
Abstract
Background: 5-Fluorouracil (5-FU) has been used as a standard first-line treatment for colorectal cancer (CRC) patients. Although 5-FU-based chemotherapy and immune checkpoint blockade (ICB) have achieved success in treating CRC, drug resistance and low response rates remain substantial limitations. Thus, it is necessary to construct a 5-FU resistance-related signature (5-FRSig) to predict patient prognosis and identify ideal patients for chemotherapy and immunotherapy. Methods: Using bulk and single-cell RNA sequencing data, we established and validated a novel 5-FRSig model using stepwise regression and multiple CRC cohorts and evaluated its associations with the prognosis, clinical features, immune status, immunotherapy, neoadjuvant therapy, and drug sensitivity of CRC patients through various bioinformatics algorithms. Unsupervised consensus clustering was performed to categorize the 5-FU resistance-related molecular subtypes of CRC. The expression levels of 5-FRSig, immune checkpoints, and immunoregulators were determined using quantitative real-time polymerase chain reaction (RT‒qPCR). Potential small-molecule agents were identified via Connectivity Map (CMap) and molecular docking. Results: The 5-FRSig and cluster were confirmed as independent prognostic factors in CRC, as patients in the low-risk group and Cluster 1 had a better prognosis. Notably, 5-FRSig was significantly associated with 5-FU sensitivity, chemotherapy response, immune cell infiltration, immunoreactivity phenotype, immunotherapy efficiency, and drug selection. We predicted 10 potential compounds that bind to the core targets of 5-FRSig with the highest affinity. Conclusion: We developed a valid 5-FRSig to predict the prognosis, chemotherapeutic response, and immune status of CRC patients, thus optimizing the therapeutic benefits of chemotherapy combined with immunotherapy, which can facilitate the development of personalized treatments and novel molecular targeted therapies for patients with CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the leading cause of death worldwide, with its incidence and mortality rates ranking third and second among malignant tumors, respectively [1]. In recent years, the incidence and mortality rates of CRC have increased significantly [2]. Surgical treatment, radiation therapy, and systemic chemotherapy are the main therapeutic approaches for treating CRC [3]. Surgery is usually the treatment of choice for early-stage CRC. Chemotherapy drugs such as 5-fluorouracil (5-FU), fluorouracil (capecitabine), and calcium oxide (oxaliplatin) can be used as adjuvant treatments to reduce tumor volume, control postoperative recurrence, or provide remission therapy in advanced stages [4,5,6].
Since the 1990s, fluorouracil (FU)-based adjuvant chemotherapy has been an essential option for treating advanced CRC [7]. The use of adjuvant fluorouracil-based chemotherapy in patients with stage III colon cancer is thought to be standard care; however, it is not routinely recommended for patients with stage II colon cancer [8, 9]. Some patients develop resistance to chemotherapy drugs, which is one of the main causes of tumor treatment failure [10]. Once chemoresistance emerges, tumors tend to relapse and metastasize, causing the death of 70 to 80% of cancer patients; thus, chemoresistance is one of the greatest challenges in the long-term management of incurable metastatic disease [11]. 5-FU, an intravenous synthetic fluorouracil analog, is currently the most important chemical for treating CRC [12]. Using 5-FU can effectively reduce tumor recurrence and metastasis and improve the survival rates. However, cancer cells gradually develop resistance during chemotherapy, leading to the failure of chemotherapy drugs [13]. Resistance to 5-FU can result from various factors, including metabolic enzymes and cancer stemness. Some studies have suggested that mutations in genes and changes in the expression levels of genes involved in metabolic pathways associated with 5-FU may contribute to the development of resistance [14]. Thymidylate synthase polymorphism is now an emerging focus of interest responsible for 5-FU resistance [15,16,17,18]. In addition, cancer stem cells have long been associated with chemotherapy resistance [19]. However, because of the complexity of the tumor microenvironment, the underlying mechanism leading to chemotherapy resistance remains unclear.
A tremendous benefit has been achieved with immunotherapy in cancer treatment in recent years. The FDA approved immune checkpoint regimens in 2017 for CRC patients with defective mismatch repair (dMMR) or high microsatellite instability (MSI-H) levels. However, immunotherapy is inefficient for tumors that are proficient in mismatch repair (pMMR), microsatellite stable (MSS), or have low levels of microsatellite instability (MSI-L), which account for a large proportion of CRCs [20]. Although chemotherapy and immunotherapy have achieved unexpected efficacy in treating CRC, with the development of precision therapy, the limitations of monotherapy, especially chemotherapy resistance and a low rate of immunotherapy response, have gradually emerged. Recent research suggests that there are complex interactions between the immune system and chemotherapy. While immunosuppressive effects of chemotherapeutic agents have been reported [21, 22], studies have shown that chemotherapy can enhance the immunogenicity of tumor cells, activate immune effectors, and alleviate tumor-induced immunosuppression [23]. However, there are currently no effective biomarkers for determining the prognosis of patients with CRC or for predicting their response to chemotherapy and immunotherapy.
In this study, we first identified genes associated with 5-FU resistance in CRC patients and constructed a novel 5-FU resistance-related signature (5-FRSig) according to these genes. We systematically investigated and validated the prognostic value, chemotherapeutic response, immune landscape, and immunotherapy predictive power of the signature. Our study demonstrated that the 5-FRSig can be used as an independent prognostic factor to predict the response to chemotherapy and immunotherapy in CRC patients. This study is expected to lead to more accurate and effective treatment strategies for patients with CRC, including chemotherapy, immunotherapy, targeted therapy, and combination therapy, providing guidance strategies for the precise diagnosis and treatment of CRC.
Materials and methods
Data sources and processing
The transcriptomic data of parental and 5-FU-resistant cells were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE196900 [24]. Differentially expressed genes (DEGs) between 5-FU-resistant cells and parental cells in the HCT116 and SW480 cell lines were analyzed with the R package “DESeq2” [25]. Genes with an absolute value of log2 (fold change (FC)) > 2 and adjusted P value < 0.05 were considered DEGs. DEGs with consistent trends in both cell lines were considered 5-FU resistance-related candidate genes. The transcriptomic and clinical data of The Cancer Genome Atlas (TCGA) colon adenocarcinoma and rectum adenocarcinoma datasets were downloaded from the GDC data portal (https://portal.gdc.cancer.gov/). A total of 597 CRC samples with accessible clinical and survival data were enrolled in the training cohort. External validation was performed using the GSE37892 [26], GSE17537 [27], GSE192667 [28], and GSE29621 [29] datasets, all of which were downloaded from the GEO database. This study used consensus molecular subtypes (CMSs) obtained from the Colorectal Cancer Subtyping Consortium Synapse [30].
Construction of the 5-FRSig
Univariate Cox regression was conducted for 5-FU resistance-related candidate genes in the TCGA cohort. Subsequently, multivariate stepwise regression was conducted using the R package “MASS.” An optimized risk model associated with 5-FU resistance was then established, including thirteen 5-FU resistance-related genes (5-FRGs). For each patient, the risk score was calculated as follows: \(\text{R}\text{i}\text{s}\text{k} \,\text{s}\text{c}\text{o}\text{r}\text{e}={\sum }_{i=1}^{13}\text{E}\text{x}\text{p}\text{r}\text{e}\text{s}\text{s}\text{i}\text{o}\text{n} \left({\text{m}\text{R}\text{N}\text{A}}_{i}\right)\times \text{C}\text{o}\text{e}\text{f}\text{f}\text{i}\text{c}\text{i}\text{e}\text{n}\text{t}\left({\text{m}\text{R}\text{N}\text{A}}_{i}\right)\).
The correlation between the risk score and the expression level of the thirteen genes was analyzed using Spearman’s correlation. The R package “ComplexHeatmap” was applied to depict the results [31].
Prognostic analysis and construction of the nomogram
Patients were divided into high- and low-risk groups according to the median value of the risk score. The prognostic value of the 5-FRSig was evaluated by Kaplan‒Meier (K‒M) survival analysis, multivariate Cox regression analysis, and time-dependent receiver operating characteristic (ROC) curve analysis. A nomogram model integrating all independent prognostic factors, including risk factors, was established with the R package “rms” to further improve the prediction power. Moreover, calibration curves were generated. Overall survival (OS) and progression-free survival (PFS) rates were analyzed in the TCGA cohort. In addition, associations between the risk score, stage, and CMS were analyzed.
Functional enrichment analysis
DEGs between the high- and low-risk groups were identified using the R package “DESeq2” with a threshold of adjusted P < 0.05 and an absolute value of log2FC > 0.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses, along with gene set enrichment analysis (GSEA), were conducted using the R package “clusterProfiler” [32].
Tumor immune microenvironment analysis
First, we assessed the immune scores of risk groups using the Estimation of STromal and Immune cells in MAlignant Tumors using Expression data (ESTIMATE) algorithm, including estimate, immune, and stromal scores [33]. In addition, multiple deconvolution algorithms were used to evaluate immune subsets, including ESTIMATE [33], Tumor Immune Estimation Resource (TIMER) [34], Cell-type Identification By Estimating Relative Subsets of RNA Transcripts (CIBERSORT) [35], Estimating the Proportion of Immune and Cancer cells (EPIC) [36], xCELL [37], Microenvironment Cell Populations-counter (MCP-counter) [38], Quantification of the Tumor Immune Contexture from Human RNA-seq Data (quanTIseq) [39] and gene set variation analysis (GSVA) [40]. In addition, the expression levels of immune-related genes were extracted and compared between the high- and low-risk groups.
Prediction of the response to immunotherapy and chemotherapy
The correlation between the gene signature and somatic mutations and neoantigens was analyzed. The mutation annotation format (MAF) for the TCGA cohort was obtained from the TCGA data portal (https://portal.gdc.cancer.gov) and analyzed with the R package “maftools.” The numbers of somatic mutations and neoantigens were retrieved from The Cancer Immunome Atlas (TCIA) (https://tcia.at) [40]. Patient responses to immune checkpoint inhibitors were predicted using tumor immune dysfunction and exclusion (TIDE, http://tide.dfci.harvard.edu) [41], a well-developed and accurate method for predicting the efficacy of immunotherapy. Four independent cohorts with immunotherapy information, namely, the IMvigor210 (n = 298) [42], CheckMate 025 (n = 281) [43], GSE176307 (n = 88) [44], and GSE78220 (n = 27) [45] cohorts, were retrieved to further validate the results in samples with treatment response data. Receiver operating characteristic (ROC) curves were used to assess the ability of the 5-FRSig to predict the response to 5-FU. The data of three GEO cohorts, GSE39582 [46], GSE106584 [47], and GSE103479 [48], were retrieved, and only samples from patients who received adjuvant 5-FU treatment were included in the analysis. For GSE39582, GSE103479, and GSE106584, 82, 66, and 35 samples were retained, respectively. Patients who experienced relapse were considered nonresponders to 5-FU treatment. The R package “oncoPredict” [49] was used to analyze the sensitivity of patients to commonly used drugs. This analysis was conducted using transcriptomic data and drug sensitivity data downloaded from Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org/) [50], The Cancer Therapeutics Response Portal (CTRP, https://portals.broadinstitute.org/ctrp.v2.1/) [51], and Profiling Relative Inhibition Simultaneously in Mixtures (PRISM, https://www.theprismlab.org/) [52]. The half-maximal inhibitory concentration (IC50) in the GDSC and the area under the dose‒response curve (AUC) in the CTRP and PRISM cohorts were negatively correlated with drug sensitivity.
Consensus clustering analysis
Sixty-five prognostic genes were identified in the univariate Cox regression analysis. Unsupervised consensus clustering was conducted using the R package “ConsensusClusterPlus” with 50 iterations and a resampling rate of 80% [53]. K‒M survival, tumor immune microenvironment, DEG, and functional enrichment analyses were conducted between subclusters to explore the biological properties. In addition, the correlation between the gene signature and clusters was analyzed.
Real-time quantitative PCR validation
Forty-five pairs of CRC and adjacent normal tissue samples were collected from the Affiliated Hospital of Qingdao University for RT–qPCR validation. The ethical considerations and the criteria for inclusion and exclusion were used, as previously described [54]. Total RNA was extracted using an RNeasy kit (Beyotime, Shanghai, China, R0027) according to the manufacturer´s instructions. Then, 1 µg of total RNA was reverse transcribed with SuperScript II reverse transcriptase (Takara, Japan, RR047). Quantitative PCR analysis was performed with SYBR Green Mix (Takara, Japan, RR820) using an ABI 7900 HT Real-Time PCR System. GAPDH was used as an internal control. The primers used in this study are listed in Supplementary Table 1.
Identification and verification of candidate small molecules to reverse 5-FU resistance
To improve the clinical application of our signature, we used a Connectivity Map (CMap) to predict candidate small molecules that might reverse 5-FU resistance. CMap is a public resource comprising a comprehensive catalog of cellular signatures representing systematic perturbation with genetic and pharmacologic interference. The connectivity score, as calculated by CMap, indicates that the molecule could enhance a biological property if positive and reverse the biological property if negative [55]. In this study, the hub genes were divided into upregulated and downregulated groups and imported into the CMap database. Then, a list of small molecules was obtained, and these molecules were ranked by the connectivity score. Small molecules with negative connectivity scores, an FDR q value < 0.05, a specific mechanism of action (MoA), and targets were considered to have reliable potential to reverse 5FU resistance. Molecular docking was used to verify the reliability of these small molecules in reversing 5FU resistance. First, the 3D structures of the target proteins were downloaded from the RCSB Protein Database Bank (PDB, http://www.rcsb.org/) or the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/). The proteins were dehydrated and/or ligand-removed with PyMOL 2.5 software and saved in PDB format. The processed target protein was then imported into AutoDock Tools 1.5.6 software for hydrogenation and charge calculations and stored in PDBQT format. Second, the 3D structures of small molecules in SDF format were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and saved in mol2 format with Open Babel 2.3.1 software. Mol2 files of small molecule drugs were imported into AutoDock Tools, the total charge was detected, the charge was assigned, and flexible rotatable bonds were viewed and saved in PDBQT format. The grid box was obtained by using the GetBox Plugin in PyMOL. Finally, AutoDock Vina 1.1.2 was used to conduct molecular docking for 10 potential molecules and thirteen target proteins. The binding strength was evaluated according to the docking binding energy. The results were visualized with PyMOL 2.5 software.
Single-cell RNA sequencing analysis
We used the scRNA-seq cohort GSE178318 [56], which contains single-cell expression profiling of CRC liver metastases from treated patients and untreated patients. The R packages “Seurat” [57] and “Harmony” [58] were used to read sample data and remove batch effects between the samples. We then used the t-SNE method for dimension reduction processing to obtain the clusters and performed cell type annotation through the R package “SingleR” [59]. The score and distribution of the 5-FRSig in the single-cell samples were calculated using two methods, namely, Ucell and singscore. The Ucell algorithm uses the Mann–Whitney U statistic to calculate the gene set enrichment score for a single sample based on the gene expression ranking, while the singscore algorithm employs a gene enrichment score that is calculated based on the gene expression ranking of a single sample. This score assesses the distance of the gene set from the center. Both of these algorithms can be implemented through the R package “irGSEA.”
Statistical analysis
R software (version 4.1.3) and GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, United States) were used for data analysis and visualization. The continuous variables were analyzed using the Wilcoxon or Kruskal‒Wallis tests. Categorical variables were analyzed using the chi-square test (χ2) or Fisher’s exact test. Relationships between the risk scores and the expression levels of different genes were examined by Spearman’s correlation analysis. A P value < 0.05 was considered to indicate significance.
Results
Construction of a prognostic 5-FRSig
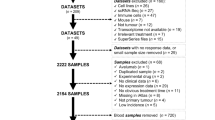
The flowchart provides an overview of the primary design of the current investigation (Fig. 1). We selected DEGs between 5-FU-resistant cells and parental cells in the HCT116 and SW480 cell lines from GSE196900. A total of 565 DEGs with a consistent trend in the two cell lines were identified; of these 5-FRGs, 513 were identified in the TCGA cohort (Fig. 2A). Then, we used univariate Cox regression analysis and obtained 65 of the 513 5-FRGs associated with prognosis (P < 0.05). Multivariate Cox analysis was subsequently applied, and 13 5-FRGs with independent prognostic value were identified. Figure 2B shows the results of the univariate Cox regression analysis of 13 5-FRGs. Then, an optimized risk model with 13 5-FRGS was established stepwise, including ALPK3 (alpha kinase 3), CPA4 (carboxypeptidase A4), DNAH7 (dynein axonemal heavy chain 7), FGF2 (fibroblast growth factor 2), HOXD13 (homeobox D13), NRG1 (neuregulin 1), PPP1R3F (protein phosphatase 1 regulatory subunit 3 F), SIX2 (SIX homeobox 2), SLC39A8 (solute carrier family 39 member 8), TMEM139 (transmembrane protein 139), TNFRSF19 (TNF receptor superfamily member 19), ZDHHC2 (zinc finger DHHC-type palmitoyltransferase 2), and ZNF607 (zinc finger protein 607). The patients were divided into high- and low-risk groups using the median risk score. Figure 2C displays the distribution of risk scores among TCGA-CRC patients. Additionally, the expression levels of twelve of the thirteen genes were significantly correlated with the risk score. Notably, SLC39A8 showed the strongest negative correlation (r = − 0.485, Fig. 2D). As shown in Fig. 2E, HOXD13, NRG1, SIX2, DNAH7, CAP4, ALPK3, and FGF2 were significantly upregulated in 5-FU-resistant cells, and TNFRSF19, TMEM139, PPP1R3F, SLC39A8, ZNF607, and ZDHHC were significantly downregulated. In addition, we analyzed the correlation between the risk score and clinicopathological features; the risk score was significantly correlated with status, T stage, N stage, and tumor stage (Fig. 2F). We then used three external datasets for validation and found that the risk scores of the GSE17537, GSE39582, and GSE37892 datasets differed significantly at various stages (Fig. 2G).

Flowchart of the entire study

Construction and validation of the 5-FU resistance-related signature. A. The DEGs of two cell lines (HCT116 and SW480) in the GSE196900 dataset. B. Univariate Cox analysis of TCGA-OS data for the 13 5-FU resistance-related genes used to construct the signature. C. Distribution of risk score (high and low) and status (dead and alive) in the TCGA-CRC cohort; D. The correlation between risk score and thirteen genes; E. Expression profiles of the genes in the normal and resistant groups in GSE196900. F. Correlation heatmap between risk groups and clinical characteristics. G. Validation of the signature in the GSE17537 and GSE37892 datasets. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001
Prognosis analysis of the 5-FRSig and construction of a nomogram
We then evaluated the prognostic capacity of the signature using the TCGA cohort. According to K‒M survival curves, patients in the high-risk group had significantly worse OS (P < 0.0001) and PFS (P < 0.001, Fig. 3A and B) rates. The risk score was confirmed to be an independent prognostic factor for both OS (HR = 3.136, 95% CI = 1.997–4.924, P < 0.001) and PFS (HR = 1.418, 95% CI = 1.002–2.007, P = 0.049). This result was validated in four external datasets, namely, GSE192667, GSE29621, GSE17537, and GSE37892 (Fig. 3C–F). To improve discrimination and make the model more applicable, we established a prognostic nomogram integrating the signature and independent risk factors (Fig. 4A–C). To confirm the superiority of the nomogram, calibration curves and ROC analysis were used to validate the nomogram’s prognostic accuracy and specificity. The results indicated that the 5-FRSig score and nomogram were superior to the stage in predicting OS or PFS outcomes in the TCGA cohort and GSE39582 cohort (Fig. 4D–E). These results indicate that our risk signature is an independent prognostic factor reliable for predicting survival probability. Furthermore, the nomogram integrating the risk score and clinicopathological characteristics was more reliable and accurate in predicting survival outcomes.

Prognostic analysis and validation of the 5-FU resistance-related signature. K‒M survival curve and multivariate analysis of the TCGA-OS (A), TCGA-PFS (B), GSE192667-OS (C), GSE29621-OS (D), GSE17537-OS (E), and GSE37892 (F) cohorts

Construction and validation of a nomogram. Nomograms constructed with three independent prognostic factors for 1-, 3-, and 5-year OS rates in the TCGA cohort (A), PFS rate in the TCGA cohort (B), and OS rate in the GSE39582 cohort (C). Calibration curves and receiver operating characteristic (ROC) curves showing the predictive accuracy of the risk score and nomogram in the TCGA-OS (D), TCGA-PFS (E), and GSE39582-OS (F) cohorts
The 5-FRSig predicts patient response to 5-FU therapy
Given that this model was constructed according to 5-FU resistance-related genes, we wondered whether it could discriminate patients’ responses to 5-FU treatment. We validated this idea using CRC samples treated with 5-FU. The risk score was calculated, and the association between the risk score and patient response to 5-FU treatment was analyzed. The results revealed that the recurrence rates were significantly greater in the high-risk group, and patients who experienced relapse had significantly greater risk scores. In addition, the AUCs of the ROC curves were 0.782, 0.755, and 0.944, all of which were greater than 0.75, demonstrating the signature’s good discriminative ability (Fig. 5A–C).

The risk signature predicts patient response to 5-FU therapy. The 5-FU resistance-related signature predicts the recurrence of patients who received 5-FU treatment and the relationship between the risk score and recurrence in the GSE39582 (A), GSE103479 (B), and GSE106584 (C) datasets. ∗∗∗P < 0.001
Analysis of the underlying biological pathway of the 5-FRSig
We used GSEA and GO and KEGG analyses to explore the underlying molecular mechanism of the 5-FRSig. First, the DEGs between subgroups in the TCGA cohort were analyzed. The results are displayed in a volcano plot (Fig. 6A). A total of 1,057 upregulated genes and 664 downregulated genes were identified with an adjusted P value < 0.05 and abs(logFC) > 0.5. GO analysis revealed that the DEGs were mainly enriched in inflammation-related pathways, including acute inflammatory response and chemokine activity. In addition, cancer-related pathways, such as ERK1 and ERK2 cascade, and immune-related pathways, such as granulocyte migration, were also found to be enriched in the analysis (Fig. 6B). GSEA further revealed that cancer-related pathways, such as epithelial mesenchymal transition, apical junction, KRAS signaling, WNT beta-catenin signaling, Hedgehog signaling, angiogenesis and hypoxia, were enriched in the high-risk group (Fig. 6C). KEGG analysis revealed that the upregulated genes were enriched in cancer-related pathways, including the PI3K-Akt signaling pathway, MAPK signaling pathway, Wnt signaling pathway, Rap1 signaling pathway, gastric cancer pathway, and TGF-beta signaling pathway (Fig. 6D), while the downregulated genes were enriched in immune-related pathways, including cytokine‒cytokine receptor interaction, the IL-17 signaling pathway, viral protein interaction with cytokine and cytokine receptor, rheumatoid arthritis and the TNF signaling pathway (Fig. 6E). The enrichment analysis demonstrated that cancer-immunity interactions may explain the prognostic power of our signature.

Analysis of the underlying biological pathways of the 5-FU resistance-related signature. A. Volcano plot of differentially expressed genes in the high- and low-risk groups with a threshold of FDR < 0.05 and absolute log2(FC) > 0.5. B. GO enrichment of differentially expressed genes. C. GSEA enrichment of differentially expressed genes. KEGG enrichment of the high- (D) and low-risk groups (E)
Immune profile analysis of the 5-FRSig
Multiple computational methods were used to determine the degree of immune cell infiltration in each sample to further investigate the relationship between the signature and the immune system. A heatmap of the cells that differed significantly between the subgroups is shown in Fig. 7A. According to the heatmap, the low-risk group exhibited increased levels of antitumor immune cells, such as CD4 + T cells (EPIC, xCELL), CD8 + T cells (EPIC, xCELL), neutrophils (EPIC, xCELL) and B cells (EPIC, xCELL), while the high-risk group exhibited increased levels of cancer-associated fibroblasts (MCPcounter, EPIC, xCELL) and macrophages (CIBERSORT, xCELL). We plotted radar charts to better visualize the differences between subgroups (Fig. 7B), demonstrating that the cytotoxicity score, immune score, number of neutrophils, number of CD4 + T cells, number of CD8 + T cells, number of B cells and number of NK cells were significantly greater in the low-risk group, while the stroma score and the numbers of cancer-associated fibroblasts and macrophages were significantly greater in the high-risk group. In addition, the analyses of immune-related genes demonstrated that the low-risk group had significantly greater immunostimulator levels and moderately greater cytotoxicity and immune inhibitor levels (Fig. 7C). Given these findings, we concluded that the low-risk group had more immune cell infiltration and greater antitumor activity, explaining the better outcomes in this group.

Immune profile analysis of the 5-FU resistance-related signature in the TCGA-CRC cohort. The heatmap (A) and radar map (B) show immune cell infiltration in the high- and low-risk groups calculated by multiple algorithms. C. Expression of immunomodulators in the high- and low-risk groups. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001
Mutation and immunotherapy response analysis of the 5-FRSig
Since immune cells are a prerequisite for immunotherapy, we hypothesized that the low-risk group would have a greater response rate to immune checkpoint inhibitors. We validated this hypothesis from different perspectives. First, we investigated the mutational landscape. We identified the top 20 mutated genes in the TCGA cohort (Fig. 8A). Significantly greater mutation rates were detected in two genes in each of the high- and low-risk groups, while no differences were detected in other genes (Fig. 8B). Moreover, there was no significant difference in the number of somatic mutations between the subgroups (Fig. 8C). Thus, there were no significant differences in mutations between the subgroups. However, there were more neoantigens in the low-risk group. Then, we analyzed the expression of the immune checkpoints PD-1, PD-L1, PD-L2, and CTLA4. The results showed that the expression levels of CD274 and CTLA4 were significantly greater in the low-risk group (Fig. 8D). Subsequently, we applied the TIDE algorithm to predict patient response to immune checkpoint blockade therapy. We observed a significantly greater response rate in the low-risk group and a significantly lower risk score for responders. Consistent with this finding, a lower TIDE score/dysfunction score/exclusion score and higher MSI score were found in the low-risk group, all of which indicated a greater response rate. Immunosuppressive MDSCs and CAFs were significantly more abundant in the high-risk group (Fig. 8E). We then analyzed four datasets with immunotherapeutic information to validate the above results. Consistent results were obtained, including significantly greater response rates (CR/PR/SD) in the low-risk group and significantly lower risk scores for responders (Fig. 8F-I). Therefore, we directly demonstrated the association between a low risk score and a high response rate to immunotherapy. These results suggested that the 5-FRSig could predict immunotherapy efficacy.

Mutation and immunotherapy response analysis of the 5-FU resistance-related signature. A. The top 20 mutated genes in the high- and low-risk groups. B. Comparison of the mutation ratio in the high- and low-risk groups. C. Comparison of mutations and neoantigens between the high- and low-risk groups. D. Expression of key immune checkpoint molecules in the high- and low-risk groups. E. The distribution of different responders and calculated scores in the high- and low-risk groups in the TCGA cohort, calculated by the TIDE algorithm. The distribution of different responders in the high- and low-risk groups in the IMvigor210 (F), CheckMate 025 (G), GSE176307 (H), and GSE78220 (I) datasets. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001
Construction and prognosis analysis of 5-FU resistance-related clusters
The 65 5-FRGs associated with prognosis were subjected to unsupervised cluster analysis. The ideal number of clusters was found to be two using the consensus CDF curve. After unsupervised clustering, we identified two clusters within the TCGA cohort (Fig. 9A). Figure 9B displays the distribution of clusters and status. Subsequently, we investigated the prognostic value of 5-FU resistance-related clusters. The results of the K‒M survival curve and multivariate Cox regression analyses showed that patients in Cluster 1 had a better survival probability, and this cluster was a significant independent prognostic factor for CRC (Fig. 9C and D). As shown in Fig. 9E, the cluster was significantly correlated with status, MSI status, and CMS status. In addition, we conducted functional enrichment analyses of the clusters, including GSEA and KEGG and GO analyses. The DEGs in Clusters 1 and 2 are shown in Supplementary Fig. 1A. We found that the pathways enriched in the clusters were mainly inflammatory and immune-related pathways (Supplementary Fig. 1B-D). Similarly, we analyzed the immune landscape of the clusters, including immune cell infiltration and the expression of immunomodulators. We found that a greater proportion of inhibitory immune cells infiltrated Cluster 2, including CAFs, Tregs, and MDSCs (Supplementary Fig. 2A–C). Interestingly, in the low-risk group, the proportion of patients in Cluster 1 was greater than that in Cluster 2, and the risk score of patients in Cluster 1 was significantly lower than that in Cluster 2 (Fig. 9F and G). A Sankey diagram was constructed to show the connections among status, risk score, and cluster (Fig. 9H). In addition, we analyzed the ability of the clusters to predict immunotherapy efficacy. We found that the response rate of patients in Cluster 1 was slightly greater than that of patients in Cluster 2 (Fig. 9I), and the TIDE score of patients in Cluster 1 was significantly greater than that of patients in Cluster 2 (Fig. 9J), indicating that patients in Cluster 1 were more sensitive to immunotherapy.

Construction and prognosis analysis of 5-FU resistance-related clusters. A. Consensus CDF curve of unsupervised cluster analysis. B. Distribution of the cluster (Cluster 1 and Clusters) and status (dead and alive) in the TCGA-CRC cohort. C. Kaplan‒Meier survival curves of clusters in the TCGA cohort. D. Multivariable analysis of clusters in the TCGA cohort. E. Heatmap showing the correlations between clusters and clinical characteristics. F. The distribution of clusters in the high- and low-risk groups. G. The difference in the risk score between Cluster 1 and Cluster 2. H. Sankey diagram combining OS, cluster, and risk score data. I. The distribution of different responses in Cluster 1 and Cluster 2. J. The TIDE algorithm, including the TIDE score, dysfunction score, and CAF score, was used to predict patient response to immunotherapy in different clusters. ∗P < 0.05, ∗∗∗P < 0.001
Clinical validation of the 5-FRSig
The expression levels of the 13 5-FRGs were measured by RT‒qPCR, and the risk score was calculated according to the formula for each patient in the external cohort. Spearman’s test was used to assess the correlation between the risk score and the 13 5-FRGs. As shown in Fig. 10A, the expression levels of FGF2 and CPA4 were positively correlated with the risk score, while the expression levels of ZNF607, SIX2, DNAH7, and TMEM139 were negatively correlated. The patients were divided into two subgroups according to the median risk score. The risk score significantly correlated with N stage. Patients with N0 stage tumors had significantly lower risk scores, and the low-risk group had a greater percentage of N0-stage tumors (Fig. 10B). Subsequently, we examined CD8A and CD8B expression levels in high-risk and low-risk samples, and we found no significant differences between the two groups of patients (Supplementary Fig. 3). However, the low-risk group had increased levels of cytolytic factors, including GZMA and GZMB. Moreover, the low-risk group had increased levels of immune stimulators, including HHLA2, CD28, and CD40LG (Fig. 10C). In particular, the low-risk group had significantly greater levels of CTLA4 (Fig. 10D). These data demonstrated that the low-risk group had significantly greater antitumor immune function, which was consistent with the better outcome and the predicted greater response rate to immunotherapy in this group.

Clinical validation of the 5-FU resistance-related signature. A. Correlations between the risk score and the expression of thirteen genes in tumor samples. B. The difference and distribution of risk scores in different clinical stages. C. Expression of immunomodulators in high- and low-risk groups in tumor samples. D. Expression of four key immune checkpoint molecules in tumor samples from the high- and low-risk groups. ∗P < 0.05, ∗∗P < 0.01
scRNA-seq analysis of the 5-FRSig
The scRNA-seq cohort GSE178318 was used to conduct scRNA-seq analysis of the 5-FRSig. After t-SNE reduction and cell annotation, we obtained nine cell clusters, including B cells, CAFs, cancer cells, endothelial cells, mast cells, myeloid cells, NK cells, plasma cells, and T cells (Supplementary Fig. 4A). Supplementary Fig. 5A shows the proportions of different cells in each sample. The cell markers used for cell annotation are displayed in Supplementary Fig. 4B and Supplementary Fig. 5B. We also analyzed the distribution of cells in diverse types of tissues (Supplementary Fig. 4C and 4D). Next, we explored the cellular distribution of the 13 5-FRGs used to construct the signature (Supplementary Fig. 5C). Subsequently, we used two methods to analyze the distribution of risk scores in the cells and found high-risk cells mainly in the tumor cell and myeloid cell populations (Supplementary Fig. 4E). Therefore, we speculated that the poor prognosis of patients in the high-risk group was closely related to these cells.
Drug sensitivity analysis in the high- and low-risk groups
We next assessed the correlation between the signature score and drug sensitivity. The GDSC analysis showed that the low-risk group was more sensitive to chemotherapy, including camptothecin, cisplatin, fluorouracil, oxaliplatin, irinotecan and irinotecan. Similarly, the low-risk group was more sensitive to drugs targeting EGFR signaling, including gefitinib, afatinib, erlotinib, lapatinib, AZD3759 and osimertinib. The low-risk group was also more sensitive to drugs targeting ERK MAPK signaling and RTK signaling, with one or two exceptions (Fig. 11A). In addition, we screened patients in the high-risk group for sensitivity to drugs in the CTRP and PRISM databases (Fig. 11B and C). The above results demonstrate that our model has good discriminative ability for 5-FU adjuvant therapy and that the low-risk group is more sensitive to most chemotherapeutic and targeted drugs.

Drug sensitivity analysis in the high- and low-risk groups. A. Compounds in the GSDC database that target chemotherapy, EGFR signaling, ERK MAPK signaling, and RTK signaling. B. Compounds in the PRISM database. C. Compounds in the CTRP database. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001
Candidate small molecules to reverse 5-FU resistance
The top 10 small molecules with negative connectivity with specific targets were screened out (Fig. 12A). These small molecules have the potential to reverse 5-FU resistance by inhibiting the upregulation of hub proteins. We verified the binding energy of these small molecules to the hub proteins, and the results are shown in Fig. 12B. Normally, a binding energy less than 0 indicates spontaneous binding, and the lower the binding energy is, the greater the possibility of interaction. As shown in Fig. 12B, the binding energies of the 10 potential molecules and the hub proteins were typically below − 5 kcal/mol, demonstrating their potential interaction. For each protein, the small molecule with the lowest binding energy was selected for docking visualization (Fig. 12C). In the Fig. 12C, the amino acids to which the small molecule binds are labeled, and the dotted lines show hydrogen bonds. We screened 10 small molecules that have the potential to reverse 5-FU resistance and validated the binding potential between the small molecules and the hub proteins.

Candidate small molecules that reverse 5-FU resistance. A. The structures of the top 10 compounds predicted by the CMap website. B. Heatmap displaying the binding energy of the small molecule drug and the hub protein. C. Small molecule drug docking targets with the lowest binding energy
Discussion
5-FU is a classic chemotherapy drug that has long played a key role in the treatment of cancer, especially CRC and breast cancer [60]. Despite the remarkable achievements of 5-FU in cancer treatment, some patients develop resistance to the drug, likely due to individuals’ unique genetic and epigenetic makeup [61]. Therefore, there is an urgent need for molecular identification to guide clinical chemotherapy. In this study, we constructed a 5-FRSig model using 5-FU-resistant CRC cell lines to explore the related mechanisms and provide more evidence for the precise treatment of CRC patients.
The 5-FRSig was constructed from thirteen 5-FRGs, including ALPK3, CPA4, DNAH7, FGF2, HOXD13, NRG1, PPP1R3F, SIX2, SLC39A8, TMEM139, TNFRSF19, ZDHHC2, and ZNF607. In a preliminary study, ALPK3 was shown to be associated with metastasis in osteosarcoma patients [62]. The prognostic value of CPA4 in non-small cell lung cancer (NSCLC), pancreatic cancer, and bladder cancer has also been reported [63,64,65]. In addition, mutations in DNAH7 were found to benefit CRC patients receiving immune checkpoint inhibition therapy [66]. However, FGF2 and SIX2 have been found to promote the development of NSCLC and breast cancer [67,68,69,70]. Similarly, HOXD13 has been found to promote the malignant progression of colon cancer [71], and the overexpression of NRG1 promotes the progression of gastric cancer [72]. Interestingly, studies have shown that PPP1R3F and TNFRSF19 are associated with the prognosis of CRC patients [73, 74]. Liu et al. reported that SLC39A8 suppressed the progression of clear cell renal cell carcinoma [75]. Zhang et al [76]. reported that TMEM139 prevents NSCLC metastasis by inhibiting lysosomal degradation of E-cadherin. Moreover, it has been reported that ZDHHC2 and ZNF607 expression levels are reduced and increased, respectively, in gastric adenocarcinoma patients [77, 78].
In this study, the 5-FRSig constructed from 5-FRGs was found to be an independent prognostic factor for CRC. Our signature also allowed better patient risk stratification. Using the ROC curve to predict patient survival, we can see that the AUC for risk score is higher than that for TNM staging in the TCGA cohort. Compared with traditional TNM staging to predict the prognosis of patients, our nomogram model based on the risk score has better predictive power. Furthermore, the ROC curves for predicting the therapeutic effect of 5-FU demonstrate that all the AUC values are above 0.7, indicating that the model exhibits high specificity and sensitivity in predicting the efficacy of 5-FU. In the clinical validation of the 5-FRSig, we further demonstrated that patients with high risk scores had worse outcomes. In addition, as the development of 5-FU resistance is an important cause of cancer treatment failure, our signature can predict the responsiveness of patients to 5-FU treatment and immunotherapy well, providing a basis for the precise treatment of patients with CRC.
It has been reported that 5-FU resistance involves various complex factors, including noncoding RNA regulation, tumor stem cells, tumor cell autophagy, epigenetics, and ATP-binding protein overexpression [79,80,81,82,83]. In this study, when we explored the mechanism underlying differences in the prognosis of patients in different risk groups through pathway enrichment, the results showed many pathways associated with tumor immune pathways. We speculated that the occurrence of 5-FU resistance might be related to changes in the patients’ immune microenvironment. Further analysis of immune cell infiltration and immune regulatory factor expression showed that patients in the high-risk group presented an immunosuppressive tumor microenvironment. The immunotherapy results also demonstrated that low-risk patients were more sensitive to immunotherapy. In addition, through scRNA-seq, we found that myeloid cells had greater risk scores than other immune cells, which could provide a new target for the future study of 5-FU resistance. Overall, our study links 5-FU resistance to the immune microenvironment, providing additional evidence for a combination of chemotherapy and immunotherapy in patients with CRC.
The results showed that our 5-FRSig has good potential for diagnosing and treating CRC. Nonetheless, our research still has certain limitations. This study provides a solid theoretical basis for subsequent research. We obtained many novel research results through in-depth data mining. Although we used our own clinical cohort for validation, we lack sufficient clinical data to validate the ability of the 5-FRsig to predict the response to chemotherapy and immunotherapy in patients with CRC, and further studies are needed. Furthermore, the TCGA database lacks patient treatment information, preventing direct inspection of the signature’s predictive ability for 5-FU efficacy using TCGA data. Instead, the efficacy of the signature for predicting 5-FU treatment outcomes was validated using the GEO dataset, which introduces a limitation to our results. Finally, the inclusion of an internal cohort, including a combination of chemotherapy and immunotherapy, to validate the hypotheses of this study will be the focus of future research. Although we conducted a comprehensive analysis of multiple independent cohorts and obtained some clinically promising conclusions, the molecular mechanisms of the 5-FRSig in CRC prognosis, chemotherapy, and immunotherapy need to be further validated in vivo and in vitro.
Conclusion
In this study, 5-FU resistance in CRC was comprehensively analyzed using various methods, and a novel 5-FRSig was successfully constructed. This signature could be used for risk stratification, prognosis prediction, 5-FU sensitivity prediction, and immunotherapy prediction in CRC patients. In addition, it was found that the underlying mechanism was related to tumor immune pathways. Finally, we used drug sensitivity analysis and molecular docking technology to explore 10 suitable drugs for CRC patients. This study provided a new perspective on 5-FU resistance in CRC patients and a theoretical basis for improvements in chemotherapy, immunotherapy, targeted therapy, combination therapy, and individualized antitumor therapy.
Data availability
The original contributions presented in the study are included in the article and Supplementary Material, and further inquiries can be directed to the corresponding author.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global Cancer statistics 2018: Globocan estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492. Epub 2018/09/13
Yang Y, Wang HY, Chen YK, Chen JJ, Song C, Gu J (2020) Current status of Surgical treatment of rectal Cancer in China. Chin Med J (Engl) 133(22):2703–2711 Epub 2020/09/06. https://doi.org/10.1097/cm9.0000000000001076
Costa AF, Campos D, Reis CA, Gomes C (2020) Targeting glycosylation: a New Road for Cancer Drug Discovery. Trends Cancer 6(9):757–766 Epub 2020/05/10. https://doi.org/10.1016/j.trecan.2020.04.002
André T, Quinaux E, Louvet C, Colin P, Gamelin E, Bouche O et al (2007) Phase Iii Study comparing a semimonthly with a monthly regimen of Fluorouracil and Leucovorin as Adjuvant treatment for stage ii and iii Colon cancer patients: final results of Gercor C96.1. J Clin Oncol 25(24):3732–3738 Epub 2007/08/21. https://doi.org/10.1200/jco.2007.12.2234
Gray R, Barnwell J, McConkey C, Hills RK, Williams NS, Kerr DJ (2007) Adjuvant chemotherapy Versus Observation in patients with colorectal Cancer: a randomised study. Lancet 370(9604):2020–2029 Epub 2007/12/18. https://doi.org/10.1016/s0140-6736(07)61866-2
de Gramont A, Bosset JF, Milan C, Rougier P, Bouché O, Etienne PL et al (1997) Randomized trial comparing monthly low-dose leucovorin and Fluorouracil Bolus with Bimonthly High-Dose Leucovorin and Fluorouracil Bolus Plus continuous infusion for Advanced Colorectal Cancer: A French Intergroup Study. J Clin Oncol 15(2):808–815 Epub 1997/02/01. https://doi.org/10.1200/jco.1997.15.2.808
Allen WL, Johnston PG (2005) Role of genomic markers in Colorectal Cancer Treatment. J Clin Oncol 23(20):4545–4552 Epub 2005/07/09. https://doi.org/10.1200/jco.2005.19.752
Benson Ar, Schrag D, Somerfield MR, Cohen AM, Figueredo AT, Flynn PJ et al (2004) American Society of Clinical Oncology Recommendations on adjuvant chemotherapy for stage ii Colon cancer. J Clin Oncol 22(16):3408–3419
Figueredo A, Charette ML, Maroun J, Brouwers MC, Zuraw L (2004) Adjuvant therapy for stage ii Colon Cancer: a systematic review from the Cancer Care Ontario Program in evidence-based Care’s gastrointestinal Cancer Disease Site Group. J Clin Oncol 22(16):3395–3407
El Zarif T, Yibirin M, De Oliveira-Gomes D, Machaalani M, Nawfal R, Bittar G et al (2022) Overcoming therapy resistance in Colon cancer by Drug Repurposing. Cancers (Basel) 14(9). https://doi.org/10.3390/cancers14092105. Epub 2022/05/15
Vasan N, Baselga J, Hyman DM (2019) A view on Drug Resistance in Cancer. Nature 575(7782):299–309 Epub 2019/11/15. https://doi.org/10.1038/s41586-019-1730-1
Akalovich S, Portyanko A, Pundik A, Mezheyeuski A, Doroshenko T (2021) 5-Fu resistant colorectal Cancer cells possess improved invasiveness and Β(Iii)-Tubulin expression. Exp Oncol 43(2):111–117 Epub 2021/07/01. https://doi.org/10.32471/exp-oncology.2312-8852.vol-43-no-2.16314
Phipps O, Brookes MJ, Al-Hassi HO (2021) Iron Deficiency, Immunology, and Colorectal Cancer. Nutr Rev 79(1):88–97 Epub 2020/07/18. https://doi.org/10.1093/nutrit/nuaa040
Zheng H-C (2017) The Molecular mechanisms of Chemoresistance in Cancers. Oncotarget 8(35):59950
Van der Zee J, Van Eijck C, Hop W, van Dekken H, Dicheva B, Seynhaeve A et al (2012) Expression and prognostic significance of Thymidylate synthase (Ts) in pancreatic head and Periampullary Cancer. Eur J Surg Oncol (EJSO) 38(11):1058–1064
Sulzyc-Bielicka V, Domagala P, Bielicki D, Safranow K, Domagala W (2014) Thymidylate Synthase expression and P21 Waf1/P53 phenotype of Colon cancers identify patients who May Benefit from 5-Fluorouracil Based Therapy. Cell Oncol 37:17–28
Nagaraju GP, Alese OB, Landry J, Diaz R, El-Rayes BF (2014) Hsp90 Inhibition Downregulates Thymidylate Synthase and Sensitizes Colorectal Cancer Cell Lines to the Effect of 5fu-Based Chemotherapy. Oncotarget 5(20):9980
Matsuoka K, Nakagawa F, Kobunai T, Takechi T (2018) Trifluridine/Tipiracil overcomes the resistance of human gastric 5-Fluorouracil-refractory cells with high Thymidylate synthase expression. Oncotarget 9(17):13438
Chang JC (2016) Cancer Stem Cells: Role in Tumor Growth, Recurrence, Metastasis, and Treatment Resistance. Medicine 95(Suppl 1)
Kather JN, Halama N, Jaeger D (2018) Genomics and emerging biomarkers for Immunotherapy of Colorectal Cancer. Semin Cancer Biol 52(Pt 2):189–197 Epub 2018/03/05. https://doi.org/10.1016/j.semcancer.2018.02.010
Rasmussen L, Arvin A, Chemotherapy-Induced, Immunosuppression (1982) Environ Health Perspect 43:21–25 Epub 1982/02/01. https://doi.org/10.1289/ehp.824321
Schwartz RS (1968) Are immunosuppressive anticancer drugs Self-Defeating? Cancer Res 28(7):1452–1454 Epub 1968/07/01
Ghiringhelli F, Apetoh L (2014) The interplay between the Immune System and Chemotherapy: emerging methods for optimizing therapy. Expert Rev Clin Immunol 10(1):19–30 Epub 2013/12/07. https://doi.org/10.1586/1744666x.2014.865520
Roy S, Zhao Y, Yuan YC, Goel A (2022) Metformin and Icg-001 Act Synergistically to Abrogate Cancer Stem Cells-Mediated Chemoresistance in Colorectal Cancer by Promoting Apoptosis and Autophagy. Cancers (Basel) 14(5). Epub 2022/03/11. https://doi.org/10.3390/cancers14051281
Love MI, Huber W, Anders S (2014) Moderated estimation of fold Change and Dispersion for Rna-Seq Data with Deseq2. Genome Biol 15(12):550. https://doi.org/10.1186/s13059-014-0550-8
Laibe S, Lagarde A, Ferrari A, Monges G, Birnbaum D, Olschwang S (2012) A seven-gene signature aggregates a subgroup of stage ii Colon cancers with Stage Iii. Omics 16(10):560–565 Epub 2012/08/25. https://doi.org/10.1089/omi.2012.0039
Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A et al (2010) Experimentally derived Metastasis Gene expression Profile predicts recurrence and death in patients with Colon cancer. Gastroenterology 138(3):958–968 Epub 2009/11/17. https://doi.org/10.1053/j.gastro.2009.11.005
Sugawara T, Miya F, Ishikawa T, Lysenko A, Nishino J, Kamatani T et al (2022) Immune subtypes and Neoantigen-Related Immune Evasion in Advanced Colorectal Cancer. iScience 25(2):103740. https://doi.org/10.1016/j.isci.2022.103740. Epub 2022/02/08
Chen DT, Hernandez JM, Shibata D, McCarthy SM, Humphries LA, Clark W et al (2012) Complementary strand micrornas Mediate Acquisition of metastatic potential in Colonic Adenocarcinoma. J Gastrointest Surg 16(5):905–912 discussion 912 – 903. Epub 2012/03/01. https://doi.org/10.1007/s11605-011-1815-0
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C et al (2015) The Consensus Molecular subtypes of Colorectal Cancer. Nat Med 21(11):1350–1356 Epub 2015/10/13. https://doi.org/10.1038/nm.3967
Gu Z, Eils R, Schlesner M (2016) Bioinformatics 32(18):2847–2849 Epub 2016/05/22. https://doi.org/10.1093/bioinformatics/btw313. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z et al (2021) Clusterprofiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data Innov (Camb) 2(3):100141 Epub 2021/09/25. https://doi.org/10.1016/j.xinn.2021.100141.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W et al (2013) Inferring Tumour Purity and Stromal and Immune Cell admixture from expression data. Nat Commun 4:2612 Epub 2013/10/12. https://doi.org/10.1038/ncomms3612
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS et al (2017) Timer: a web server for Comprehensive Analysis of Tumor-infiltrating Immune cells. Cancer Res 77(21):e108–e110 Epub 2017/11/03. https://doi.org/10.1158/0008-5472.CAN-17-0307
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA (2018) Profiling Tumor infiltrating Immune cells with Cibersort. Methods Mol Biol 1711:243–259 Epub 2018/01/19. https://doi.org/10.1007/978-1-4939-7493-1_12
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D (2017) Simultaneous Enumeration of Cancer and Immune Cell Types from Bulk Tumor Gene Expression Data. Elife 6. Epub 2017/11/14. https://doi.org/10.7554/eLife.26476
Aran D, Hu Z, Butte AJ, Xcell (2017) Digitally portraying the tissue Cellular Heterogeneity Landscape. Genome Biol 18(1):220 Epub 2017/11/17. https://doi.org/10.1186/s13059-017-1349-1
Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F et al (2016) Estimating the population abundance of tissue-infiltrating Immune and stromal cell populations using gene expression. Genome Biol 17(1):218 Epub 2016/10/22. https://doi.org/10.1186/s13059-016-1070-5
Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H et al (2019) Molecular and pharmacological modulators of the Tumor Immune Contexture revealed by Deconvolution of Rna-Seq Data. Genome Med 11(1):34 Epub 2019/05/28. https://doi.org/10.1186/s13073-019-0638-6
Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D et al (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18(1):248–262 Epub 2017/01/05. https://doi.org/10.1016/j.celrep.2016.12.019
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X et al (2018) Signatures of T cell dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat Med 24(10):1550–1558 Epub 2018/08/22. https://doi.org/10.1038/s41591-018-0136-1
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y et al (2018) Tgfbeta attenuates Tumour response to Pd-L1 blockade by contributing to Exclusion of T cells. Nature 554(7693):544–548 Epub 2018/02/15. https://doi.org/10.1038/nature25501
Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J et al (2020) Interplay of somatic alterations and Immune Infiltration modulates response to Pd-1 blockade in Advanced Clear Cell Renal Cell Carcinoma. Nat Med 26(6):909–918 Epub 2020/05/31. https://doi.org/10.1038/s41591-020-0839-y
Rose TL, Weir WH, Mayhew GM, Shibata Y, Eulitt P, Uronis JM et al (2021) Fibroblast growth factor receptor 3 alterations and response to Immune Checkpoint Inhibition in Metastatic Urothelial Cancer: a Real World Experience. Br J Cancer 125(9):1251–1260 Epub 2021/07/24. https://doi.org/10.1038/s41416-021-01488-6
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S et al (2016) Genomic and transcriptomic features of response to anti-pd-1 therapy in metastatic melanoma. Cell 165(1):35–44 Epub 2016/03/22. https://doi.org/10.1016/j.cell.2016.02.065
Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L et al (2013) Gene expression classification of Colon cancer into Molecular subtypes: characterization, validation, and Prognostic Value. PLoS Med 10(5):e1001453 Epub 20130521. https://doi.org/10.1371/journal.pmed.1001453
Zhu J, Deane NG, Lewis KB, Padmanabhan C, Washington MK, Ciombor KK et al (2016) Evaluation of frozen tissue-derived prognostic gene expression signatures in Ffpe Colorectal Cancer samples. Sci Rep 6:33273 Epub 2016/09/15. https://doi.org/10.1038/srep33273
Allen WL, Dunne PD, McDade S, Scanlon E, Loughrey M, Coleman H et al (2018) Transcriptional Subtyping and Cd8 Immunohistochemistry Identifies Poor Prognosis Stage Ii/Iii Colorectal Cancer Patients Who Benefit from Adjuvant Chemotherapy. JCO Precis Oncol 2018. Epub 2018/08/09. https://doi.org/10.1200/po.17.00241
Maeser D, Gruener RF, Huang RS (2021) Oncopredict: An R Package for Predicting in Vivo or Cancer Patient Drug Response and Biomarkers from Cell Line Screening Data. Brief Bioinform 22(6). Epub 2021/07/15. https://doi.org/10.1093/bib/bbab260
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S et al (2013) Genomics of Drug Sensitivity in Cancer (Gdsc): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res 41(Database issue):D955-961. Epub 2012/11/28. https://doi.org/10.1093/nar/gks1111
Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S et al (2016) Correlating Chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol 12(2):109–116 Epub 2015/12/15. https://doi.org/10.1038/nchembio.1986
Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG et al (2020) Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 1(2):235–248 Epub 2020/07/03. https://doi.org/10.1038/s43018-019-0018-6
Wilkerson MD, Hayes DN (2010) Consensusclusterplus: a Class Discovery Tool with confidence assessments and item Tracking. Bioinformatics 26(12):1572–1573 Epub 2010/04/30. https://doi.org/10.1093/bioinformatics/btq170
Hou Y, Zhang R, Zong J, Wang W, Zhou M, Yan Z et al (2022) Comprehensive Analysis of a Cancer-immunity cycle-based signature for Predicting Prognosis and Immunotherapy Response in patients with colorectal Cancer. Front Immunol 13:892512 Epub 2022/06/18. https://doi.org/10.3389/fimmu.2022.892512
Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X et al (2017) A Next Generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell 171(6):1437–1452 e1417. Epub 2017/12/02. https://doi.org/10.1016/j.cell.2017.10.049
Che LH, Liu JW, Huo JP, Luo R, Xu RM, He C et al (2021) A single-cell Atlas of Liver metastases of Colorectal Cancer reveals reprogramming of the Tumor Microenvironment in Response to Preoperative Chemotherapy. Cell Discov 7(1):80 Epub 2021/09/08. https://doi.org/10.1038/s41421-021-00312-y
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell Transcriptomic Data across different conditions, technologies, and species. Nat Biotechnol 36(5):411–420. https://doi.org/10.1038/nbt.4096
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K et al (2019) Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16(12):1289–1296. https://doi.org/10.1038/s41592-019-0619-0
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A et al (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172 Epub 2019/01/16. https://doi.org/10.1038/s41590-018-0276-y
Sethy C, Kundu CN (2021) 5-Fluorouracil (5-Fu) Resistance and the New Strategy to enhance the sensitivity against Cancer: implication of DNA repair inhibition. Biomed Pharmacother 137:111285 Epub 2021/01/24. https://doi.org/10.1016/j.biopha.2021.111285
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V (2020) 5-Fluorouracil and other Fluoropyrimidines in Colorectal Cancer: past, Present and Future. Pharmacol Ther 206:107447 Epub 2019/11/23. https://doi.org/10.1016/j.pharmthera.2019.107447
Dong S, Huo H, Mao Y, Li X, Dong L (2019) A risk score model for the prediction of Osteosarcoma Metastasis. FEBS Open Bio 9(3):519–526 Epub 2019/03/15. https://doi.org/10.1002/2211-5463.12592
Sun L, Wang Y, Yuan H, Burnett J, Pan J, Yang Z et al (2016) Cpa4 is a Novel Diagnostic and prognostic marker for human non-small-cell Lung Cancer. J Cancer 7(10):1197
Sun L, Burnett J, Guo C, Xie Y, Pan J, Yang Z et al (2016) Cpa4 is a Promising Diagnostic serum biomarker for pancreatic Cancer. Am J cancer Res 6(1):91
Wei C, Zhou Y, Xiong Q, Xiong M, Hou Y, Yang X et al (2021) Comprehensive Analysis of Cpa4 as a poor prognostic biomarker correlated with Immune cells infiltration in bladder Cancer. Biology 10(11):1143
Yang W, Shen Z, Yang T, Wu M (2022) Dnah7 mutations benefit Colorectal Cancer patients receiving Immune Checkpoint inhibitors. Ann Transl Med 10(24):1335 Epub 2023/01/21. https://doi.org/10.21037/atm-22-6166
Berger W, Setinek U, Mohr T, Kindas-Mügge I, Vetterlein M, Dekan G et al (1999) Evidence for a role of Fgf‐2 and Fgf Receptors in the proliferation of non‐small cell Lung Cancer cells. Int J Cancer 83(3):415–423
Giulianelli S, Riggio M, Guillardoy T, Pérez Piñero C, Gorostiaga MA, Sequeira G et al (2019) Fgf2 induces breast Cancer growth through ligand-independent activation and recruitment of Erα and Prbδ4 isoform to Myc Regulatory sequences. Int J Cancer 145(7):1874–1888
Hou H, Yu X, Cong P, Zhou Y, Xu Y, Jiang Y (2019) Six2 promotes non–small cell Lung Cancer Cell Stemness Via transcriptionally and epigenetically regulating E-Cadherin. Cell Prolif 52(4):e12617
Wang C-A, Drasin D, Pham C, Jedlicka P, Zaberezhnyy V, Guney M et al (2014) Homeoprotein Six2 promotes breast Cancer Metastasis Via Transcriptional and Epigenetic Control of E-Cadherin expression. Cancer Res 74(24):7357–7370
Yin J, Guo Y (2021) Hoxd13 promotes the malignant progression of Colon cancer by upregulating Ptprn2. Cancer Med 10(16):5524–5533
Han M-E, Kim H-J, Shin DH, Hwang S-H, Kang C-D, Oh S-O (2015) Overexpression of Nrg1 promotes progression of gastric Cancer by regulating the Self-Renewal of Cancer Stem cells. J Gastroenterol 50:645–656
Wei W, Xi Y, Jiamin X, Jing Z, Shuwen H (2019) Screening of molecular targets and construction of a Cerna Network for Oxaliplatin Resistance in Colorectal Cancer. RSC Adv 9(54):31413–31424
Schön S, Flierman I, Ofner A, Stahringer A, Holdt LM, Kolligs FT et al (2014) Β-Catenin regulates Nf‐Κb Activity Via Tnfrsf19 in Colorectal Cancer cells. Int J Cancer 135(8):1800–1811
Liu L, Hou Y, Hu J, Zhou L, Chen K, Yang X et al (2021) Slc39a8/Zinc suppresses the progression of Clear Cell Renal Cell Carcinoma. Front Oncol 11:651921
Zhang S, He Y, Xuan Q, Ling X, Men K, Zhao X et al (2022) Tmem139 prevents Nsclc Metastasis by inhibiting lysosomal degradation of E-Cadherin. Cancer Sci 113(6):1999–2007
Yan S-M, Tang J-J, Huang C-Y, Xi S-Y, Huang M-Y, Liang J-Z et al (2013) Reduced expression of Zdhhc2 is Associated with Lymph Node Metastasis and poor prognosis in gastric adenocarcinoma. PLoS ONE 8(2):e56366
Dastsooz H, Alizadeh A, Habibzadeh P, Nariman A, Hosseini A, Mansoori Y et al (2022) Lncrna–Mirna–Mrna Networks of gastrointestinal cancers representing common and specific Lncrnas and Mrnas. Front Genet 12:791919
Najafi M, Mortezaee K, Majidpoor J (2019) Cancer Stem Cell (Csc) Resistance Drivers. Life Sci 234:116781. Epub 2019/08/21. https://doi.org/10.1016/j.lfs.2019.116781
Onorati AV, Dyczynski M, Ojha R, Amaravadi RK (2018) Targeting Autophagy in Cancer. Cancer 124(16):3307–3318 Epub 2018/04/20. https://doi.org/10.1002/cncr.31335
Recillas-Targa F (2022) Cancer epigenetics: an overview. Arch Med Res 53(8):732–740 Epub 2022/11/22. https://doi.org/10.1016/j.arcmed.2022.11.003
Chen B, Dragomir MP, Yang C, Li Q, Horst D, Calin GA (2022) Targeting non-coding rnas to Overcome Cancer Therapy Resistance. Signal Transduct Target Ther 7(1):121 Epub 2022/04/15. https://doi.org/10.1038/s41392-022-00975-3
Zhao R, Xia D, Chen Y, Kai Z, Ruan F, Xia C et al (2023) Improved diagnosis of Colorectal Cancer using combined biomarkers including Fusobacterium Nucleatum, Fecal Occult blood, transferrin, Cea, Ca19-9, gender, and Age. Cancer Med 12(13):14636–14645 Epub 2023/05/10. https://doi.org/10.1002/cam4.6067
Acknowledgements
The authors are thankful for the publicly available databases.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) Grant (2023-I2M-2-009) and the Natural Science Foundation of China (NSFC) Grant (No. 81773750).
Author information
Authors and Affiliations
Contributions
Writing – original draft: FZ, YH and MY. Writing – review & editing: MY, ZM, ZY, TL, WG, SL, LY, ZZ and WZ. Conceptualization: MY and YH. Investigation: YH, ZY and SL. Software: YH and TL. Data curation: JZ, WG and ZZ. Methodology: FZ, YH, ZY and SL. Supervision: MY. Formal Analysis: YH, FZ and WZ. Validation: YH, FZ and LY. Funding acquisition: MY. Resources: JZ and WG. Visualization: FZ and TL.
Corresponding author
Ethics declarations
Ethical statement
The studies involving human participants were reviewed and approved by the Research Ethics Committee of The Affiliated Hospital of Qingdao University. The patients/participants provided their written informed consent to participate in this study.
Competing of interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hou, Y., Zhang, F., Zong, J. et al. Integrated analysis reveals a novel 5-fluorouracil resistance-based prognostic signature with promising implications for predicting the efficacy of chemotherapy and immunotherapy in patients with colorectal cancer. Apoptosis 29, 1126–1144 (2024). https://doi.org/10.1007/s10495-024-01981-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-024-01981-2