10.1016/B978-0-12-374984-0.01589-8
Bao J, Li XD, Yu H, Jiang HB (2018) Respiratory metabolism responses of Chinese mitten crab, eriocheir sinensis and Chinese grass shrimp, Palaemonetes sinensis, subjected to environmental hypoxia stress. Front Physiol 9:1559. https://doi.org/10.3389/fphys.2018.01559
Camacho-Jiménez L, Peregrino-Uriarte A, Martnez-Quintana JA, Yepiz-Plascencia G (2018) The glyceraldehyde-3-phosphate dehydrogenase of the shrimp Litopenaeus vannamei: Molecular cloning, characterization and expression during hypoxia. Mar Environ Res 138:65–75. https://doi.org/10.1016/j.marenvres.2018.04.003
Carlos AN, Cesar GP, Jenny C, Hugo DL, & Moisés W (2012). The ubiquitin-activating enzyme (E1) of the early-branching eukaryote Giardia intestinalis shows unusual proteolytic modifications and play important roles during encystation. Acta Tropica, 123 (1), 39–46. https://doi.org/10.1016/j.actatropica.2012.03.012
Chen WJ, Moore MJ (2015) Spliceosomes (Article). Curr Biol 25(5):R181–R183. https://doi.org/10.1016/j.cub.2014.11.059
Chen SF, Zhou YQ, Chen YR, Gu J (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17):884–890. https://doi.org/10.1093/bioinformatics/bty560
Cheng W, Liu CH, Kuo CM (2003) Effects of dissolved oxygen on hemolymph parameters of freshwater giant prawn, Macrobrachium rosenbergii (de Man). Aquaculture 220(1):843–856. https://doi.org/10.1016/S0044-8486(02)00534-3
Cindrova-Davies T, Spasic-Boskovic O, Jauniaux E, Charnock-Jones OS, Burton BJ (2007) Nuclear factor-kappa B, p38, and stress-activated protein kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to oxidative stress: effects of antioxidant. American Journal of Pathology: Official Publication of the American Association of Pathologists 170(5):1511–1520. https://doi.org/10.2353/ajpath.2007.061035
Conrad, P. W., Millhorn, D. E., & Beitner-Johnson, D. (2000). Hypoxia differentially regulates the mitogen- and stress-activated protein kinases: role of Ca2+/CaM in the activation of MAPK and p38γ. Adv. Exp. Med. Biol.
Cota-Ruiz K, Leyva-Carrillo L, Peregrino-Uriarte AB et al (2016) Role of HIF-1 on phosphofructokinase and fructose 1, 6-bisphosphatase expression during hypoxia in the white shrimp Litopenaeus vannamei. Comparative Biochemistry and Physiology, Part A 198:1–7. https://doi.org/10.1016/j.cbpa.2016.03.015
David T, Hanna L, Maria CF, Eduard CH (1991) The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rRNA processing in yeast. EMBO J 10(3):573–583
Deng YN, Xia ZJ, Zhang P, Ejaz S, Liang SF (2020) Transcription factor RREB1: from target genes towards biological functions. Int J Biol Sci 16(8):1463–1473. https://doi.org/10.7150/ijbs.40834
Dominik A, Michael P, Lisa K, Ingrid R, Mathias L, Melanie P et al (2019) Inhibiting eukaryotic ribosome biogenesis. BMC Biol 17(1):46. https://doi.org/10.1186/s12915-019-0664-2
Galic N, Hawkins T, Forbes VE (2019) Adverse impacts of hypoxia on aquatic invertebrates: a meta-analysis. Sci Total Environ 652:736–743. https://doi.org/10.1016/j.scitotenv.2018.10.225
Gao H, Xue B, Zhao L, Lai XF, Yan BL et al (2017) Cloning of the ANT gene and its expression profiles at different developmental stages and post-molting times in the ridgetail white prawn Exopalaemon carinicauda. Fish Sci 83(4):553–561. https://doi.org/10.1007/s12562-017-1094-0
GB17378.4-2007, The state oceanic administration of China: The specification for marine monitoring (in Chinese). Ocean Press, Beijing, China.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652. https://doi.org/10.1038/nbt.1883
Hopkins JS, Sandifer PA, Browdy CL (1994) Sludge management in intensive pond culture of shrimp: effect of management regime on water quality, sludge characteristics, nitrogen extinction, and shrimp production. Aquac Eng 13(1):11–30. https://doi.org/10.1016/0144-8609(94)90022-1
Horn DM, Mason SL, Karbstein K (2011) A novel nuclease for 18S ribosomal RNA production. J Biol Chem 286(39):34082–34087. https://doi.org/10.1074/jbc.M111.268649
Hou ZS, Wen HS, Li JF, He F, Li Y, Qi X (2020) Environmental hypoxia causes growth retardation, osteoclast differentiation and calcium dyshomeostasis in juvenile rainbow trout (Oncorhynchus mykiss). Sci Total Environ 705:135–272. https://doi.org/10.1016/j.scitotenv.2019.135272
Huang ZH, Ma AJ, Yang SS, Liu XF, Zhao TT, Zhang JS et al (2020) Transcriptome analysis and weighted gene co-expression network reveals potential genes responses to heat stress in turbot Scophthalmus maximus. Comparative biochemistry and physiology Part D, Genomics & proteomics 33:100632. https://doi.org/10.1016/j.cbd.2019.100632
Iurlaro R, Munoz-Pinedo C (2016) Cell death induced by endoplasmic reticulum stress. FEBS J 283:2640–2652. https://doi.org/10.1111/febs.13598
Jayasundara N, Tomanek L, Dowd WW, Somero GN (2015) Proteomic analysis of cardiac response to thermal acclimation in the eurythermal goby fish Gillichthys mirabilis. J Exp Biol 218(9):1359–1372. https://doi.org/10.1242/jeb.118760
Jia RK, Zhao HX, Jia MW (2020) Identification of co-expression modules and potential biomarkers of breast cancer by WGCNA. Gene 750:144757. https://doi.org/10.1016/j.gene.2020.144757
Jiang H, Li FH, Xie YS, Huang BX, Zhang JK, Zhang JQ, Zhang CS et al (2009) Comparative proteomic profiles of the hepatopancreas in Fenneropenaeus chinensis response to hypoxic stress. Proteomics 9(12):3353–3367. https://doi.org/10.1002/pmic.200800518
Jiang WX, Sequeira JM, Nakayama Y, Lai SC, Quadros EV (2010) Characterization of the promoter region of TCblR/CD320 gene, the receptor for cellular uptake of transcobalamin-bound cobalamin. Gene 466(1-2):49–55. https://doi.org/10.1016/j.gene.2010.07.004
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Keni CR, Uriarte AB, Portillo MF, Quintana JA, Plascencia GY (2015) Expression of fructose 1,6-bisphosphatase and phosphofructokinase is induced in hepatopancreas of the white shrimp Litopenaeus vannamei by hypoxia. Mar Environ Res 106:1–9. https://doi.org/10.1016/j.marenvres.2015.02.003
Kent O, Talbot KF, Halushka MK (2013) RREB1 repressed miR-143/145 modulates KRAS signaling through downregulation of multiple targets. Oncogene 32(20):2576–2585. https://doi.org/10.1038/onc.2012.266
Kim A, Yoon D, Lim Y, Roh HJ, Kim S, Park C et al (2020) Co-expression network analysis of spleen transcriptome in rock bream (Oplegnathus fasciatus) naturally infected with rock bream iridovirus (RBIV). Int J Mol Sci 21(5):1707. https://doi.org/10.3390/ijms21051707
Kniffin CD, Burnett LE, Burnett KG (2014) Recovery from hypoxia and hypercapnic hypoxia: impacts on the transcription of key antioxidants in the shrimp Litopenaeus vannamei. Comparative Biochemistry and Physiology, Part B 170:43–49. https://doi.org/10.1016/j.cbpb.2014.01.006
Kodama K, Rahman MS, Horiguchi T, Thomas P (2012) Upregulation of hypoxia-inducible factor (HIF)-1a and HIF-2a mRNA levels in dragonet Callionymus valenciennei exposed to environmental hypoxia in Tokyo Bay. Mar Pollut Bull 64:1339–1347. https://doi.org/10.1016/j.marpolbul.2012.05.002
Lambert-Smith IA, Saunders DN, Yerbury JJ (2020) The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int J Biochem Cell Biol 123:105746. https://doi.org/10.1016/j.biocel.2020.105746
Larkin G, Closs GP, Peake B (2008) Tolerance and behaviour of the mysid shrimp Tenagomysis novae-zealandiae to low dissolved oxygen. N Z J Mar Freshw Res 41(3):317–323. https://doi.org/10.1080/00288330709509919
Li TD, Brouwer M (2013) Gene expression profile of hepatopancreas from grass shrimp Palaemonetes pugio exposed to cyclic hypoxia. Comparative Biochemistry and Physiology - Part D: Genomics and Proteomics, 2013 8(1):1–10. https://doi.org/10.1016/j.cbd.2012.10.003
Li YH, Wei L, Cao JR et al (2016) Oxidative stress, DNA damage and antioxidant enzyme activities in the pacific white shrimp (Litopenaeus vannamei) when exposed to hypoxia and reoxygenation. Chemosphere 144:234–240. https://doi.org/10.1016/j.chemosphere.2015.08.051
Li JT, Lv JJ, Liu P, Chen P, Wang JJ, Li J (2019) Genome survey and high-resolution backcross genetic linkage map construction of the ridgetail white prawn Exopalaemon carinicauda applications to QTL mapping of growth traits. BMC Genomics 20(1):598. https://doi.org/10.1186/s12864-019-5981-x
Lin B, Xu J, Feng DG, Wang F, Wang JX, Zhao H (2018) DUSP14 knockout accelerates cardiac ischemia reperfusion (IR) injury through activating NF-kB and MAPKs signaling pathways modulated by ROS generation. Biochem Biophys Res Commun 501(1):24–32. https://doi.org/10.1016/j.bbrc.2018.04.101
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. https://doi.org/10.1186/s13059-014-0550-8
Ma HK, Sun JQ, Xu WY, Gao W, Hu GW, Lai XF et al (2020) Cloning and functional study of lipocalin: retinol-binding protein-like gene family of the ridgetail white prawn, Exopalaemon carinicauda. Mol Genet Genomics 295(2):453–464. https://doi.org/10.1007/s00438-019-01633-0
Magagnin MG, Sergeant K, Beucken T, Rouschop KM, Jutten B, Seigneuric R et al (2007) Proteomic analysis of gene expression following hypoxia and reoxygenation reveals proteins involved in the recovery from endoplasmic reticulum and oxidative stress. Radiother Oncol 83(3):340–345. https://doi.org/10.1016/j.radonc.2007.04.027
Miller Neilan R, Rose K (2014) Simulating the effects of fluctuating dissolved oxygen on growth, reproduction, and survival of fish and shrimp (Article). J Theor Biol 343:54–68. https://doi.org/10.1016/j.jtbi.2013.11.004
Morshed S, Mochida T, Shibata R, Ito K, Mostofa MG, Rahman MA, Ushimaru T (2019) Def1 mediates the degradation of excess nucleolar protein Nop1 in budding yeast. Biochem Biophys Res Commun 519(2):302–308. https://doi.org/10.1016/j.bbrc.2019.09.002
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628. https://doi.org/10.1038/nmeth.1226
Nakielny S, Fischer U, Michael WM, Dreyfuss G (1997) RNA transport. Annu Rev Neurosci 20:269–301. https://doi.org/10.1146/annurev.neuro.20.1.269
Ning XH, Sun L (2020) Gene network analysis reveals a core set of genes involved in the immune response of Japanese flounder (Paralichthys olivaceus) against Vibrio anguillarum infection. Fish & shellfish immunology 98:800–809. https://doi.org/10.1016/j.fsi.2019.11.033
Nuñez-Hernandez DM, Felix-Portillo M, Peregrino-Uriarte AB, Yepiz-Plascencia G (2018) Cell cycle regulation and apoptosis mediated by p53 in response to hypoxia in hepatopancreas of the white shrimp Litopenaeus vannamei. Chemosphere 190:253–259. https://doi.org/10.1016/j.chemosphere.2017.09.131
Nuñez-Hernandez DM, Camacho-Jiménez L, González-Ruiz R, Mata-Haro V, Ezquerra-Brauer JM, Yepiz-Plascencia G (2019) Cyclin-dependent kinase 2 (Cdk-2) from the white shrimp Litopenaeus vannamei: Molecular characterization and tissue-specific expression during hypoxia and reoxygenation. Comparative Biochemistry and Physiology. Part A, Molecular & Integrative Physiology 230:56–63. https://doi.org/10.1016/j.cbpa.2018.12.013
Parrilla-Taylor DP, Zenteno-Savín T (2011) Antioxidant enzyme activities in Pacific white shrimp (Litopenaeus vannamei) in response to environmental hypoxia and reoxygenation. Aquaculture 318(3-4):379–383. https://doi.org/10.1016/j.aquaculture.2011.05.015
Pillet M, Dupont-Prinet A, Chabot D, Tremblay R, Audet C (2016) Effects of exposure to hypoxia on metabolic pathways in northern shrimp (Pandalus borealis) and Greenland halibut (Reinhardtius hippoglossoides). J Exp Mar Biol Ecol 483:88–96. https://doi.org/10.1016/j.jembe.2016.07.002
Rzymski T, Milani M, Singleton DC, Harris AL (2009) Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8(23):3838–3847. https://doi.org/10.4161/cc.8.23.10086
Seibel BA, Luu BE, Tessier SN, Towanda T, Storey KB (2018) Metabolic suppression in the pelagic crab, Pleuroncodes planipes, in oxygen minimum zones. Comparative Biochemistry and Physiology Part - B: Biochemistry and Molecular Biology 224:88–97. https://doi.org/10.1016/j.cbpb.2017.12.017
Silva MD, Labas V, Nys Y, R´ehault-Godbert, S. (2017) Investigating proteins and proteases composing amniotic and allantoic fluids during chicken embryonic development. Poult Sci 96(8):2931–2941. https://doi.org/10.3382/ps/pex058
Simón BA, Piñón M, Racotta R, Racotta IS (2018) Neuroendocrine and metabolic responses of Pacific whiteleg shrimp Penaeus vannamei exposed to hypoxia stress. Lat Am J Aquat Res 46(2):364–376. https://doi.org/10.3856/vol46-issue2-fulltext-12
Sun SM, Wu Y, Fu HT, Ge XP, You HZ, Wu XG (2019) Identification and characterization of four autophagy-related genes that are expressed in response to hypoxia in the brain of the oriental river prawn (Macrobrachium nipponense). Int J Mol Sci 20(8):1856. https://doi.org/10.3390/ijms20081856
Sun JL, Zhao LL, Liao L, Tang XH, Cui C, Liu Q, He K, Ma JD, Jin L, Yan T, Zhou J, Yang S (2020a) Interactive effect of thermal and hypoxia on largemouth bass (Micropterus salmoides) gill and liver: aggravation of oxidative stress, inhibition of immunity and promotion of cell apoptosis. Fish & Shellfish Immunology 98:923–936. https://doi.org/10.1016/j.fsi.2019.11.056
Sun JL, Zhao LL, Wu H, Liu Q, Liao L, Luo J, Lian WQ, Cui C, Jin L, Ma JD, Li MZ, Yang S (2020b) Acute hypoxia changes the mode of glucose and lipid utilization in the liver of the largemouth bass (Micropterus salmoides). Sci Total Environ 713:135–157. https://doi.org/10.1016/j.scitotenv.2019.135157
Thomsen MD, Koerber JT, Wells JA (2013) Structural snapshots reveal distinct mechanisms of procaspase-3 and -7 activation. Proc Natl Acad Sci U S A 110(21):6. https://doi.org/10.1073/pnas.1306759110
Thomson E, Ferreira-Cerca S, Hurt E (2013) Eukaryotic ribosome biogenesis at a glance. J Cell Sci 126(21):4815–4821. https://doi.org/10.1242/jcs.111948
Tian CX, Lin XH, Saetan W, Huang Y, Shi HJ, Jiang DN et al (2020) Transcriptome analysis of liver provides insight into metabolic and translation changes under hypoxia and reoxygenation stress in silver sillago (Sillago sihama). Comparative Biochemistry and Physiology Part D: Genomics and Proteomics 36:100715. https://doi.org/10.1016/j.cbd.2020.100715
Tokumoto M, Nagahama Y, Tokumoto T (2000) Molecular cloning of cDNA encoding a ubiquitin-activating enzyme (E1) from gold¢sh (Carassius auratus) and expression analysis of the cloned gene. Biochim Biophys Acta 1492(1):259–263. https://doi.org/10.1016/S0167-4781(00)00091-9
Trasviña-Arenas CH, Garcia-Triana A, Peregrino-Uriarte AB, Yepiz-Plascencia G (2013) White shrimp Litopenaeus vannamei catalase: gene structure, expression and activity under hypoxia and reoxygenation. Comparative Biochemistry and Physiology. Part B, Biochemistry & Molecular Biology 164(1):44–52. https://doi.org/10.1016/j.cbpb.2012.10.004
Venugopal A, Mondal S, Ranganatha KS, Datta D, Kumar NS, Swamy NJ (2020) Purification and biochemical/biophysical characterization of two hexosaminidases from the fresh water mussel, Lamellidens corrianus. Int J Biol Macromol 149:754–766. https://doi.org/10.1016/j.ijbiomac.2020.01.241
Wang XQ, Sudha K, Mei C, Shen M (2010) Effects of low salinity and low temperature on survival, growth, and energy budget of juvenile Exopalaemon carinicauda. J Shellfish Res 29(4):1035–1041. https://doi.org/10.2983/035.029.0405
Wang RH, Zhang P, Li JH, Guan HZ, Shi GJ (2016) Ubiquitination is absolutely required for the degradation of hypoxia-inducible factor - 1 alpha protein in hypoxic conditions. Biochem Biophys Res Commun 470(1):117–122. https://doi.org/10.1016/j.bbrc.2016.01.005
Wilson RE, Sutherland RM (1989) Enhanced synthesis of specific proteins, RNA, and DNA caused by hypoxia and reoxygenation. Int J Radiat Oncol Biol Phys 16(4):957–961
Yang BY, Xu Y, Hu YG, Luo YW, Lu X et al (2016) Madecassic Acid protects against hypoxia-induced oxidative stress in retinal microvascular endothelial cells via ROS-mediated endoplasmic reticulum stress. Biomed Pharmacother 84:845–852. https://doi.org/10.1016/j.biopha.2016.10.015
Yang YJ, Fu Q, Wang XZ, Liu Y, Zeng QF, Li Y et al (2018) Comparative transcriptome analysis of the swimbladder reveals expression signatures in response to low oxygen stress in channel catfish, Ictalurus punctatus (Article). Physiol Genomics 50(8):636–647. https://doi.org/10.1016/j.biopha.2016.10.015
Zhou LQ, Liu ZH, Dong YH, Sun XJ, Wu B, Yu T et al (2019) Transcriptomics analysis revealing candidate genes and networks for sex differentiation of yesso scallop (Patinopecten yessoensis). BMC Genomics 20:671. https://doi.org/10.1186/s12864-019-6021-6
Data availability statement
The data that support the results of this present study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
Funding
This research was funded by The Jiangsu Provincial Agricultural Major New Varieties Creation Project (PZCZ201747), The fifth “226 Project” cultivation fund support project of Nantong, The Jiangsu Provincial Agricultural Science and Technology Independent Innovation Project in 2018 (CX18-2010), Science and Technology Plan Project of Nantong in 2019 (JC2019057).
Author information
Authors and Affiliations
Contributions
W. S., P. W., and X. W. designed the study. R. H., H. S., H. L., L. W., Y. Q., G. J., and J. C. performed the experiment. W. S., P. W., X. W., and Z. Y. analyzed the data. W. S. and P. W. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics statement
All samples and methods used in the present study were conducted in accordance with the Laboratory Animal Management Principles of China. All experimental protocols were approved by the Ethics Committee for Animal Experiments of the Jiangsu Institute of Marine Fisheries. All shrimps handling performed under ice anesthesia.
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Pierre Boudry
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shi, W., Wang, P., Hu, R. et al. Transcriptome analysis reveals hub genes in the hepatopancreas of Exopalaemon carinicauda in response to hypoxia and reoxygenation. Aquacult Int 29, 1785–1811 (2021). https://doi.org/10.1007/s10499-021-00717-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-021-00717-2