
Abstract
Leishmaniasis is a neglected tropical disease that affects millions of people worldwide. Current therapies mainly rely on antimonial drugs that are inadequate because of their high toxicity and increased drug resistance. An urgent need exists to discover new, more effective, more affordable, and more target-specific drugs. Pathways that are associated with apoptosis-like cell death have been identified in unicellular eukaryotes, including protozoan parasites. In the present study, we studied the mechanism of cell death that is induced by A3K2A3 against L. amazonensis. A3K2A3 is a dibenzylideneacetone that has an acyclic dienone that is attached to aryl groups in both β-positions, which is similar to curcuminoids and chalcone structures. This compound was previously shown to be safe with regard to cytotoxicity and active against the parasite. Biochemical and morphological approaches were used in the present study. The results suggested that A3K2A3 caused mitochondrial dysfunction in L. amazonensis promastigotes, leading to mechanisms of cell death that share some common phenotypic features with metazoan apoptosis, such as an increase in reactive oxygen species production, a decrease in the adenosine triphosphate ratio, phosphatidylserine exposure, a decrease in cell volume, caspase production, and DNA fragmentation. Altogether, these findings indicate that apoptosis can indeed be triggered by chemotherapeutic agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leishmaniasis remains endemic in several parts of the world and is a serious health problem in numerous developing countries. It occurs as a complex and clinically diverse illness that is caused by protozoan Leishmania species that are transmitted through the bite of sandflies. Despite recent advances, the treatment of leishmaniasis is still problematic because of the high toxicity and adverse side effects of therapeutic drugs. The need to identify new molecular targets to improve therapy is clearly justified. Target-identification and mechanism-of-action studies play important roles in drug discovery. The putative target should be either absent in the host or markedly different from the host homolog so that it can be exploited as a drug target [1].
Interestingly, programmed cell death in protists appears to share some morphological features with apoptosis in multicellular organisms, including cell shrinkage, the loss of mitochondrial membrane potential, and the externalization of phosphatidylserine [2, 3]. A better understanding of the mechanistic machinery of apoptosis-like programmed cell death in protists would thus prove immensely beneficial in the design of rational chemotherapeutic interventions in a target-dependent manner [4].
1,5-Diarylpentanoid dibenzylideneacetone is the parent compound of a class of compounds that have an acyclic dienone that is attached to aryl groups in both β-positions. These structures resemble those of curcuminoids (1,7-diarylheptanes) and chalcones (1,3-diarylpropanes), which are very important bioactive natural compounds that are found in many plant species [5]. Dibenzylideneacetones (DBAs) and their analogues have antiproliferative, antiinflammatory, and apoptotic effects through the regulation of multiple signaling pathways in cancer cell lines [6–10]. Dibenzylideneacetone has been reported to enhance TRAIL-induced (cytokine tumor necrosis factor (TNF)-related apoptosis-inducing ligand) apoptosis by regulating cell-survival proteins and pro-apoptotic proteins through the activation of reactive oxygen species (ROS) and CHOP (C/EBP homologous protein) in colon cancer cells [11]. The good bioavailability of some dibenzylideneacetones and their derivatives, which is required for bioactivity, and their mode of cross linking have raised chemists’ interest in their synthesis [5].
Our previous study reported the anti-leishmanial and anti-trypanosomal effects of A3K2A3 (2d) [5]. Considering its anti-leishmanial activity and selectivity for parasites, the aim of the present study was to better characterize the biochemical alterations that are induced by this compound against promastigote forms of L. amazonensis and elucidate the possible mechanism of action of A3K2A3 that is involved in the cell death of this protozoan.
Materials and methods
Chemicals
Actinomycin D, antimicyn A, bovine serum albumin, carbonyl cyanide m-chlorophenylhydrazone (CCCP), digitonin, dimethylsulfoxide (DMSO), rhodamine 123 (Rh123), 2′,7′-dichlorofluorescin diacetate (H2DCFDA), and Nile Red were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) was obtained from Invitrogen (Grand Island, NY, USA). Annexin-V FITC and propidium iodide (PI) were obtained from Invitrogen (Eugene, OR, USA). Diphenyl-1-pyrenylphosphine (DPPP), the APO-BrdU terminal deoxynucleotidyl transferase enzyme mediated dUTP end labeling (TUNEL) Assay Kit, and the EnzChek Caspase-3 Assay Kit were purchase from Molecular Probes (Eugene, Oregon, USA). CellTiter-Glo was obtained from Promega (Madison, WI, USA). All of the other reagents were of analytical grade.
Synthesis and preparation of A3K2A3
A3K2A3 was synthesized as previously described by Ud Din et al. [5]. Stock solutions were aseptically prepared in DMSO and diluted in culture medium so that the DMSO concentration did not exceed 1 % in the experiments. The concentrations of A3K2A3 that were used in the assays were 3.4 and 9.3 µM, representing the IC50 and IC90, respectively, as described previously [5].
Parasites
Leishmania amazonensis promastigotes (MHOM/BR/Josefa) were maintained at 25 °C in Warren’s medium (brain–heart infusion plus hemin and folic acid; pH 7.2) supplemented with 10 % heat-inactivated FBS.
In vitro antiproliferative assay
Promastigotes in the logarithmic phase (1 × 106 parasites/ml) were grown in 24-well culture microplates at 25 °C in Warren’s medium supplemented with 10 % FBS. Parasites were then incubated in the presence of different concentrations of A3K2A3 (1–100 μM). Anti-leishmanial activity was determined by directly counting free-living parasites in a Neubauer chamber daily until 72 h of incubation, and the concentration vs. percentage of growth inhibition was plotted each day, in order to determine de inhibition concentration of 50 % of parasites (IC50).
Activity against intracellular amastigotes
Peritoneal macrophages were collected from BALB/c mice by washing with cold phosphate-buffered saline (PBS) supplemented with 3 % FBS. The animal protocol was approved by the Ethical Committee of the State University of Maringá (approval no. 029/2014). Sterile glass coverslips were placed in the wells of a 24-well microplate, and 5 × 105 cells/ml were added to each well in RPMI 1640 medium supplemented with 10 % FBS. The microplate was incubated for 2 h at 37 °C in a 5 % CO2-air mixture to adhere macrophages. The macrophage monolayer was infected with promastigote forms at a 7:1 (parasite: macrophage) ratio. After 4 h of interaction at 34 °C in a 5 % CO2-air mixture, the microplate was washed with RPMI 1640 medium to remove non-interiorized parasites. Afterward, the infected macrophages were treated with A3K2A3 at different concentrations (1, 2.5, 5.0, and 10 µM) and incubated for 48 h. The percentage of infected macrophages was evaluated after Giemsa staining by microscopically counting the number of amastigotes per macrophage [12].
Hemolytic activity
Another way to assess the safety of a drug is to measure its hemolytic activity. Defibrinated sheep blood was washed in glycosylated saline to remove any free hemoglobin from the defibrinization process. Red blood cells were inoculated in 96-well plates at 3 % in glycosylated saline with different concentrations of A3K2A3 (10-2000 µM). The plates were incubated for 3 h at 37 °C, and the supernatant was read at 550 nm. To calculate the percentage of hemolysis, 1 % Triton X-100 was used as a positive control [13].
Transmission electron microscopy
For the ultrastructural analysis, promastigote forms were treated for 72 h at 25 °C with concentrations of A3K2A3 that corresponded to the IC50 and IC90. After washing with PBS, the parasites were fixed in 2.5 % glutaraldehyde in 0.1 M sodium cacodylate buffer at 4 °C and post-fixed in a solution that contained 1 % osmium tetroxide, 0.8 % potassium ferrocyanide, and 5 mM calcium chloride. The parasites were dehydrated in an acetone series and embedded in Epon resin for 72 h at 60 °C. Ultrathin sections were stained with 5 % uranyl acetate and lead citrate and examined in a JEOL JEM 1400 transmission electron microscope.
Detection of cytoplasmic lipid bodies by Nile Red staining
Promastigote forms of L. amazonensis were treated with concentrations of A3K2A3 that corresponded to the IC50 and IC90 as described previously in the antiproliferative assay. The treated parasites were then harvested, washed twice in PBS, and directly stained with 10 μg/ml Nile Red (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature. Cytoplasmic lipid bodies in the parasites were detected with an epifluorescence microscope (Olympus BX51) equipped with a WG filter. Parasites were photographed using an Olympus UC30 camera. The samples were also analyzed in a fluorescence microplate reader (Victor X3, PerkinElmer, Finland) at an excitation wavelength of 515–560 nm and emission wavelength >590 nm.
Determination of mitochondrial transmembrane potential (ΔΨm)
Promastigotes (1 × 107 cells/ml) were treated with 3.4 and 9.3 µM A3K2A3 for 24 h at 37 °C, harvested, and washed with PBS. The parasites were incubated with 1 ml (5 mg/ml in ethanol) of Rh123, a fluorescent probe that accumulates in mitochondria, for 15 min, resuspended in 0.5 ml PBS, and incubated for an additional 30 min. The assay was conducted according to the manufacturer’s instructions. The parasites were analyzed using a BD FACSCalibur flow cytometer and CellQuest Pro software (Becton Dickinson; Franklin Lakes, NJ, USA). A total of 10,000 events were acquired in the region that corresponded to the parasites. CCCP (100 µM) was used as a positive control [14].
Measurement of reactive oxygen species
Promastigotes (1 × 107 cells/ml) were treated or untreated with 3.4 and 9.3 µM A3K2A3 for 24 h, centrifuged, washed, and resuspended in PBS, pH 7.4. Afterward, these parasites were loaded with 10 µM of the permeant probe H2DCFDA in the dark for 45 min [15]. Reactive oxygen species were measured as an increase in fluorescence that is caused by the conversion of the nonfluorescent dye to highly florescent 20,70-dichlorofluorescein. Parasites were analyzed using a BD ACCURI™ C6 flow cytometer with a total of 10,000 events acquired in the pre-determined region that corresponded to the parasites. Additionally, fluorescence was also measured in a microplate reader (Victor X3, PerkinElmer, Finland) at an excitation wavelength of 488 nm and emission wavelength of 530 nm. Hydrogen peroxide (20 mM) was used as a positive control.
Estimation of lipid peroxidation by diphenyl-1-pyrenylphosphine assay
Promastigote forms of L. amazonensis (1 × 106 cells/ml) were treated with 3.4 and 9.3 µM A3K2A3 for 24 h at 25 °C. The cells were washed twice, resuspended in PBS, and loaded with DPPP [16] for 15 min. During the labeling procedure, the cell suspension was kept in the dark. After incubation, fluorescence intensities of the samples were measured with a fluorescence microplate reader (Victor X3, PerkinElmer, Finland) at an excitation wavelength of 355 nm and emission wavelength of 460 nm. Hydrogen peroxide (200 µM) was used as a positive control.
Intracellular adenosine triphosphate determination
Promastigotes of L. amazonensis (1 × 106 cells/ml) were treated with 3.4 and 9.3 µM A3K2A3 for 24 h. After incubation at 25 °C, the promastigotes were centrifuged, washed, and resuspended in PBS. In white 96-well plates, equal volumes of CellTiter-Glo reagent and an aliquot of each sample were added, mixed, and incubated for 10 min. The luminescence intensity was quantified using a luminescence microplate reader (VICTOR X3, PerkinElmer). CCCP (100 µM) was used as a positive control.
Determination of cellular membrane integrity
Promastigotes (1 × 107 cells/ml) were treated with 3.4 and 9.3 µM A3K2A3 for 24 h at 32 °C, harvested, and washed with PBS. The parasites were incubated with 50 µl of 2 mg/ml PI for 5 min according to the manufacturer’s instructions. Immediately thereafter, the parasites were analyzed using a BD FACSCalibur flow cytometer equipped with CellQuest software. A total of 10,000 events were acquired in the region that corresponded to the parasites. Digitonin (40.0 μM) was used as a positive control [17].
Promastigotes and intracellular amastigotes scanning electron microscopy
Promastigotes (1 × 106 cells/ml) and peritoneal macrophages adhered to the surface of small glass coverslips previously infected with promastigotes for 4 h at 34 °C (as described in 2.4. item) were prepared. After the infection period plates were incubated at 34 °C for 24 h, and just then cells were treated with 3.4 and 9.3 µM A3K2A3 for 48 h and 24 h at the same temperature, in order to assess the morphological alterations in promastigotes and intracellular amastigotes, respectively. After treatment cells were fixed in 2.5 % glutaraldehyde in 0.1 M sodium cacodylate buffer for 1–3 h. Subsequently, they were dehydrated in increasing concentrations of ethanol and critical-point dried in CO2. For intracellular amastigotes observation, the slides were placed in an appropriate apparat and infected macrophages were fractured with adhesive tape. All samples were coated with gold, and observed in a Shimadzu SS-550 (Japan) scanning electron microscope [18, 19].
Determination of cell volume of parasites
Promastigotes (1 × 107 cells/ml) were treated with 3.4 and 9.3 µM A3K2A3 for 24 h at 25 °C, harvested, and washed with PBS. Subsequently, the parasites were analyzed using a BD FACSCalibur flow cytometer and CellQuest Pro software. Histograms were generated, and FSC-H represented the cell volume. A total of 10,000 events were acquired in the region that corresponded to the parasites. Actinomycin D (20.0 mM) was used as a positive control [18].
Determination of phosphatidylserine exposure
Promastigotes (1 × 106 cells/ml) were treated with 3.4 and 9.3 A3K2A3 for 24 h at 25 °C. Afterward, the parasites were washed and resuspended in 100 µl of binding buffer (140 mM NaCl, 5 mM CaCl2, and 10 mM HEPES-Na, pH 7.4), followed by the addition of 5 µl of the calcium-dependent phospholipid binding protein annexin-V FITC for 15 min at room temperature. Binding buffer (400 µl) was then added. Antimycin A (125.0 µM) was used as a positive control. Data acquisition was performed using a fluorescence microplate reader (Victor X3, PerkinElmer, Finland) at an excitation wavelength of 488 nm and emission wavelength of 520 nm. Additionally, cells were counterstained with PI (0.2 µg/mL) and images were acquired by an epifluorescence microscope (Olympus BX51) using an Olympus UC30 camera. Cells that were stained with annexin-V were considered apoptotic [20].
Caspase detection
Caspase-3/7 protease activity was measured using the EnzChek Caspase-3 Assay Kit (Molecular Probes). The assay was performed according to the manufacturer’s instructions with the following minor modifications. Briefly, 106 parasites were incubated with 3.4, 9.3, and 18.6 µM A3K2A3 for 24 h, washed, and lysed with Triton X-100 in 1 % PBS for 30 min, followed by the addition of kit reagents (i.e., reaction buffer, dithiothreitol, and 5 mM Z-DEVD-AMC substrate) and incubation for 30 min. When the reactions were completed, the increase in fluorescence, which is indicative of cleavage of the Z-DEVD-AMC substrate, was fluorometrically read at excitation and emission wavelengths of 342 and 441 nm, respectively. The reactions were performed with and without the Ac-DEVD-CHO inhibitor (1 mM). Camptothecin (20 μM) was used as a positive control.
Determination of DNA fragmentation
In addition to determining DNA fragmentation and nuclease activity, treated L. amazonensis promastigote cells were fixed with 4 % paraformaldehyde and incubated with 0.2 % Triton X-100 for 10 min for permeabilization and then layered with a TdT reaction mixture that contained BrdUTP for 2 h at 37 °C according to the manufacturer’s instructions with minor modifications (APO-BrdU TUNEL Assay Kit, with Alexa Fluor 488 Anti-BrdU). Cells that were linked with BrdU were detected using a green-fluorescent Alexa Fluor 488 dye-labeled anti-BrdU antibody for 30 min. Fluorescence was fluorometrically quantified in a microplate reader at an excitation wavelength of 485 nm and emission wavelength of 520 nm. Camptothecin (10 μM) was used as a positive control.
Statistical analysis
The data that are presented in the graphs are expressed as the mean ± standard deviation (SD) of at least three independent experiments. The data were analyzed using one- and two-way analysis of variance (ANOVA). Significant differences among means were identified using the Tukey or Bonferroni post hoc test. Values of p ≤ 0.05 were considered statistically significant. The statistical analyses were performed using Prism 5 software (GraphPad, San Diego, CA, USA).
Results
In a previous study, A3K2A3 (2k) presented interesting activity against promastigotes of L. amazonensis, with an IC50 of 3.4 ± 0.07 µM, demonstrating its effects against this protozoan parasite. In the present study, we further investigated this activity and mechanism of action.
To confirm the previously reported effects of A3K2A3 (Fig. 1) on the cellular proliferation of L. amazonensis, daily parasite counts were performed, and growth inhibition curves were constructed for each day of the experiment. A3K2A3 inhibited the growth of promastigotes at a similar IC50 (2.6 ± 0.3 µM) after 72 h of treatment. Interestingly, with 24 h incubation, A3K2A3 had an IC50 of 2.7 ± 0.7 µM, indicating potent activity even after only a few hours of incubation (Fig. 2). For intracellular amastigotes, A3K2A3 also presented potent activity, with an IC50 of 2.9 ± 0.5 µM.

Chemical structure of (1E,4E)-2-methyl-1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one (A3K2A3)

Effect of A3K2A3 on promastigote forms of Leishmania amazonensis. Promastigotes were treated with 1, 5, 10, 50, and 100 μM A3K2A3 and counted daily in a Neubauer chamber until 72 h of incubation. The graph of concentration vs. percentage growth inhibition was plotted each day, and inhibitory concentrations were determined. Symbols represent the mean ± standard deviation of at least two independent experiments, which were performed in duplicate
The cytotoxicity effects of A3K2A3 on J774A1 macrophages were tested as described previously (CC50 = 43.0 ± 4.24 µM). To corroborate its non-cytotoxic profile, the hemolytic activity of this substance was assayed. A3K2A3 up to a concentration of 2000 µM did not present hemolytic activity. The percentage of hemolysis was less than 6 % (data not shown).
To further investigate and identify the organelles that might be potential targets of A3K2A3 in promastigotes, transmission electron microscopy (TEM) was performed as described previously. A3K2A3 caused significant ultrastructural alterations (Fig. 3), including the disorganization of Golgi complex (disintegration of the slacks of cisternae and formation of clusters of tubules and vesicles dispersed in the cytoplasm) and nuclei and accumulation of cytoplasmic lipid bodies. A3K2A23 also caused severe damage in parasite mitochondria, reflected by extensive swelling and disorganization of the inner mitochondrial membrane, intense cytoplasmic vacuolization, and the presence of concentric membrane structures inside the organelle. These changes were observed in greater proportions when the cells were treated with the IC90 of A3K2A3. Control parasites had an apparently normal ultrastructure (Fig. 3a).

Ultrathin sections of Leishmania amazonensis promastigotes without treatment that presented a normal ultrastructure (a) and promastigotes that were treated with A3K2A3 at concentrations that corresponded to the IC50 (b–d) and IC90 (e–f). White arrows indicate swollen mitochondria. Black arrows represent lipid-storage bodies. Black arrowheads indicate the presence of vesicles inside the flagellar pocket. White arrowheads indicate DNA disorganization in nuclei. The star indicates the presence of concentric membranous structures. The asterisk indicates Golgi complex disorganization. n nucleus, m mitochondrion, k kinetoplast, f flagellum, fp flagellar pocket. Scale bar 0.5 μm
To confirm the accumulation of lipid bodies in treated promastigotes that was observed by TEM, we evaluated the existence of this structure by Nile Red staining, which stains neutral lipids. Fluorescence microscopy revealed an increase in the presence of lipid bodies in promastigotes after treatment with the IC50 and IC90 of A3K2A3 for 24 h (Fig. 4) compared with non-treated cells. This finding was confirmed by fluorimetry, with a 2.7-fold increase in fluorescence in cells that were treated with the IC90 of A3K2A3 compared with control cells (i.e., without treatment).

a Differential interference contrast microscopy (DIC) and fluorescence microscopy with Nile Red staining of Leishmania amazonensis promastigotes without treatment (a, b) and treated with A3K2A3 at concentrations of 3.4 µM (c, d) and 9.3 µM (e, f) for 24 h. In treated promastigotes, images suggest accumulation of lipid-storage bodies in the cytoplasm (b, d, f). Scale bar 10 μm. b Accumulation of lipid-storage bodies in promastigote forms of L. amazonensis treated with A3K2A3 at the IC50 and IC90 for 24 h using the fluorescent probe Nile Red. The data are expressed as the mean fluorescence (in arbitrary units) ± SD of at least three independent experiments. *p ≤ 0.05, significant difference compared with the negative control group (i.e., untreated parasites)
Transmission electron microscopy also revealed a pronounced effect of A3K2A3 on L. amazonensis mitochondria. Therefore, the ΔΨm was evaluated by flow cytometry in treated parasites. Histograms showed a marked decrease in total Rh123 fluorescence intensity in promastigotes after 24 h treatment at all of the concentrations tested compared with the control group, indicating mitochondrial depolarization (Fig. 5). This loss of ΔΨm was higher at the IC90 (40.11 %) than at the IC50 (27.00 %). The positive control, CCCP, induced a 72.96 % decrease in ΔΨm.

Mitochondrial membrane potential assay in promastigote forms of L. amazonensis treated with A3K2A3 at concentrations of 3.4 µM (b) and 9.3 µM (c) for 24 h and stained with Rh123, which accumulates in mitochondria. a Positive control (CCCP). The gray area corresponds to the negative control group (i.e., untreated parasites). Typical histograms of at least three independent experiments are shown
Given the ΔΨm results, to investigate possible oxidative damage that is caused by A3K2A3 in promastigotes, the production of total ROS was investigated using the fluorescent probe H2DCFDA, which primarily detects H2O2 and hydroxyl radicals [21]. A3K2A3 increased total ROS production at all of the concentrations tested compared with the control group (Fig. 6a), with 12.00 and 45.87 % increases at the IC50 and IC90 of A3K2A3, respectively. The positive control (H2O2) increased the fluorescence intensity by 89.74 %. These results were also confirmed by flow cytometry analysis (Fig. 6b), by which it was possible to identify that at a populational level, 93.8 and 98.9 % of the analyzed cells presents some evidence of oxidative stress after IC50 and IC90 treatment, respectively, against only 3.98 % of positivity at untreated populations.

a Total ROS production in promastigote forms of L. amazonensis treated with A3K2A3 at concentrations of 3.4 and 9.3 µM for 24 h using the fluorescent probe H2DCFDA. The data are expressed as the mean fluorescence (in arbitrary units) ± SD of at least three independent experiments. Hydrogen peroxide (20 mM) was used as a positive control (PC). *p ≤ 0.05, significant difference compared with the negative control group (i.e., untreated parasites). b Flow Cytometry histograms showing the total ROS production in promastigote forms of L. amazonensis treated with A3K2A3 at concentrations of 3.4 and 9.3 µM for 24 h using the fluorescent probe H2DCFDA. Control untreated cells: light gray filled area, 3.4 µM treated cells: dashed-line surrounded gray filled area; 9.3 µM solid-line surrounded gray filled area
Our previous experiment showed that A3K2A3 caused mitochondrial damage and oxidative imbalance in L. amazonensis, leading to enhanced ROS production. Thus, we expected that A3K2A3 may trigger molecular and structural alterations in the parasite through oxidative reactions. Lipid peroxidation was assessed by measuring total fluorescent lipid peroxidation products in leishmanial cells after treatment with A3K2A3 using DPPP, which stoichiometrically reacts with lipid hydroperoxides to yield the fluorescent product DPPP oxide. A3K2A3 treatment significantly increased lipid peroxides after 24 h of treatment at both concentrations tested (Fig. 7).

Formation of hydroperoxides in promastigotes treated with the IC50 and IC90 of A3K2A3. DPPP-labeled cells were evaluated by measuring the fluorescence intensity after 24 h of treatment. Hydrogen peroxide (200 µM) was used as a positive control (PC). The data are expressed as the mean ± SD of at least three independent experiments in triplicate. *p ≤ 0.05, significant difference compared with the negative control group (i.e., untreated parasites)
Because the disruption of mitochondrion function translates into a reduction of ATP generation, the levels of ATP were measured after exposure to A3K2A3, using CellTiter-Glo reagent, which is detected by luminescence. A gradual decrease in ATP levels was observed as the A3K2A3 concentration increased after 24 h of treatment (Fig. 8).

Determination of the level of intracellular ATP using CellTiter-Glo reagent after treatment with A3K2A3 at concentrations of 3.4 and 9.3 µM for 24 h. CCCP (100 µM) was used as a positive control. The data are expressed as the mean ± SD of three independent experiments. *p ≤ 0.05, significant difference compared with the negative control group (i.e., untreated parasites)
To determine the possible mechanism of cell death that is triggered by A3K2A3, alterations in cell membrane integrity were evaluated using PI staining. A3K2A3-treated, PI-labeled promastigotes did not exhibit significant permeabilization of the plasma membrane compared with untreated parasites. A3K2A3 at concentrations of 3.4 and 9.3 μM showed 4.75 and 6.16 %, respectively, of the cells PI-positive. The negative control showed a PI fluorescence intensity of 4.15 %, and digitonine (i.e., the positive control) showed a PI fluorescence intensity of 57.12 % (data not shown).
Scanning electron microscopy of promastigotes was used to investigate the mechanism of cell death by identifying morphological alterations that were induced by A3K2A3 in the parasite. The photomicrographs showed that untreated protozoa exhibited typical characteristics, with an elongated shape and terminal flagellum. In contrast, A3K2A3 dose-dependently altered the size and shape of the parasites, including a rounding and overall reduction of the cell body (Fig. 9). Alterations were also observed in intracellular amastigote forms treated with both concentrations of A3K2A3 in scanning electron mycroscopy, damage and disruption to the amastigotes cell membrane and a reduction in amastigotes number per cell (Fig. 10).

Scanning electron microscopy images of promastigote forms of L. amazonensis incubated in the absence (A) or presence of A3K2A3 at concentrations of 3.4 µM (B) and 9.3 µM (C) for 72 h. Scale bar 20 µm (a–c) and 5 µm (a–c)

Scanning electron microscopy images of intracellular amastigotes forms of L. amazonensis incubated in the absence (a and d) or presence of A3K2A3 at concentrations of 3.4 µM (b and e) and 9.3 µM (c and f) for 72 h. Scale bar 10 µm (a–f)
Additional experiments were performed to evaluate cell shrinkage, a hallmark of apoptotic death. As shown in Fig. 11 decrease in cell volume was observed in cells that were treated with 3.4 and 9.3 μM A3K2A3 after 24 h and analyzed by flow cytometry, with reductions of 22.26 and 27.66 %, respectively. The positive control, actinomycin D, decreased cell volume by 47.20 %.

Cell volume in promastigote forms of L. amazonensis treated with A3K2A3 at concentrations of 3.4 µM (b) and 9.3 µM (c) for 24 h. Actinomycin D (20.0 mM) was used as a positive control (a). FSC-H was considered a function of cell size. The gray area corresponds to the negative control group (i.e., untreated parasites). Typical histograms of at least three independent experiments are shown
To determine whether the mechanism of cell death that is triggered by A3K2A3 involves apoptosis, we evaluated the externalization of phosphatidylserine, an apoptotic marker that is present in the outer leaflet of plasmalemma [22], in promastigotes that were treated with A3K2A3 for 24 h and stained with FITC-conjugated annexin-V. As shown in Fig. 12, A3K2A3 increased the annexin-V fluorescence intensity more than 8 times at both the IC50 and IC90 compared with the negative control group, indicating phosphatidylserine exposure. Moreover, fluorescence microscopy revealed parasites labeled by annexin-V-FITC after treatment with both concentrations of A3K2A3 (3.4 and 9.3 µM). At the higher concentration (9.3 µM) of drug tested, it is also possible to identify some PI staining at the nucleus, indicating some compromise of cell membrane integrity, probably due to a later apoptotic process.

a Phosphatidylserine exposure in promastigote forms of L. amazonensis treated with A3K2A3 at concentrations of 3.4 and 9.3 µM for 24 h using annexin V-FITC. Negative control (NC; i.e., untreated parasites). Antimycin A (125.0 µM) was used as a positive control (PC). A typical graph of at least three independent experiments is shown. b Differential interference contrast microscopy (DIC) in a.1, b.1, c.1 and fluorescence microscopy of Leishmania amazonensis promastigotes after Annexin-V-FITC (a.2; b.2 and c.2) and Propidium Iodide staining (a.3; b.3 and c.3). Control parasites were kept untreated (a) and the treated were incubated with A3K2A3 at concentrations of 3.4 µM (b) and 9.3 µM (c) for 24 h. Bars 10 µm
A major feature of apoptotic death is the involvement of cysteine aspartate proteases (caspases) that mediate events downstream of the mitochondria, most notably the executioners caspase 3 and 7 [23, 24]. To further substantiate the existence of these proteases in L. amazonensis, a fluorometric assay using EnzChek Caspase-3 Assay Kit was performed. The substrate DEVD-AFC was added to this extract, and the release of AFC was used as a measure of mature caspase-3-like protease activity in A3K2A3-treated cells. A significant increase in these proteases was observed after treatment with A3K2A3 for 24 h compared with untreated cells. This effect was more pronounced in promastigotes that were treated with the IC50 (Fig. 13).

Activity of caspase-3/7-like proteases in promastigotes that were treated with A3K2A at concentrations of 33.4 and 9.3 µM for 24 h using the fluorescent EnzCheck Caspase-3 Assay kit. Camptothecin (20 μM) was used as a positive control (PC). The specificity of activity was tested by preincubating the cells with a caspase-3-specific inhibitor prior to the analysis of caspase-3/7 activity. The data are expressed as the mean ± SD from three independent experiments. *p ≤ 0.05, significant difference compared with the negative control (NC) group (i.e., untreated parasites)
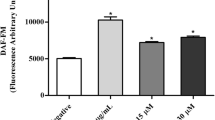
The TUNEL assay detects apoptosis at the single-cell level and thus permits a better evaluation of the apoptotic cell fraction [22]. As shown in Fig. 14, promastigotes that were treated with different concentrations of A3K2A3 and subjected to the TUNEL assay exhibited a significant increase in fluorescence intensity at both concentrations tested. The IC50 (3.4 µM) promoted a less than twofold increase in fluorescence compared with untreated parasites (i.e., the negative control), thus indicating DNA fragmentation. Camptothecin, the positive control, caused increase of more than ninefold in fluorescence intensity.

DNA fragmentation in promastigotes treated with A3K2A3 at the IC50 and IC90 for 24 h using the TUNEL assay. Camptothecin (20 μM) was used as a positive control (PC). The data are expressed as the mean ± SD from three independent experiments. *p ≤ 0.05, significant difference compared with the negative control (NC) group (i.e., untreated parasites)
Discussion
Drug therapy for leishmaniasis has not significantly advanced since the beginning of the twentieth century, and adequate treatment remains a problem. There continues to be an urgent need to develop more rational and effective therapeutic drugs [25].
Dibenzylideneacetone derivatives are a class of substances that have an acyclic dienone that is attached to aryl groups in both β-positions, with a broad spectrum of biological activity, especially antitumor activity [11, 26]. A3K2A3 has been previously reported to have interesting anti-trypanosomal and anti-leishmanial activity, and it has been shown to be more selective to parasites than to mammalian and macrophage cells [5]. A better understanding of the mechanism of cell death would be helpful for developing therapeutic interventions against parasites. In the present study, we evaluated the anti-leishmanial activity of A3K2A3 to delineate its putative mechanism of action.
This compound at low micromolar concentrations presented significant activity against promastigote and intracellular amastigote forms of L. amazonensis and did not affect the viability of red blood cells. These results are very interesting because amastigote forms of Leishmania are responsible for clinical manifestations in the vertebrate host. These amastigotes are the major target of chemotherapy for leishmaniasis [27].
Unicellular kinetoplastid parasites have special organelles that are involved in essential metabolic pathways, with steps that differ from their mammalian counterparts, thus making them attractive targets for new chemotherapeutic agents. Ultrastructural studies can be very helpful for achieving this goal [4, 28]. To obtain information about the mechanism of action of A3K2A3 against L. amazonensis, promastigotes were analyzed by TEM. Photomicrographs of A3K2A3-treated cells exhibited important ultrastructural alterations, such as lipid body accumulation and Golgi complex disorganization. Extensive degradation of the Golgi complex and endoplasmic reticulum that is caused by the accumulation of intracellular lipid bodies in the cytoplasm, indicated by fluorescence microscopy and fluorimetry (Nile Red staining), may indicate alterations in phospholipids and sterol content and may also be related to exocytic activity and autophagy. Some studies have reported that this exocytic activity might occur as a result of the secretion of abnormal lipids into this region, which accumulate as a consequence of drug action or might indicate a process of exacerbated protein production by the cells [29–31].
Furthermore, ultrastructural analyses of the treated parasites revealed that the mitochondrion was one of the most affected organelles, which could also be related to possible alterations in lipid composition. Therefore, we focused on investigating mitochondrial alterations and their consequences, especially with regard to parasite death. A3K2A3 may exert its anti-leishmanial activity by affecting mitochondrial function in the parasite, demonstrated by TEM, a decrease in ΔΨm, and an increase in mitochondrial ROS production.
Electrons move through the mitochondrial respiratory chain during oxidative phosphorylation, and a proton gradient is established across the inner mitochondrial membrane as an energy source for ATP. A decrease in Rh123 fluorescence intensity suggests an increase in proton permeability across the inner mitochondrial membrane, which can decrease ATP synthesis and result in parasite death [13, 32, 33], especially because the parasite depends mainly on oxidative phosphorylation for ATP production [34]. The depletion of ATP levels was observed after treating the promastigotes with A3K2A3. For organisms with a single mitochondrion, such as Leishmania, there is no possibility of compensating for injured mitochondria. Therefore, survival depends on the proper functioning of a single organelle [35].
Our results are consistent with a previous study that reported that ATP levels gradually decreased after the loss of ΔΨm during treatment with H2O2 [36]. Adenosine triphosphate is a key molecule for chromatin condensation, nuclear fragmentation and regulation, and the maintenance of ion homeostasis during apoptosis. Therefore, we can assume that ATP levels were generated prior to the loss of ΔΨm, and ATP that was supplied by glycolysis was sufficient to perform these cellular activities and propagate programmed cell death in leishmanial cells [37].
We also found a significant increase in ROS production after A3K2A3 treatment in promastigotes. Mitochondrial ROS formation is essential for various signaling processes and strongly involved in the degenerative process through damage to macromolecules, mainly proteins, lipids, and DNA [38–40]. Promastigotes that were treated with A3K2A3 exhibited an increase in the ratio of lipid peroxidation. Earlier studies found that Camptothecin induced the formation of ROS inside leishmanial cells and also increased the level of lipid peroxidation [22].
In multicellular and unicellular organisms, mitochondria serve as an important cellular source of ROS, which are critical for the induction of apoptosis. The production of ROS during the early phase of apoptosis usually follows an imbalance in cellular redox homeostasis [41]. Indeed, previous studies have shown that antimony exerts its anti-leishmanial effect by generating ROS and depleting thiols within both parasites and macrophages [42–44].
A3K2A3 treatment also induced the externalization of phosphatidylserine, which was visualized by annexin-V staining. The translocation of phosphatidylserine from inside the plasma membrane to the outer layer is a common alteration that occurs during programmed cell death [45–47]. In addition to biochemical alterations, apoptosis also causes morphological alterations [48]. Based on this, we evaluated cell shrinkage (i.e., a hallmark of apoptotic death). Our SEM observations revealed swelling and overall rounding of A3K2A3-treated cells, which was also confirmed by flow cytometry. Membrane integrity was also evaluated, but it was not significantly altered by A3K2A3 treatment, reflected by a low ratio of PI staining. Therefore, we could exclude the possibility that necrosis was the main pathway of cell death.
The apoptotic process is associated with signaling cascades that involve mitochondria (intrinsic pathway) or death receptors (extrinsic pathway) [49]. Stimulation of the intrinsic mitochondrial apoptotic pathway by ROS and mitochondrial DNA damage promotes outer membrane permeabilization and the mitochondrion-to-cytosol translocation of cytochrome c, AIF (apoptosis-inducing factor), and Smac/Diablo, which trigger caspase-dependent or caspase-independent cytosolic signaling events [50].
Immediately after the loss of ΔΨm, protons are released into the cytosol from mitochondria, and this contributes to intracellular acidification in leishmanial cells, similar to mammalian cells [51]. Changes in pH modulate the apoptotic responsiveness of the cell and amplify the apoptotic program by regulating the activity of caspase-like proteases [35].
It is well known that caspases (i.e., a family of cysteine proteases) are involved in orchestrating apoptosis in metazoan. Caspase homologues known as metacaspases (MCAs) in Trypanosoma and Leishmania have been reported to play distinct roles in Programmed Cell Death [52, 53]. The role of MCAs is subject to debate: roles in cell cycle control, in cell death or even in cell survival have been suggested. It was shown, using a Leishmania major MCA-deficient strain, that L. major MCA (LmjMCA) not only had a role similar to caspases in cell death but also in autophagy and this through different domains [54].
In caspase-independent programmed cell death, the increase in intracellular calcium increases mitochondrial calcium and causes further mitochondrial membrane depolarization, the generation of ROS, and the activation of endonucleases [55]. In the present study, we observed an increase in caspase-3/-7-like protease levels in cells following A3K2A3 exposure. Pretreatment with the caspase-3 inhibitor Ac-DEVD-CHO diminished cellular caspase-3/-7-like protease activity. Oligonucleosomal DNA fragmentation represents a late event of this programmed cell death pathway, leading to the passive release of DNA in the cytoplasm of apoptotic cells [56]. This also occurred in promastigotes that were treated with A3K2A3. Thus, DNA nicking in promastigotes strongly suggests that the anti-leishmanial effect is mediated by apoptosis-like cell death.
Conclusion
In conclusion, the present study found that the anti-leishmanial effect of A3K2A3 appears to be a consequence of the induction of apoptosis-like cell death that is caused by an increase in ROS levels inside cells. This increase is likely responsible for the collapse of mitochondrial membrane potential and depletion of ATP levels. A3K2A3 treatment also triggered apoptosis-like changes in L. amazonensis promastigotes, characterized by phosphatidylserine externalization, cell shrinkage, caspase activation, the induction of DNA fragmentation, and an increase in the number of cytoplasmic lipid droplets. This work provides some clues regarding the pathway by which Leishmania undergoes apoptosis and may pave the way for designing chemotherapeutic compounds against leishmaniasis.
References
Rajasekaran R, Chen Yi-Ping P (2015) Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discov Today 20(8):958–968. doi:10.1016/j.drudis.2015.04.006
Duszenko M, Figarella K, Macleod ET, Welburn SC (2006) Death of a trypanosome: a selfish altruism. Trends Parasitol 22(11):536–542. doi:10.1016/j.pt.2006.08.010
Arambage SC, Grant KM, Pardo I, Ranford-Cartwright L, Hurd H. (2009) Malaria ookinetes exhibit multiple markers for apoptosis-like programmed cell death in vitro. Parasit Vector 2(1):32. doi:10.1186/1756-3305-2-32
Kathuria M, Bhattacharjee A, Sashidhara KV, Singh SP, Mitra K (2014) Induction of mitochondrial dysfunction and oxidative stress in Leishmania donovani by orally active clerodane diterpene. Antimicrob Agents Chemother 58(10):5916–5928. doi:10.1128/AAC.02459-14
Din ZU, Fill TP, de Assis FF, Lazarin-Bidóia D, Kaplum V, Garcia FP, Nakamura CV, Oliveira KT, Rodrigues-Filho E (2014) Unsymmetrical 1, 5-diaryl-3-oxo-1, 4-pentadienyls and their evaluation as antiparasitic agents. Bioorg Med Chem 22(3):1121–1127. doi:10.1016/j.bmc.2013.12.020
Shi M, Cai Q, Yao L, Mao Y, Ming Y, Ouyang G (2006) Antiproliferation and apoptosis induced by curcumin in human ovarian cancer cells. Cell Biol Int 30(3):221–226. doi:10.1016/j.cellbi.2005.10.024
Aher RB, Wanare G, Kawathekar N, Kumar RR, Kaushik NK, Sahal D, Chauhan VS (2011) Dibenzylideneacetone analogues as novel Plasmodium falciparum inhibitors. Bioorg Med Chem Lett 21(10):3034–3036. doi:10.1016/j.bmcl.2011.03.037
Prakobwong S, Gupta SC, Kim JH, Sung B, Pinlaor P, Hiraku Y, Wongkham S, Sripa B, Pinlaor S, Aggarwal BB (2011) Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis 32(9):1372–1380. doi:10.1093/carcin/bgr032
Prakobwong S, Khoontawad J, Yongvanit P, Pairojkul C, Hiraku Y, Sithithaworn P, Pinlaor P, Aggarwa BB, Pinlaor S (2011) Curcumin decreases cholangiocarcinogenesis in hamsters by suppressing inflammation-mediated molecular events related to multistep carcinogenesis. Int J Cancer 129(1):88–100. doi:10.1002/ijc.25656
Yu HJ, Shin JA, Nam JS, Kang BS, Cho SD (2013) Apoptotic effect of dibenzylideneacetone on oral cancer cells via modulation of specificity protein 1 and Bax. Oral Dis 19(8):767–774. doi:10.1111/odi.12062
Prasad S, Yadav VR, Ravindran J, Aggarwal BB (2011) ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res 71(2):538–549. doi:10.1158/0008-5472
Britta EA, Scariot DB, Falzirolli H, Ueda-Nakamura T, Silva CC, Dias Filho BP, Borsali R, Nakamura CV (2014) Cell death and ultrastructural alterations in Leishmania amazonensis caused by new compound 4-Nitrobenzaldehyde thiosemicarbazone derived from S-limonene. BMC Microbiol 14(1):236. doi:10.1186/s12866-014-0236-0
Rodrigues JH da S, Ueda-Nakamura T, Corrêa AG, Sangi DP, Nakamura CV (2014) A quinoxaline derivative as a potent chemotherapeutic agent, alone or in combination with benznidazole, against Trypanosoma cruzi. PLoS One 9(1):e85706. doi:10.1371/journal.pone.0085706
Menna-Barreto RF, Goncalves RL, Costa EM, Silva RS, Pinto AV, Oliveira MF, de Castro SL (2009) The effects on Trypanosoma cruzi of novel synthetic naphthoquinones are mediated by mitochondrial dysfunction. Free Radical Bio Med 47(5):644–653. doi:10.1016/j.freeradbiomed.2009.06.004
Shukla AK, Patra S, Dubey VK (2012) Iridoid glucosides from Nyctanthes arbortristis result in increased reactive oxygen species and cellular redox homeostasis imbalance in Leishmania parasite. Eur J Med Chem 54:49–58. doi:10.1016/j.ejmech.2012.04.034
Takahashi M, Shibata M, Niki E (2001) Estimation of lipid peroxidation of live cells using a fluorescent probe, diphenyl-1-pyrenylphosphine. Free Radical Bio Med 31(2):164–174. doi:10.1016/S0891-5849(01)00575-5
Lazarin-Bidóia D, Desoti VC, Ueda-Nakamura T, Dias Filho BP, Nakamura CV, Silva SO (2013) Further evidence of the trypanocidal action of eupomatenoid-5: confirmation of involvement of reactive oxygen species and mitochondria owing to a reduction in trypanothione reductase activity. Free Radical Bio Med 60:17–28. doi:10.1016/j.freeradbiomed.2013.01.008
Britta EA, Silva AB, Ueda-Nakamura T, Dias-Filho BP, Silva CC, Sernaglia RL, Nakamura CV (2012) Benzaldehyde thiosemicarbazone derived from limonene complexed with copper induced mitochondrial dysfunction in Leishmania amazonensis. PLOS One 7(8):1–12. doi:10.1371/journal.pone.0041440
Stefanello TF, Panice MR, Ueda-Nakamura T, Sarragiotto MH, Auzély-Velty R, Nakamura CV (2014) N-butyl-[1-(4-Methoxy)phenyl-9H–carboline]–3-carboxamide prevents cytokinesis in Leishmania amazonensis. Antimicrob Agents Chemother 58(12):7112–7120. doi:10.1128/AAC.03340-14
Jimenez V, Paredes R, Sosa MA, Galanti N (2008) Natural programmed cell death in T. cruzi epimastigotes maintained in axenic cultures. J Cell Biochem 105(3):688–698. doi:10.1002/jcb.21864
Kaur J, Singh BK, Tripathi RP, Singh P, Singh N (2009) Leishmania donovani: a glycosyl dihydropyridine analogue induces apoptosis like cell death via targeting pteridine reductase 1 in promastigotes. Exp Parasitol 123:258–264. doi:10.1016/j.exppara.2009.07.009
Sen N, Das BB, Ganguly A, Mukherjee T, Tripathi G, Bandyopadhyay S, Rakshit S, Sen T, Majumder HK (2004) Camptothecin induced mitochondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ 11(8):924–936. doi:10.1038/sj.cdd.4401435
Arnoult D, Akarid K, Grodet A, Petit PX, Estaquier J, Ameisen JC (2002) On the evolution of programmed cell death: apoptosis of the unicellular eukaryote Leishmania major involves cysteine proteinase activation and mitochondrion permeabilization. Cell Death Differ 9:65–81. doi:10.1038/sj.cdd.4400951
Kulkarni MM, McMaster WR, Kamysz W, McGwire BS (2009) Antimicrobial peptide-induced apoptotic death of Leishmania results from calcium-dependent, caspase-independent mitochondrial toxicity. J Biol Chem 284(23):15496–15504. doi:10.1074/jbc.M809079200
Goto H, Lindoso, JAL (2012) Cutaneous and mucocutaneous leishmaniasis. Infect Dis Clin N Am 26(2):293–307. doi:10.1016/j.idc.2012.03.001
Bhandarkar SS, Bromberg J, Carrillo C, Selvakumar P, Sharma RK, Perry BN, Govindarajan B, Fried L, Sohn A, Reddy K, Arbiser JL (2008) Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth In vitro and In vivo. Clin Cancer Res 14(18):5743–5748. doi:10.1158/1078-0432.CCR-08-0405
Vendrametto MC, Santos AO, Nakamura CV, Dias Filho BP, Cortez DAG, Ueda-Nakamura T (2010) Evaluation of antileishmanial activity of eupomatenoid-5, a compound isolated from leaves of Piper regnellii var. pallescens. Parasitol Int 59(2):154–158. doi:10.1016/j.parint.2009.12.009
Fernandes-Rodrigues JC, Souza WD (2008) Ultrastructural alterations in organelles of parasitic protozoa induced by different classes of metabolic inhibitors. Curr Pharm Des 14(9):925–938. doi:10.2174/138161208784041033
Kafetzis DA, Velissariou IM, Stabouli S, Mavrikou M, Delis D, Liapi G (2005) Treatment of paediatric visceral leishmaniasis: amphotericin B or pentavalent antimony compounds? Int J Antimicrob Agents 25:26–30. doi:10.1016/j.ijantimicag.2004.09.01131
Tiuman TS, Ueda-Nakamura T, Cortez DAG, Dias-Filho BP, Morgado-Díaz JA, Souza W, Nakamura CV (2005) Antileishmanial activity of parthenolide, a sesquiterpene lactona isolated from Tanacetum parthenium. Antimicrob Agents Chemother 49:176–182. doi:10.1128/AAC.49.11.176-182.2005
Ueda-Nakamura T, Mendonça-Filho RR, Morgado-Díaz JA, Maza PK, Dias-Filho BP, Cortez DAG et al (2006) Antileishmanial activity of Eugenol-rich essential oil from Ocimum gratissimum. Parasitol Int 55(2):99–105. doi:10.1016/j.parint.2005.10.006
Inácio JDF, Canto-Cavalheiro MM, Menna-Barreto RFS, Almeida-Amaral EE (2012) Mitochondrial damage contribute to epigallocatechin-3-gallate induced death in Leishmania amazonensis. Exp Parasitol 132(2):151–155. doi:10.1016/j.exppara.2012.06.008
Medina JM, Rodrigues JCF, De Souza W, Atella GC, Barrabim H (2012) Tomatidine promotes the inhibition of 24-alkylated sterol biosynthesis and mitochondrial dysfunction in Leishmania amazonensis promastigotes. Parasitology 139(10):1253–1265. doi:10.1017/S0031182012000522
Bringaud F, Rivière L, Coustou V (2006) Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasite 149:1–9. doi:10.1016/j.molbiopara.2006.03.017
Sen N, Das BB, Ganguly A, Mukherjee T, Bandyopadhyay S, Majumder HK (2004) Camptothecin-induced imbalance in intracellular cation homeostasis regulates programmed cell death in unicellular hemoflagellate Leishmania donovani. J Biol Chem 279(50):52366–52375. doi:10.1074/jbc.M406705200
Mukherjee SB, Das M, Sudhandiran G, Shaha C (2002) Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem 277(27):24717–24727. doi:10.1074/jbc.M201961200
Chowdhury S, Mukherjee T, Chowdhury SR, Sengupta S, Mukhopadhyay S, Jaisankar P, Majumder HK (2014) Disuccinyl betulin triggers metacaspase-dependent endonuclease G-mediated cell death in unicellular protozoan parasite Leishmania donovani. Antimicrob Agents Chemother 58(4):2186–2201. doi:10.1128/AAC.02193-13
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE (2009) Mitochondria and reactive oxygen species. Free Radical Bio Med 47(4):333–343. doi:10.1016/j.freeradbiomed.2009.05.004
Smirlis D, Duszenko M, Ruiz AJ, Scoulica E, Bastien P, Fasel N, Soteriadou K (2010) Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasite Vector 3:107. doi:10.1186/1756-3305-3-107
Fonseca-Silva F, Inacio JD, Canto-Cavalheiro MM, Almeida-Amaral EE (2011) Reactive oxygen species production and mitochondrial dysfunction contribute to quercetin induced death in Leishmania amazonensis. PLoS One 6(2):e14666. doi:10.1371/journal.pone.0014666
Islamuddin M, Chouhan G, Tyagi M, Abdin MZ, Sahal D, Afrin F (2014) Leishmanicidal activities of Artemisia annua leaf essential oil against visceral leishmaniasis. Front Microbiol 5:626. doi:10.3389/fmicb.2014.00626
Mehta A, Shaha C (2006) Mechanism of metalloid-induced death in Leishmania spp.: role of iron, reactive oxygen species. Ca2+ and glutathione. Free Radical Bio Med 40:1857–1868. doi:10.1016/j.freeradbiomed.2006.01.024
Mandal G, Wyllie S, Singh N, Sundar S, Fairlamb AH, Chatterjee M (2007) Increased levels of thiols protect antimony unresponsive Leishmania donovani field isolates against reactive oxygen species generated by trivalent antimony. Parasitology 134(12):1679–1687. doi:10.1017/S0031182007003150
Sarkar A, Mandal G, Singh N, Sundar S, Chatterjee M (2009) Flow cytometric determination of intracellular non-protein thiols in Leishmania promastigotes using 5-chloromethyl fluorescein diacetate. Exp Parasitol 122(4):299–305. doi:10.1016/j.exppara.2009.04.012
Koonin EV, Aravind L (2002) Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ 9(4):394–404. doi:10.1038/sj/cdd/4400991
Mehta A, Shaha C (2004) Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition complex II inhibition results in increased pentamidine citotoxicity. J Biol Chem 279(12):11798–11813. doi:10.1074/jbc.M309341200
Dutta A, Ghoshal A, Mandal D, Mondal NB, Banerjee S, Sahu NP, Mandal C (2007) Racemoside A, an anti-leishmanial, water-soluble, natural steroidal saponin, induces programmed cell death in Leishmania donovani. J Med Microbiol 56(9):1196–1204. doi:10.1099/jmm.0.47114-0
Kroemer G, Galluzzi L, Vandenabeele P et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1):3–11. doi:10.1038/cdd.2008.150
Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2(4):277–288. doi:10.1038/nrc776
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM (2007) Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9(1):49–89. doi:10.1089/ars.2007.9.49
Facompre M, Goossens JF, Bailly C (2001) Apoptotic response of HL-60 human leukemia cells to the antitumor drug NB-506, a glycosylated indolocarbazole inhibitor of topoisomerase 1. Biochem Pharmacol 61(3):299–310. doi:10.1016/S0006-2952(00)00553-0
Kosec G, Alvarez VE, Agüero F, Sánchez D, Dolinar M, Turk B, Turk V, Cazzulo JJ (2006) Metacaspases of Trypanosoma cruzi: possible candidates for programmed cell death mediators. Mol Biochem Parasitol 145:18–28. doi:10.1016/j.molbiopara.2005.09.001
Lee N, Gannavaram S, Selvapandiyan A, Debrabant A (2007) Characterization of metacaspases with trypsin-like activity and their putative role in programmed cell death in the protozoan parasite Leishmania. Eukaryot Cell 6:1745–1757. doi:10.1128/EC.00123-07
Casanova M, Gonzalez IJ, Sprissler C, Zalila H, Dacher M, Basmaciyan L, Späth GF, Azas N, Fasel N (2015) Implication of different domains of the Leishmania major metacaspase in cell death and autophagy. Cell Death Dis 6(10):e1933. doi:10.1038/cddis.2015.288
Martinvalet D, Zhu P, Lieberman J (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22(3):355–370. doi:10.1016/j.immuni.2005.02.004
Zangger H, Mottram JC, Fasel N (2002) Cell death in Leishmania induced by stress and differentiation: programmed cell death or necrosis? Cell Death Differ 9(10):1126–1139. doi:10.1038/sj.cdd.4401071
Acknowledgments
This work was supported through grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq, Capacitação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES, Financiadora de Estudos e Projetos—FINEP, Programa de Pós-Graduação em Ciências Biológicas da Universidade Estadual de Maringá and Complexo de Centrais de Apoio a Pesquisa COMCAP—UEM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All of the animal procedures were performed in accordance with guidelines established by the Universidade Estadual de Maringá ethical committee (Protocol No. 029/2014).
Rights and permissions
About this article

Cite this article
Garcia, F.P., Henrique da Silva Rodrigues, J., Din, Z.U. et al. A3K2A3-induced apoptotic cell death of Leishmania amazonensis occurs through caspase- and ATP-dependent mitochondrial dysfunction. Apoptosis 22, 57–71 (2017). https://doi.org/10.1007/s10495-016-1308-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-016-1308-4