
Abstract
In this study, we reported a Gram-stain-negative, rod-shaped, atrichous, and aerobic bacterial strain named YMD87T, which was isolated from the intertidal zone sediment of Chinese Yellow Sea. Growth of strain YMD87T occurred at 10.0–40.0 °C (optimum, 25–30 °C), pH 4.0–12.0 (optimum, 8.0) and with 0–6.0% (w/v) NaCl (optimum, 0.0–2.0%). Phylogenetic tree analysis based on 16S rRNA gene sequence indicated that strain YMD87T belonged to the genus Tropicibacter and was closely related to Tropicibacter alexandrii LMIT003T (97.2% sequence similarity). Genomic analysis indicated that strain YMD87T contains a circular chromosome of 3,932,460 bp with G + C content of 63.8% and three circular plasmids of 116,492 bp, 49,209 bp and 49,673 bp, with G + C content of 64.3%. Genomic functional analysis revealed that strain YMD87T is potential a novel sulfur-metabolizing bacteria. The predominant respiratory quinone of YMD87T was ubiquinone-10 (Q-10). The major polar lipids of YMD87T contained phosphatidylglycerol, phosphatidylethanolamine, five unidentified lipids, five unidentified phospholipids, phosphatidylcholine, unidentified glycolipid and five unidentified aminolipids. The major fatty acids of strain YMD87T contained C12:1 3-OH, C16:0, and summed feature 8 (C18:1 ω7c or/and C18:1 ω6c). Phylogenetic, physiological, biochemical and morphological analyses suggested that strain YMD87T represents a novel species of the genus Tropicibacter, and the name Tropicibacter oceani sp. nov is proposed. The type strain is YMD87T (= MCCC 1K08473T = KCTC 92856 T).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The genus Tropicibacter, belonging to the family Rhodobacteraceae, was first proposed by Harwati et al. in 2009 (Harwati et al 2009). At the time of writing, there are only two species in this genus, Tropicibacter naphthalenivorans, isolated from seawater obtained from Semarang Port in Indonesia (Harwati et al 2009), and Tropicibacter alexandrii LMIT003T, isolated from a liquid culture of the dinoflagellate Alexandrium minutum (Wang et al. 2020). Four species previously affiliated with this genus were reclassified into other genera based on genomic analyses. Tropicibacter multivorans was reclassified in the genus Epibacterium, while Tropicibacter mediterraneus, and Tropicibacter litoreus were reclassified in the genus Ruegeria, and Tropicibacter phthalicicus was reclassified in the genus Pelagimonas. Strains of the genus Tropicibacter are Gram-stain-negative, aerobic, oxidase-positive and rod-shaped. Chemotaxonomic characteristics include the predominant lipoquinone ubiquinone-10 (Q-10), and a G + C content of 61.9–64.6 mol% (Harwati et al 2009; Wang et al. 2020). The common major fatty acids identified in this genus include C16:0 and summed feature 8 (C18:1 ω7c or/and C18:1 ω6c) (Harwati et al 2009; Wang et al. 2020).
The intertidal zone is a buffer area between sea and land that is rich in sulfur compounds, nitrogen-containing compounds, and toxins, and has been influenced by complex environmental factors and human activities (Cao and Wong 2007; Pan and Wang 2012). The intertidal zone is also featured by high abundant and diverse microbial communities, and the fluctuation of intertidal environmental factors significantly influence the abundance and contribution of the microbial communities (Wilms et al. 2006). Sulfur metabolism bacteria, ubiquitous in intertidal zones, play important roles in the global cycling of sulfur and carbon (Pester et al. 2012; Rabus et al. 2015). The diversity of sulfate-reducing bacteria community in intertidal zones have been revealed in the surface sediments of Yangtze Estuary and Shenzhen Bay (Wu et al. 2019). In this study, a bacterial strain named YMD87T was isolated from the intertidal zone sediment of Chinese Yellow Sea. Based on the polyphasic taxonomic features, we proposed that strain YMD87T represents a novel species of the genus Tropicibacter, for which the name Tropicibacter oceani sp. nov., is proposed.
Materials and methods
Isolation and culture conditions
Strain YMD87T was isolated from the sediment collected from the intertidal zone of Chinese Yellow Sea (37°23′0″N, 121°36′0″E). For bacterial isolation, the sediment sample was tenfold serially diluted with sterilized seawater, and spread onto the marine 2216EA (Haibo, Qingdao, China) plates. After incubation at 28 ℃ for 7 days, the pure culture of YMD87T was obtained after three successive transfers to new 2216EA plates. The strain was stored at –80 °C in marine 2216 EB (Haibo) medium supplemented with 20% (v/v) glycerol.
16S rRNA gene sequence analysis
For 16S rRNA gene and genomic sequencing, genomic DNA of YMD87T was extracted using a bacterial genomic DNA extraction kit (Tiangen, Beijing, China). The partial 16S rRNA gene sequence of YMD87T was amplified by universal primers 27F (5ʹ-AGAGTTTGATCCTGGCTCA-3ʹ) and 1492R (5ʹ-GGTTACCTTGTTACGACTT-3ʹ) (Buchan et al. 2005). The PCR product was further cloned into the pEASY-T1 simple vector (TransGen, Beijing, China), and an almost complete 16S rRNA gene sequence of strain YMD87T was obtained and submitted to GenBank (accession number OP942226). The 16S rRNA gene sequence was analyzed using EzBioCloud (http://eztaxon-e.ezbiocloud.net/) (Kim et al. 2012). The phylogenetic tree was generated using MEGA 7.0 software by neighbour-joining (Saiton et al. 1987), minimum-evolution (Rzhetsky et al. 1992) and maximum-likelihood (Guindon et al. 2003) and algorithms with bootstrap values (1000 replications) (Kumar et al. 2016).
Genome analysis
The whole genome of strain YMD87T was sequenced using PacBio Sequel and Illumina NovaSeq PE150 platforms in Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). A total of 367,182 reads were obtained, which were further assembled, corrected and authenticity ensured as described previously (Xu et al. 2021). The phylogenomic tree of strain YMD87T and related species was constructed using the up-to-date bacterial core gene (UBCG) set (Na et al. 2018). The genome sequence of T. alexandrii LMIT003T (accession number GCA_003667315.2) was obtained from NCBI database to analyze the genomic difference with strain YMD87T. EzBioCloud ANI calculator (www.ezbiocloud.net/tools/ani) (Lee et al. 2016) was used to determine the average nucleotide identity (ANI). DNA-DNA relatedness was estimated using the Genome-to- Genome Distance Calculator (GGDC) 3.0 provided by the DSMZ (http://ggdc.dsmz.de/distcalc2.php) (Meier-Kolthoff et al. 2013). The genome component and gene function were further predicted by NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al. 2016). The COG and KEGG databases were also used to predict the functional genes and pathways. The secretory proteins were predicted with SignalP (Version 4.1) and TMHMM (Version 2.0c) (Petersen et al. 2011). The secondary metabolic gene clusters were analyzed with antiSMASH (version 2.0.2) (Medema et al. 2011). The integrins were predicted with INTEGRALL database (http://integrall.bio.ua.pt/). The virulence factors were predicted with pathogen host interactions database and virulence factors of pathogenic bacteria database (Chen et al. 2012; Martin et al. 2015). Antibiotic resistance genes were analyzed with antibiotic resistance genes database and comprehensive antibiotic research database (Jia et al. 2017; Liu and Pop 2009).
Physiological and chemotaxonomic characterization
The type strain T. alexandrii LMIT003T (KCTC 62895 T) was obtained from Korean Collection for Type Cultures (KCTC, Korean), and used as a reference strain for biochemistry and fatty acid analysis. For cell morphology analysis, strain YMD87T was grown on marine 2216 EB for 24 h at 28 °C, after harvesting and washing with distilled water, cells were negatively stained with 1% phosphotungstic acid and observed by transmission electron microscopy HT-7700 (Hitachi, Tokyo, Japan). Gram staining was performed using a Gram-staining kit (Haibo). Growth temperature of 4, 10, 15, 20, 25, 30, 35, 37, 40 and 45 °C were evaluated on 2216 EB to measure the growth range of YMD87T. The NaCl range for growth was tested by using NaCl-free 2216 EB with different NaCl concentrtions (0–15.0%, at intervals of 1.0%) at 28 °C. The optimal pH was determined by growing at pH 4.0–12.0 (at intervals of 1.0 pH unit) using the buffer system described by Xu et al. (2005). In order to investigate anaerobic growth, strain YMD87T was cultured at 28 °C on 2216 EB and supplemented with resazurin (0.02%, w/v) as an indicator of anaerobic condition. The plates were incubated in an anaerobic incubator YQX (Yuejin, Shanghai, China) filled with nitrogen. Oxidase activity was evaluated with the oxidase reagent (Haibo), and catalase activity was tested by the production of oxygen bubbles in 3% (v/v) H2O2 solution. Hydrolysis of starch, casein, tween 20, and 80 was examined on 2216EA plates with the corresponding substrate. Nitrate reductase, gelatinase, urease activities and other enzyme productions were examined using the API 20NE and API ZYM system (bioMérieux, Marcy-l'Étoile, France) in 28 °C, according to the instructions. Acid production from different carbohydrates was performed using the API 50CH system (bioMérieux). For fatty acid methyl esters (FAME) analysis, bacteria were grown on 2216EA until the late of the exponential growth phase (2 days) at 28 °C, and the cells were harvested and analyzed as reported by Sasser (Sasser et al. 1990). Polar lipids were extracted according to the protocol of Minnikin et al. and examined using two-dimensional TLC (Minnikin et al. 1984). Respiratory quinones of strain YMD87T was analyzed using freeze-dried cells as reported previously (Sasser et al. 1990). Antibiotic susceptibility was determined on 2216 EA plates using antibiotic discs containing the following (µg per disc unless otherwise stated): polymyxin B (300 IU), furazolidone (300), piperacillin (100), cefoperazone (75), midecamycin (30), minocycline (30), doxycycline (30), tetracycline (30), neomycin (30), amikacin (30), ceftriaxone (30), ceftazidime (30), cefuroxime (30), cefradine (30), cefamezin (30), cephalexin (30), penicillin (10 U), gentamicin (10), norfloxacin (10), ciprofloxacin (5), ofloxacin (5), clindamycin (2), sulfamethoxazole (1.25).
Results and discussion
Phylogeny analysis of 16S rRNA gene sequence
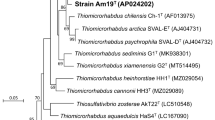
The 16S rRNA gene sequence analysis showed that strain YMD87T belonged to the genus Tropicibacter and was most closely related to T. alexandrii LMIT003T with 97.18% sequence similarities, and the identities between strain YMD87T and other close members were all below 97.0%. The NJ phylogenetic tree showed that strain YMD87T formed a group with T. alexandrii LMIT003T (Fig. S1). The corresponding ML and ME trees showed similar topologies (Fig. S1), which supported the proposal that strain YMD87T belonged to the genus Tropicibacter.
Physiological and chemotaxonomic analysis
The morphological, cultural, physiological and biochemical characteristics of strain YMD87T are given in the species descriptions (Table 1, S1, and Fig. 1). FAME analysis showed that the major fatty acid (≥ 5% of the total fatty acids) detected in strain YMD87T was C16:0 (9.5%), C12:1 3-OH (5.8%), and summed feature 8 (72.4%) (Table S2). The reference strain, T. alexandrii LMIT003T showed different profile of FAME with strain YMD87T in some aspects, and contained C18:0 (9.5%) as the major fatty acids, but C16:0 and C12:1 3-OH were not the major fatty acids (Table S2). The polar lipids of YMD87T contained phosphatidylglycerol (PG), phosphatidylethanolamine (PE), five unidentified lipids (L), five unidentified phospholipids (PL), phosphatidylcholine (PC), unidentified glycolipid (GL), and five unidentified aminolipids (AL) (Fig. S2). The PC, PE, PG, AL, and PL were also detected in T. alexandrii LMIT003T, but L and GL were not detected. The antibiotic resistance test show that, strain YMD87T was susceptible to penicillin, piperacillin, cephalexin, cefamezin, cefradine, cefuroxime, ceftazidime, ceftriaxone, cefoperazone, amikacin, neomycin, doxycycline, minocycline, midecamycin, norfloxacin, ofloxacin, ciprofloxacin, polymyxin B, sulfamethoxazole, furazolidone, weekly susceptible to clindamycin, resistant to tetracycline and gentamicin.

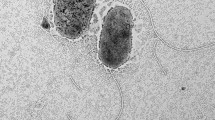
Transmission electron micrograph of the cell of strain YMD87T. Bar, 2.0 μm
Genomic features and phylogenomic analysis
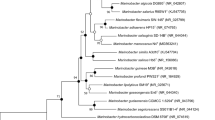
The complete genome and three plasmid sequences of strain YMD87T was obtained and submitted to GenBank (accession number CP124616-CP124619). Genomic analysis indicated that strain YMD87T contains a circular chromosome of 3,932,460 bp with G + C content of 63.8% and three circular plasmids of 116,492 bp with G + C content of 63.09%, 49,209 bp with G + C content of 66.34% and 49,673 bp with G + C content of 65.3%, respectively. The G + C content of YMD87T genomic is higher than the related reference strain T. alexandrii LMIT003T (Table 1). The predicted numbers of 5S rRNA, 16S rRNA, 23S rRNA, and tRNA sequences were 3, 3, 3, and 49, respectively (Table S3).The phylogenomic tree constructed using the 92 core genes of the genome showed the evolutionary position of strain YMD87T. According to its distribution, strain YMD87T clustered compactly with the strains T. alexandrii LMIT003T, Ponticoccus alexandrii AT2-AT (GCF_016806125.1), Ponticoccus marisrubri SJ5A-1 T (GCF_001482405.1), Mameliella alba DSM 26384 T and Maliponia aquimaris CECT 8898 T, of which YMD87T was more closely related to the type strain T. alexandrii LMIT003T (Fig. 2). Combining with the phylogenic tree analysis of 16S rRNA gene sequence, YMD87T belongs to the genus of Tropicibacter. The ANI value and the digital DDH value between strain YMD87T and T. alexandrii LMIT003T are 76.5% and 21.3%, respectively. These values are much lower than the threshold values for prokaryotic species delineation, which are 95–96% for ANI and 70% for DDH (Kim et al. 2014; Wayne et al. 1987). In addition, 10 genomics islands, 3 prophages, 2 CRISPR, and 341 secreted proteins were also detected in the genome of strain YMD87T (Table S3). Moreover, secondary metabolites analysis revealed 8 gene clusters, i.e., hserlactone (2 clusters), NRPS, RiPP-like, terpene, betalactone, ectoine, NRPS-like, and T1PKS, including a total of 186 genes (Table S1).

The phylogenomic tree based on 92 bacterial core genes showing the relationship of Tropicibacter Oceani YMD87T with other strains. Agaricicola taiwanensis CC-SBABM117T was selected as outgroup. Bootstrap values are shown at the branch points. Bar, 0.05 substitutions per nucleotide position
Functional genomics analysis
The whole genome of strain YMD87T was further analyzed to reveal its potential functional genes in intertidal environmental adaptation. The distributions of COG categories were showed in Table S4, and the most 3 abundant categories were amino acid transport and metabolism (343 genes), carbohydrate transport and metabolism (236 genes), and energy production and conversion (230 genes). Different from the reference strain T. alexandrii LMIT003T, which inhabit in the phycosphere, and capable of transforming DMSP to methylmercaptopropionate by the DMSP demethylase (dmdA) gene, dmdA was not detected in strain YMD87T (Wang et al. 2020). According to KEGG pathway analysis, the functional gene were most abundant in metabolic (53 pathways), of which the top 3 pathways were microbial metabolism in diverse environments (250 genes), carbon metabolism (128 genes), and nucleotide metabolism (96 genes), which conducive to utilize the complex nutrients of the intertidal environment. For sulfur metabolism pathways, 29 genes were identified, both YMD87T and the reference strain T. alexandrii LMIT003T had a complete assimilatory sulfate reduction (CysN, CysD, CysC, CysH, CysJ, and CysI) and a complete SOX system pathways (SoxB, SoxZ, SoxY, SoxX, SoxC, SoxD, and SoxB) (Fig S3). Concerning on the DNA repair pathways, a total of 37 genes were detected in YMD87T, which were categorized into three groups: DNA repair genes RadA, RadC, RecF, RecO, and RecN, the DNA mismatch repair genes MutS, MutT, and MutL, the double-strand DNA break repair genes AddB, and AddA, and DNA alkylation repair genes. Moreover, pathogenicity analysis based on PHI and VFDB databases revealed 278 and 159 genes related to pathogenicity, respectively. However, ARDB and CARD databases depended analysis only revealed two genes related to antibiotic (tetracycline and aminoglycoside) resistance, which were consistent with the results of drug sensitivity test. On the contrary, 214 genes were found to participate in the pathways of biosynthesis of antibiotics, which indicated a potential environmental adaptation mechanism of YMD87T by inhibiting the growth of other bacteria.
Conclusion
Strain YMD87T phylogenetically formed a distinct group with T. alexandrii LMIT003T, and the chemotaxonomic profiles of strain YMD87T were generally similar to T. alexandrii LMIT003T. However, some physiological and biochemical properties, such as enzyme activities, acid production substrates and antibiotic resistance, distinguished strain YMD87T from T. alexandrii LMIT003T. Therefore, strain YMD87T represents a novel species of the genus Tropicibacter, for which the name Tropicibacter oceani sp. nov. is proposed.
Description of Tropicibacter oceani sp. nov.
Tropicibacter oceani (o.ce.a’ni. L. gen. n. oceani, of the ocean).
Cells of strain YMD87T are Gram-stain-negative, rod-shaped, atrichous, and aerobic, 1.2–2.4 μm in width and 3.4–5.8 μm in length. Colonies of YMD87T on 2216EA are circular, smooth, white, and approximately 1–2 mm in diameter after incubation for 7 days at 28 °C. Growth of strain YMD87T occurred at 10.0–40.0 °C (optimum, 25–30 °C), pH 4.0–12.0 (optimum, 8.0) and with 0–6.0% (w/v) NaCl (optimum, 0–2.0%). Activities of oxidase, catalase and urease are positive, but tween-20, 40, 60, 80, starch and gelatin cannot be hydrolyzed. API ZYM reaction tests were positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidas, valine arylamidase, cystine aramidase, acid phosphatase, Naphthol-AS-BI-phosphohydrolase, α-glucosidase, negative for lipase C14, trypsin, chymotrypsin, α-galactosidase, β-galactosidase, N-acetyl-β-glucosaminidase, β-uronidase, β-glucosidas, α-fucosidase and α-mannosidase. Acid cannot be produced. The G + C content of the genomic DNA of strain YMD87T is 63.8%. The principal respiratory quinone is Q-10. The polar lipids of YMD87T contained phosphatidylglycerol, phosphatidylethanolamine, five unidentified lipids, five unidentified phospholipids, phosphatidylcholine, unidentified glycolipid, and five unidentified amino lipids. The major fatty acids of strain YMD87T contained C12:1 3-OH, C16:0, and summed feature 8 (C18:1 ω7c or/and C18:1 ω6c).
The type strain is YMD87T (= MCCC 1K08473T = KCTC 92856 T), which was isolated from the intertidal sediment obtained from the intertidal zone sediment of Chinese Yellow Sea. The GenBank accession numbers of the 16S rRNA gene sequence and the complete genome sequence of strain YMD87T are OP942226 and CP124616, respectively.
Data availability
The 16S rRNA gene and the complete genome sequences of strain YMD87T have been deposited under the GenBank accession numbers OP942226 and CP124616, respectively.
Abbreviations
- KCTC:
-
Korean collection for type cultures
- FAME:
-
Fatty acid methyl esters
- ANIP:
-
Average nucleotide identity
- GGDC:
-
Genome-to-genome distance calculator
- DSMZ:
-
Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH
- DDH:
-
DNA-DNA hybridizations
References
Buchan A, Gonzalez JM, Moran MA (2005) Overview of the marine Roseobacter lineage. Appl Environ Microbiol 71:5665–5677
Cao W, Wong MH (2007) Current status of coastal zone issues and management in China: a review. Environ Int 33:985–992
Chen L, Xiong Z, Sun L et al (2012) VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res 40(D1):D641–D645
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Guo XP, Yang Y, Niu ZS et al (2018) Characteristics of microbial community indicate anthropogenic impact on the sediments along the Yangtze estuary and its coastal area, China. Sci Total Environ 648:306–314
Harwati TU, Kasai Y, Kodama Y et al (2009) Tropicibacter naphthalenivorans gen. nov., sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from Semarang Port in Indonesia. Int J Syst Evol Microbiol 59:392–396
Hördt A, López MG, Meier-Kolthoff JP et al (2020) Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front Microbiol 11:468
Jia B, Raphenya AR, Alcock B et al (2017) CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45(Database issue):D566–D573
Kim OS, Cho YJ, Lee K et al (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62(3):716–721
Kim M, Oh HS, Park SC et al (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 70 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Lee I, Ouk Kim Y, Park SC et al (2016) OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Liu B, Pop M (2009) ARDB-antibiotic resistance genes database. Nucleic Acids Res 37(suppl 1):D443–D447
Martin U, Rashmi P, Arathi R (2015) The Pathogen-Host Interactions database (PHI-base): additions and future developments. Nucleic Acids Res 43(Database issue):D645-55
Medema MH, Blin K, Cimermancic P et al (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39(suppl 2):W339–W346
Meier-Kolthoff JP, Auch AF, Klenk HP et al (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60
Minnikin DE, Odonnell AG, Goodfellow M et al (1984) An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Meth 2:233–241
Pan K, Wang WX (2012) Trace metal contamination in estuarine and coastal environments in China. Sci Total Environ 421:3–16
Pester M, Knorr KH, Friedrich MW et al (2012) Sulfate-reducing microorganisms in wetlands–fameless actors in carbon cycling and climate change. Front Microbiol 3:72–91
Petersen TN, Brunak S, von Heijne G et al (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786
Rabus R, Venceslau SS, Woehlbrand L et al (2015) Pereira A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol 66:55–321
Rzhetsky A, Nei M (1992) A simple method for estimating and testing minimum-evolution trees. Mol Biol 9:945–967
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sasser M (1990) Identification of bacteria by gas chromatography of cellular fatty acids. USFCC News Lett 20:1–6
Tatusova T, Dicuccio M, Badretdin A et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624
Wang XL, Zhu JM, Feng JR et al (2020) Tropicibacter alexandrii sp. nov., a novel marine bacterium isolated from the phycosphere of a dinoflagellate. Alexandrium Minutum Antonie Van Leeuwenhoek 113:311–320
Wayne LG, Moore WEC, Stackebrandt E et al (1987) Report of the ad hoc Committee on reconciliation of approaches to bacterial systematics. Int J Syst Evol Microbiol 37:463–464
Wilms R, Sass H, Kopke B et al (2006) Specific bacterial, archaeal and eukaryotic communities in tidal-flat sediments along a vertical profile of several meters. Appl Environ Microbiol 72:2756–2764
Wirth JS, Whitman WB (2018) Phylogenomic analyses of a clade within the roseobacter group suggest taxonomic reassignments of species of the genera Aestuariivita, Citreicella, Loktanella, Nautella, Pelagibaca, Ruegeria, Thalassobius, Thiobacimonas and Tropicibacter, and the proposal of six novel genera. Int J Syst Evol Microbiol 68:2393–2411
Wu S, Li R, Xie S et al (2019) Shi Depth-related change of sulfate-reducing bacteria community in mangrove sediments: the influence of heavy metal contamination. Mar Pollut Bull 140:443–450
Xu P, Li WJ, Tang SK et al (2005) Naxibacter alkalitolerans gen. nov., sp nov., a novel member of the family “Oxalobacteraceae” isolated from China. Int J Syst Evol Microbiol 55:1149–1153
Xu XD, Zhang J, Sun QL et al (2021) Description of Psychrosphaera ytuae sp. Nov., isolated from the deep-sea cold seep sediment of South China Sea. Int J Syst Evol Microbiol 71(8):004983
Funding
This work is supported by the Key Laboratory of Mariculture of Ministry of Education, Ocean University of China, Development Plan of Youth Innovation Team in Colleges and Universities of Shandong Province (2022KJ269) and Yantai University Doctoral Start-up Foundation (HX20B34).
Author information
Authors and Affiliations
Contributions
DDZ: investigation, conceptualization, writing original draft. XDX: isolated the bacterium. BZZ and JXF: phylogenetic and genomic characterisation. JZ: supervision and writing-reviewing and editing, funding acquisition. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Ethical statement
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The GenBank accession number of the 16S rRNA gene sequence of strain YMD87T is OP942226. The GenBank accession number of complete genome and plasmid sequences of strain YMD87T are CP124616-CP124619.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, Dd., Xu, Xd., Zhang, Bz. et al. Tropicibacter oceani sp. nov., a novel sulfur-metabolizing bacteria isolated from the intertidal zone sediment of Chinese Yellow Sea. Antonie van Leeuwenhoek 116, 1337–1344 (2023). https://doi.org/10.1007/s10482-023-01890-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-023-01890-5