
Abstract
The genetic identity of Rhipicephalus sanguineus tick was determined for the first time in Taiwan. The phylogenetic relationships were analyzed by comparing the sequences of mitochondrial 16S ribosomal DNA gene obtained from 32 strains of ticks representing six species of Rhipicephalus, two species of Dermacentor and two outgroup species (Haemaphysalis inermis and Ixodes ricinus). Seven major clades can be easily distinguished by neighbour-joining analysis and were congruent by maximum-parsimony method. All R. sanguineus ticks of Taiwan were genetically affiliated to the tropical lineage group of R. sanguineus sensu lato with highly homogeneous sequence (99.7–100% similarity), and can be discriminated from the temperate lineage group of Rhipicephalus sp. II and R. turanicus with a sequence divergence ranging from 1.7 to 5.2%. In contrast, the nucleotide variations among other Rhipicephalus spp. and other species/genus of ticks compared with the R. sanguineus ticks of Taiwan were measured from 10.6 to 25.5%. Moreover, intra- and inter-species analysis based on the genetic distance (GD) values indicated a lower level (GD < 0.003) within tropical lineage group compared with temperate lineage group (GD > 0.055) of Rhipicephalus, as well as other (GD > 0.129) and outgroup (GD > 0.236) species. Our results provide the first genetic identification of R. sanguineus ticks collected from Taiwan and demonstrate that all these R. sanguineus of Taiwan affiliated to the tropical lineage group of R. sanguineus sensu lato.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ticks are obligate hematophagous arthropods that parasitize every class of vertebrates in almost every region of the world and it may act as vectors with the ability to transmit various pathogens including bacteria, rickettsiae, and protozoan (Balashov 1972). The medical and veterinary importance with the recent emergence of human babesiosis (Shih et al. 1997), Lyme borreliosis (Shih and Chao 1998; Chao et al. 2011) and canine babesiosis (Lee et al. 2010) in Taiwan raises the focus of research attention on vector ticks. The brown dog tick, Rhipicephalus sanguineus, is the most widespread tick species around the world and is recognized as the dominant ectoparasite of dogs that can occasionally parasitize other vertebrate hosts, including humans (Felz et al. 1996; Dantas-Torres 2010). In addition, R. sanguineus has been recognized as the primary vector for the transmission of Babesia vogeli, Ehrlichia canis, Rickettsia rickettsii, and R. conorii in humans and animals (Walker et al. 2000; Otranto et al. 2009; Eremeeva et al. 2011; Dantas-Torres et al. 2012). Although the hard tick of R. sanguineus had been identified as the incriminated vector tick for the zoonotic transmission of B. vogeli in Taiwan (Chao et al. 2016), the genetic identity of R. sanguineus collected from endemic sites of Taiwan remain undefined.
Although species determination and differentiation of Rhipicephalus ticks have traditionally been based on morphological features of the adult stages of these ticks, the taxonomic status of the R. sanguineus ticks has been repeatedly debated (Gray et al. 2013; Dantas-Torres and Otranto 2015; Nava et al. 2015). Because of the high level of morphological similarity among brown dog ticks within the R. sanguineus complex, ambiguity in taxonomy of the R. sanguineus ticks was reiterated by using molecular tools for phylogenetic analysis (Szabo et al. 2005; Burlini et al. 2010; Moraes-Filho et al. 2011; Levin et al. 2012; Liu et al. 2013). Indeed, a DNA-based approach provides the feasibility to investigate the genetic variance at the individual base-pair level and gives much more direct pathway for measuring the genetic diversity between and within species of Ixodidae (Black and Piesman 1994; Caporale et al. 1995; Black and Roehrdanz 1998). Current studies based on the mitochondrial 16S ribosomal DNA (rDNA) target region have revealed the existence of at least two separate groups (tropical vs. temperate lineage) of R. sanguineus ticks (Szabo et al. 2005; Moraes-Filho et al. 2011; Zemtsova et al. 2016). Thus, molecular analysis based on the genetic polymorphism of mitochondrial 16S rDNA gene has made possible in facilitating the identification and discrimination of taxonomically similar Rhipicephalus ticks.
It may be that the vector tick of R. sanguineus for canine babesiosis in Taiwan is a genetically distinct lineage, as compared with the existing common vector ticks of Rhipicephalus species around the world and the potential of genetic variation in relation to the geographical distribution may also exist among these R. sanguineus ticks characterized with similar morphology. Thus, the objective of this study intends to investigate the phylogenetic relationships between and within the species of R. sanguineus ticks by analyzing the mitochondrial 16S rDNA gene. The genetic divergence of R. sanguineus ticks collected from endemic sites of Taiwan was analyzed by their differential nucleotide composition, as compared with other tick species identified from various geographical sources which have been documented in GenBank.
Materials and methods
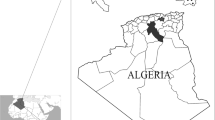
Collection and identification of tick specimen
All specimens of adult ticks including 28 strains of Rhipicephalus ticks, two strains of Dermacentor ticks, and two outgroup species (Haemaphysalis inermis and Ixodes ricinus) were used for genetic analysis in this study (Table 1). Of these, 14 strains of R. sanguineus were collected from dogs captured at various districts of Kaohsiung City (22°36′N, 120°18′E; 22°39′N, 120°17′E; 22°43′N, 120°25′E; 22°47′N, 120°22′E; 22°53′N, 120°19′E; 22°53′N, 120°28′E) in southern Taiwan (Fig. 1). All these ticks were subsequently stored in separate mesh-covered and plaster-bottomed vials. All tick specimens of R. sanguineus were identified to species level on the basis of their morphological characteristics, as described previously (Chao et al. 2016). Ultrastructural observations by stereo-microscope were used to delineate the morphological features of all stages of R. sanguineus ticks in Taiwan. Briefly, tick specimens were cleaned by sonication in 70% ethanol solution for 5–10 min and then washed twice in sterile distilled water. Afterwards, each stage of tick specimen was placed on a glass slide and photographed using a stereo-microscope (SMZ 1500, Nikon, Tokyo, Japan) equipped with a fiber lamp. The external features of the R. sanguineus ticks were recorded for species identification.

Map of Taiwan and its adjacent islands, showing the collection site for tick specimens
DNA extraction from tick specimen
Total genomic DNA was extracted from individual tick specimens used in this study. Briefly, tick specimens were cleaned by sonication for 3–5 min in ethanol solution and then washed twice in sterile distilled water. Afterwards, the individual tick specimen dissected into pieces was placed in a microcentrifuge tube filled with 180-μL lysing buffer solution supplied with a DNeasy Tissue Kit (catalogue no. 69506, Qiagen, Taipei, Taiwan) and then homogenized with a TissueLyser II (catalogue no. 85300, Qiagen, Germany), instructed by the manufacturer. The homogenate was centrifuged at room temperature and the supernatant fluid was further processed by a DNeasy Tissue Kit, as instructed by the manufacturer. After filtration, the filtrate was collected and the DNA concentration was determined spectrophotometrically with a DNA calculator (Nanovue Plus Spectrophotometer).
DNA amplification by polymerase chain reaction (PCR)
DNA samples extracted from the tick specimens were used as a template for PCR amplification. A specific primer set of 16S + 1 (5′-CTGCTCAATGATTTTTTAAATTGCTGTGG-3′) corresponding to the 3′ end of the mitochondrial 16S rDNA and 16S − 1 (5′-CCGGTCTGAACTCAGATCAAGT-3′) corresponding to the 5′ end of the mitochondrial 16S rDNA were designed to target the mitochondrial 16S rDNA gene, as described previously (Black and Piesman 1994). All PCR reagents and Taq polymerase were obtained and used as recommended by the supplier (Takara Shuzo, Japan). Briefly, a total of 0.2-μmol of the appropriate primer set and adequate amounts of template DNA were used in each 50-μl reaction mixture. In contrast, adequate amounts of sterile distilled water were added for serving as a negative control. PCR amplification was performed with a Perkin-Elmer Cetus thermocycler (GeneAmp system 9700) and was amplified for 40 cycles with the conditions of denaturation at 92 °C for 1 min, annealing at 54 °C for 35 s, and extension at 72 °C for 90 s., as described previously (Chao et al. 2009). Thereafter, amplified DNA products were electrophoresed on 2% agarose gels in Tris–Borate-EDTA (TBE) buffer and visualized under ultraviolet (UV) light after staining with ethidium bromide. A DNA ladder (1-kb plus, catalogue no. 10787-018, Invitrogen, Taipei, Taiwan) was used as the standard marker for comparison. A negative control of distilled water was included in parallel with each amplification.
Sequence alignments and phylogenetic analysis
After purification (QIAquick PCR Purification Kit, catalog No. 28104), sequencing reaction was performed with 25 cycles under the same conditions and same primer set of initial amplification by dye-deoxy terminator reaction method using the Big Dye Terminator Cycle Sequencing Kit in an ABI Prism 377-96 DNA Sequencer (Applied Biosystems, Foster City, CA, USA). The resulting sequences were initially edited by BioEdit software (V5.3) and aligned with the CLUSTAL W software (Thompson et al. 1994). Thereafter, the aligned sequences of 14 tick strains of Taiwan were further analyzed by comparing with other 18 strains of tick specimens based on the different genus and different geographical origin of Rhipicephalus ticks that are available in GenBank. Phylogenetic analysis was performed by neighbour-joining (NJ) compared with maximum parsimony (MP) methods to estimate the phylogeny of the entire alignment using MEGA 6.0 software package (Tamura et al. 2013). The genetic distance values of inter- and intra-species variations were also analyzed by the Kimura two-parameter model (Kimura 1980). All phylogenetic trees were constructed and performed with 1000 bootstrap replications to evaluate the reliability of the construction, as described previously (Felsenstein 1985).
Nucleotide sequence accession numbers
The nucleotide sequences of PCR-amplified mitochondrial 16S rDNA genes of 14 strains of R. sanguineus ticks determined in this study have been registered and assigned the following GenBank accession numbers: strains 99KHDS09EN6 (KX685412), 99KHDS04M2 (KX685413), 99KHDS09EN5 (KX685414), 99KHDS04M1 (KX685415), 98KHCJ10PEA (KX685416), 98KHCJ08M (KX685417), 100KHAL04PEA1 (KX685418), 100KHAL04M1 (KX685419), 100KHCH07PEA2 (KX685420), 100KHCH07EN1 (KX685421), 99KHYC06PEA2 (KX685422), 99KHYC06M2 (KX685423), 99KHZY01M9 (KX685424), and 99KHZY09EN2 (KX685425), respectively. For phylogenetic analysis, the nucleotide sequences of 16S rDNA genes from other 14 strains of Rhipicephalus, two strains of Dermacentor, and two outgroup ticks (i.e. H. inermis and I. ricinus) were included for comparison and their GenBank accession numbers are shown in Table 1.
Results
Sequence alignment and genetic analysis
To clarify the genetic identity of R. sanguineus ticks of Taiwan, the sequences of mitochondrial 16S rDNA fragments of 14 Taiwan strains of R. sanguineus performed by this study were aligned and compared with the downloaded sequences of eight different geographical strains of R. sanguineus, six strains of Rhipicephalus, two strains of Dermacentor, and two outgroup strains of H. inermis and I. ricinus from GenBank. Results indicate that the lengths of the aligned sequences were measured from 369 to 397 bp, and the nucleotide sequences between the 14 strains of R. sanguineus of Taiwan were highly conserved with only a few point mutations/substitutions. All these R. sanguineus ticks of Taiwan were genetically affiliated to the tropical lineage group of R. sanguineus sensu lato with highly homogeneous sequence (99.74–100% similarity), and can be distincted from the temperate lineage group of Rhipicephalus sp. II and R. turanicus with a sequence divergence ranging from 1.68 to 5.17% (Table 2). In contrast, the nucleotide variations among other Rhipicephalus ticks and other species/genus of ticks compared with the R. sanguineus ticks of Taiwan were measured from 10.59 to 25.47% (Table 2). In addition, intra- and inter-species analysis based on the genetic distance (GD) values indicated a lower level (GD < 0.003) of genetic divergence within the tropical lineage group of R. sanguineus ticks as compared with the temperate lineage group (GD > 0.055) of R. sanguineus, as well as other (GD > 0.129) and outgroup (GD > 0.236) species of ticks (Table 3).
Phylogenetic analysis of tick specimens
Phylogenetic relationships based on the sequence alignment of mitochondrial 16S rDNA were performed to demonstrate the genetic divergence among 32 strains of ticks investigated in this study. Bootstrap analysis was used to analyze the repeatability of the clustering of specimens represented in phylogenetic trees. Phylogenetic trees constructed by both NJ (Fig. 2) and MP (Fig. 3) analyses showed congruent basal topologies with seven major branch of distinguished clades (Figs. 2, 3). All these R. sanguineus ticks of Taiwan constitute a monophyletic clade closely affiliated to the tropical lineage group of R. sanguineus ticks, and can be easily discriminated from the temperate lineage group (Rhipicephalus sp. II) and R. turanicus ticks with a bootstrap value of 97 and 95 in NJ analysis (Fig. 2). The phylogenetic tree of MP analysis was identical to the NJ tree and strongly support the separation of different lineages between the R. sanguineus from Taiwan and the temperate lineage group of Rhipicephalus ticks with a bootstrap value of 97 (Fig. 3). These results reveal a lower genetic divergence within the same species of R. sanguineus ticks from Taiwan, but a higher genetic variations among different lineage or genus of Rhipicephalus ticks.

Phylogenetic relationships based on the 16S ribosomal DNA (rDNA) gene sequences between 14 strains of R. sanguineus ticks from southern Taiwan and 18 other strains belonging to five species of Rhipicephalus, one species of Dermacentor and Haemaphysalis, and one strain of Ixodes ricinus served as outgroup comparison. The trees were constructed and analyzed by neighbour-joining (NJ) method using 1000 bootstraps replicates. Numbers at the nodes indicate the percentages of reliability of each branch of the tree. Branch lengths are drawn proportional to the estimated sequence divergence

Phylogenetic relationships based on the 16S ribosomal DNA (rDNA) gene sequences between 14 strains of R. sanguineus ticks from southern Taiwan and 18 other strains belonging to five species of Rhipicephalus, one species of Dermacentor and Haemaphysalis, and one strain of Ixodes ricinus served as outgroup comparison. The trees were constructed and analyzed by maximum parsimony (MP) method using 1000 bootstraps replicates. Numbers at the nodes indicate the percentages of reliability of each branch of the tree. Branch lengths are drawn proportional to the estimated sequence divergence
Discussion
This study describes the first genetic identification of the mitochondrial 16S ribosomal gene among R. sanguineus ticks collected on Taiwan. In previous investigations, sequence analysis of the mitochondrial 16S rDNA have been used to distinguish closely related R. sanguineus ticks (Burlini et al. 2010; Moraes-Filho et al. 2011; Levin et al. 2012; Nava et al. 2012; Dantas-Torres et al. 2013; Zemtsova et al. 2016) and to assess the phylogenetic relationships of diverse species of Rhipicephalus ticks (Erster et al. 2013; Low et al. 2015; Zemtsova et al. 2016) by comparing their nucleotide variations of the mitochondrial 16S rDNA. Indeed, current investigations demonstrate that the existence of at least two distinguished groups of R. sanguineus ticks around the world. The tropical lineage group represented by R. sanguineus sensu lato collected from the countries of Brazil, Cuba, Colombia, Costa Rica, Japan, Kenya, Marshall island, Mozambique, South Africa, Thailand, and USA-FL. In contrast, the temperate lineage group includes ticks from Chile, Spain, France, Italy, Germany, Argentina, and USA-GA (Dantas-Torres et al. 2013; Zemtsova et al. 2016). Results from this study demonstrate that the nucleotide composition of the mitochondrial 16S rDNA derived from these R. sanguineus ticks of Taiwan is highly homogeneous (99.74–100% sequence similarity) with the tropical lineage group of R. sanguineus ticks. Thus, our study demonstrates the first molecular evidence confirming the genetic identity of R. sanguineus ticks collected in southern Taiwan and provides the first convincing sequences (GenBank accession numbers: KX685412~KX685425) of R. sanguineus ticks in Taiwan.
Because of the genetically high conservation and strictly maternal inheritance, the mitochondrial 16S rDNA sequences appear to provide a reliable and convenient method for distinguishing the lineages among diverse populations of Rhipicephalus ticks. In previous studies, two mitochondrial ribosomal genes, 12S and 16S rDNA, have been sequenced entirely for phylogenetic analysis of ixodid ticks focused on the family and subfamily levels (Black and Roehrdanz 1998; Campbell and Barker 1999). Indeed, genetic analysis of the mitochondrial 16S rDNA sequences of various species of Rhipicephalus ticks also permits quantitative assessment of their relatedness (Moraes-Filho et al. 2011; Nava et al. 2012; Dantas-Torres et al. 2013; Erster et al. 2013; Low et al. 2015; Zemtsova et al. 2016). Results from this study also demonstrate the closely related individuals of R. sanguineus ticks of southern Taiwan and the genetic divergence among various species of Rhipicephalus ticks based on the genetic variations of 16S rDNA (Table 2; Fig. 1). Intraspecific analysis reveals that nucleotide compositions within Taiwan and the tropical lineage group of R. sanguineus ticks averaged less than 0.3% sequence variations may fully represent a distinct species discriminated from the temperate lineage group of R. sanguineus ticks (Tables 2, 3). However, interspecific analysis also indicates the nucleotide variations between R. sanguineus ticks of Taiwan and other Rhipicephalus species or genus of ticks averaged more than 10.59% sequence variations (Tables 2, 3). Further investigation on the sequence divergence based on various targets of the mitochondrial genes of R. sanguineus ticks collected from different localities of Taiwan and its adjacent islands would be required to clarify the genetic divergence as well as the evolutionally origin among and within R. sanguineus ticks from Taiwan and its adjacent islands.
Phylogenetic relationships among Rhipicephalus ticks can be determined by analyzing the sequence heterogeneity of the mitochondrial 16S rDNA. Indeed, sequence analysis of the mitochondrial 16S rDNA among various species of Rhipicephalus ticks had been shown to be useful for evaluating the taxonomic relatedness of tick specimens collected from various geographical sources (Moraes-Filho et al. 2011; Nava et al. 2012; Dantas-Torres et al. 2013; Erster et al. 2013; Low et al. 2015; Zemtsova et al. 2016). In previous studies, two distinct lineages of R. sanguineus ticks are evident by comparing their mitochondrial 16S rDNA sequences collected from different regions of Latin America (Moraes-Filho et al. 2011; Nava et al. 2012; Zemtsova et al. 2016). Phylogenetic analysis of tick species related to the members of the Rhipicephalus complex also revealed intraspecific variation between different geographical collections (Moraes-Filho et al. 2011; Erster et al. 2013; Low et al. 2015). In this study, the phylogenetic analysis based on the mitochondrial 16S rDNA sequences among various tick species demonstrated a high genetic heterogeneity between R. sanguineus and other species of ticks (Figs. 2, 3). Although a low intraspecific variation was observed among the same species of R. sanguineus ticks, all the 14 strains of R. sanguineus ticks from Taiwan represented as a monophyletic group that can be distinguished from the temperate group of R. sanguineus and other species/genus ticks (Table 3; Fig. 2). The phylogenetic trees constructed by either NJ or MP analysis strongly support the discrimination recognizing the separation of different lineages between the R. sanguineus collected from Taiwan and the temperate group of R. sanguineus. Accordingly, these observations demonstrate that genetic identities of R. sanguineus ticks collected from southern Taiwan were verified as a unique group affiliated to the tropical lineage of R. sanguineus sensu lato.
In conclusion, this report provides the first genetic identification of the mitochondrial 16S rDNA gene of R. sanguineus ticks collected from the Taiwan area. Based on the sequence divergence of the mitochondrial 16S rDNA, all these R. sanguineus ticks of Taiwan were genetically related to a monophyletic group and were represented as a unique lineage distinguished from the temperate group of R. sanguineus ticks as well as other Rhipicephalus ticks including the common vector ticks for canine babesiosis. Further application of this molecular tool to investigate the genetic variability of R. sanguineus collected from different localities of Taiwan may help to elucidate the phylogenetic relationships among tick populations in relation to the epidemiological features of tick-borne pathogens in Taiwan.
References
Balashov YS (1972) Bloodsucking ticks (Ixodoidea)-vectors of diseases of man and animals. Misc Publ Entomol Soc Am 8:268–305
Black WC IV, Piesman J (1994) Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S ribosomal DNA sequences. Proc Natl Acad Sci USA 91:10034–10038
Black WC IV, Roehrdanz RL (1998) Mitochondrial gene order is not conserved in arthropods: prostriate and metastriate tick mitochondrial genomes. Mol Biol Evol 15:1772–1785
Burlini L, Teixeira KR, Szabo MP, Famadas KM (2010) Molecular dissimilarities of Rhipicephalus sanguineus (Acari: Ixodidae) in Brazil and its relation with samples throughout the world: is there a geographical pattern? Exp Appl Acarol 50:361–374
Campbell NJH, Barker SC (1999) The novel mitochondrial gene arrangement of the cattle tick, Boophilus microplus: fivefold tandem repetition of a coding region. Mol Biol Evol 16:732–740
Caporale DA, Rich SM, Spielman A et al (1995) Discriminating between Ixodes ticks by means of mitochondrial DNA sequences. Mol Phylogenet Evol 4:361–365
Chao LL, Wu WJ, Shih CM (2009) Molecular analysis of Ixodes granulatus, a possible vector tick for Borrelia burgdorferi sensu lato in Taiwan. Exp Appl Acarol 48:329–344
Chao LL, Chen YJ, Shih CM (2011) First isolation and molecular identification of Borrelia burgdorferi sensu stricto and Borrelia afzelii from skin biopsies of patients in Taiwan. Int J Infect Dis 15:e182–e187
Chao LL, Yeh ST, Hsieh CK, Shih CM (2016) First detection and molecular identification of Babesia vogeli from Rhipicephalus sanguineus (Acari: Ixodidae) in Taiwan. Exp Appl Acarol 68:539–551
Dantas-Torres F (2010) Biology and ecology of the brown dog tick, Rhipicephalus sanguineus. Parasites Vectors 3:26
Dantas-Torres F, Otranto D (2015) Further thoughts on the taxonomy and vector role of Rhipicephalus sanguineus group ticks. Vet Parasitol 208(1–2):9–13
Dantas-Torres F, Chomel BB, Otranto D (2012) Ticks and tick-borne diseases: a One Health perspective. Trends Parasitol 28:437–446
Dantas-Torres F, Latrofa MS, Annoscia G, Giannelli A, Parisi A, Otranto D (2013) Morphological and genetic diversity of Rhipicephalus sanguineus sensu lato from the New and Old worlds. Parasites Vectors 6:213
Eremeeva ME, Zambrano ML, Anaya L et al (2011) Rickettsia rickettsii in Rhipicephalus ticks, Mexicali, Mexico. J Med Entomol 48:418–421
Erster O, roth A, Wolkomirsky R, Leibovich B, Shkap V (2013) Comparative analysis of mitochondrial markers from four species of Rhipicephalus (Acari: Ixodidae). Vet Parasitol 198:364–370
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 52:1119–1134
Felz MW, Durden LA, Oliver JH Jr (1996) Ticks parasitizing humans in Georgia and South Carolina. J Parasitol 82:505–508
Gray J, Dantas-Torres F, Estrada-Pena A, Levin M (2013) Systematics and ecology of the brown dog tick, Rhipicephalus sanguineus. Ticks Tick Borne Dis 4:171–180
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Lee CC, Hsieh YC, Huang CC et al (2010) Sequence and phylogenetic analysis of the thrombospondin-related adhesive protein (TRAP) gene of Babesia gibsoni isolates from dogs in Taiwan. J Vet Med Sci 72:1329–1335
Levin ML, Studer E, Killmaster L, Zemtsova G, Mumcuoglu KY (2012) Crossbreeding between different geographical populations of the brown dog tick, Rhipicephalus sanguineus (Acari: Ixodidae). Exp Appl Acarol 58:51–68
Liu GH, Chen YZ, Song HQ, Lin RQ, Zhou DH, Zhu XQ (2013) Complete mitochondrial genome sequence data provides evidence that dog tick Rhipicephalus sanguineus (Acari: Ixodidae) represents a species complex. Int J Biol Sci 9:361–369
Low VL, Tay ST, Kho KL et al (2015) Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasites Vectors 8:341
Moraes-Filho J, Marcili A, Nieri-Bastos FA, Richtzenhain LJ, Labruna MB (2011) Genetic analysis of ticks belonging to the Rhipicephalus sanguineus group in Latin America. Acta Trop 117:51–55
Nava S, Mastropaolo M, Venzal JM, Mangold AJ, Guglielmone AA (2012) Mitochondrial DNA analysis of Rhipicephalus sanguineus sensu lato (Acari: Ixodidae) in the southern cone of South America. Vet Parasitol 190:547–555
Nava S, Estrada-Pena A, Petney T et al (2015) The taxonomic status of Rhipicephalus sanguineus (Latreille). Vet Parasitol 208(1–2):2–8
Otranto D, Dantas-Torres F, Breitschwerdt EB (2009) Managing canine vector-borne diseases of zoonotic concern: part one. Trends Parasitol 25:157–163
Shih CM, Chao LL (1998) Lyme disease in Taiwan: primary isolation of Borrelia burgdorferi-like spirochetes from rodents in Taiwan area. Am J Trop Med Hyg 59:687–692
Shih CM, Liu LP, Chung WC et al (1997) Human babesiosis in Taiwan: asymptomatic infection with a Babesia microti-like organism in a Taiwanese woman. J Clin Microbiol 35:450–454
Szabo MP, Mangold AJ, Joao CF, Bechara GH, Guglielmone AA (2005) Biological and DNA evidence of two dissimilar populations of the Rhipicephalus sanguineus tick group (Acari: Ixodidae) in South America. Vet Parasitol 130:131–140
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Walker JB, Keirans JE, Horak IG (2000) The genus Rhipicephalus (Acari, Ixodidae): a guide to the brown ticks of the world. Cambridge University Press, Cambridge
Zemtsova GE, Apanaskevich DA, Reeves WK, Hahn M, Snellgrove A, Levin ML (2016) Phylogeographical of Rhipicephalus sanguineus sensu lato and its relationships with climatic factors. Exp Appl Acarol 69:191–203
Acknowledgements
This work was supported in part by grants from the Kaohsiung Medical University Research Foundation (KMU-Q105001) and Research Center for Environmental Medicine (KMU-TP104A17), Kaohsiung Medical University, Kaohsiung, Taiwan, ROC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chao, LL., Shih, CM. Molecular analysis of Rhipicephalus sanguineus (Acari: Ixodidae), an incriminated vector tick for Babesia vogeli in Taiwan. Exp Appl Acarol 70, 469–481 (2016). https://doi.org/10.1007/s10493-016-0094-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-016-0094-6