
Abstract
Actinobacillus pleuropneumoniae is the cause of porcine pleuropneumonia, for which the mortality rate is high. Host peripheral blood is a body site for the immune clearance of pathogens mediated by release of inflammatory factors. However, “out of control” inflammatory factor release can contribute to host death. To further understand the changes in the transcription level of immune-related effectors, samples of peripheral blood mononuclear cells (PBMCs) collected from piglets at different stages of infection (0, 24 and 120 h) were sequenced on an Illumina HiSeq™ 4000 platform. We found 3818 differentially expressed genes (DEGs) in the 24 h-infection group compared to the 0 h-infection group (Pb24-Vs-Pb0). DEGs mainly involved in the Gene ontology and KEGG pathways that included nucleic acid metabolism regulation, cell growth, cell differentiation, and organ morphological maintenance were not significantly enriched (P > 0.05). However, DEGs associated with protein kinase activity, receptor activation, metabolism, local adhesion and immune inflammatory responses were significantly enriched in Pb120-Vs-Pb24 (P < 0.05), as were those related to the T cell receptor signalling pathway, with most being down-regulated compared to the preceding stage (Pb24-Vs-Pb0). In PBMCs there were some changes in glucose metabolism, local adhesion and the immune inflammatory response (Pb120-Vs-Pb0). In addition, up-regulated DEGs, such as IL8, IL1β, and CCL2, and were significantly enriched in immune-inflammatory related pathways compared to the uninfected stage, although they began to decline after 24 h.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine infectious pleuropneumonia is a highly contagious respiratory disease caused by Actinobacillus pleuropneumoniae (APP) and different clinical forms may be observed, from peracute to subacute or chronic (GomezLaguna et al. 2014; Sassu et al. 2017a, b). Acute pleural pneumonia shows high morbidity (Sassu et al. 2017a, b). Porcine infectious pleuropneumonia is a worldwide disease that causes huge economic losses to the pig industry. The prevalence of the disease has been on the rise, and is of serious concern to the modern pig industry (Liu et al. 2017; Sassu et al. 2017a, b).
Host peripheral blood contains a large number of immune cells, such as lymphocytes, mononuclear macrophages, and granulocytes, and is a site of immune clearance of pathogens (Mohammadi et al. 2014). Pigs produce a large number of cytokines in response to APP (Yu et al. 2013). A balance of cytokine responses is conducive to clearance of the pathogen, however, APP can induce excessive cytokine production (Auger et al. 2009; Skovgaard et al. 2009; Chen et al. 2011), which may contribute to the high mortality (Hsu et al. 2016; Reed et al. 2015; Tisoncik et al. 2012).
The latest generation of high-throughput sequencing technology has the advantage of allowing fast and accurate determination of almost all transcript information for a particular cell, tissue or organ in a given state (Chen et al. 2013; He et al. 2016; Klitgaard et al. 2012; Yu et al. 2013). In this study we have determined the transcriptome in order to understand the transcription levels of immune related molecules, such as receptors and cytokines, in peripheral blood at different infection stages by transcriptional sequencing. This helps to reveal the immune response mechanism of the host to APP.
With a view to better understanding the immune response of the host to APP, in this study, peripheral blood mononuclear cells (PBMCs) were isolated from the blood of pigs at different time points (0, 24 and 120 h) post infection and their transcriptome determined by Illumina HiSeq 4000 high-throughput sequencing. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the DEGs revealed the close relationship between the peripheral blood and inflammatory response from the perspective of molecular function, cell composition, biological processes and signalling pathways.
Materials and methods
Bacterial strains and animals
A. pleuropneumoniae serotype 5 (L20) was used for the challenge experiments. The strain was cultured using BactoTM Brain Heart Infusion (BHI) medium (Becton, Dickinson and Company, USA), and intranasal infection performed by nasal dropping 4 mL APP diluent (total containing 3.1 × 1010 CFU) into the nostrils (Yu et al. 2013).
A total of 21 specific-pathogen-free 35-day-old Landrace pigs whose body weight was approximately 10 kg were obtained from closed herds. Piglets were monitored from birth until experiments were carried out. During this period of time, the piglets were not infected with respiratory diseases. All animals were tested for the absence of APP antibodies using ELISA (the measured antigen was an APP cell lysate) (Halli et al. 2014; Wallgren and Persson 2000), and an X-ray machine was utilized to observe the respiratory system. Antibiotic-free pelleted food and water were provided ad libitum during the experiment. All animal experimental procedures were performed in strict accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China (1988.11.1).
Porcine pleural pneumonia model
The piglets were anaesthetized intraperitoneally with 25 mg/kg bw sodium pentobarbital (2.5%), and were inoculated intranasally with a bacterial suspension. Twenty-one pigs were randomly divided into seven groups (n = 3); of these, 18 pigs were experimentally infected with L20 in an intranasal infection model, and three piglets from the control group were inoculated with physiological saline by the same means. Clinical symptoms, including spirit, appetite, breath frequency, cough, nasal secretion, cyanosis, and body temperature, were continuously monitored (Reiner et al. 2014a, b). The X-ray machine was utilized to observe the respiratory system, and the lungs were observed for lesions at 0/6/12/24/48/120/168 h after infection. Broncheoalveolar lavage fluid (BALF) and lung homogenate were collected from each infection stage for colony counting. The method of BALF acquisition and colony counting was based on previous studies (Baltes et al. 2001). Lung homogenates were prepared by taking equal amounts of lung tissue from various areas of the lungs, as previously described by Reiner et al. (2014a, b). The pigs were euthanized with sodium pentobarbital (400 mg/mL at a dose of 500 µL/kg) by intravenous injection into the ear.
PBMC protocol and RNA sequencing
Peripheral blood samples were collected from the anterior vena cava of piglets at different infection stages. These piglets came from control group (Pb0:0 h), from 3 piglets at the stage where they were showing slight symptoms of infection (Pb24:24 h post-infection), and from another 3 piglets at the severe infection symptom stage (Pb120:120 h post-infection). PBMCs from the three groups of peripheral blood were extracted using the PBMC separation kit (TBDscience Biotech Co., Tianjin, China) according to the manufacturer’s instructions. Three duplicate samples of each group (0/24/120 h) were mixed in equal amounts. Then, the total RNA of the PBMCs was extracted and was treated with DNase I (Takara Bio., Dalian, China) to remove DNA. More than 20 μg of qualified total RNA from each sample (0/24/120 h) was used for RNA sequencing using an Illumina HiSeq™ 4000 platform (QL Bio., Beijing, China). For specific methods, please refer to the previous research reports (Huang et al. 2017).
Quantification and DEG analysis of transcripts
The raw data (raw reads) were first processed through quality control (QC), including removing the raw reads with an adapter or poly-N sequences and the low quality reads, in order to obtain clean data (clean reads). The Q20 parameters (the proportion of reading bases whose error was < 1%), the GC contents, sequencing randomness, etc. were evaluated after the first QC. All the following analyses were based on high-quality, clean data.
In this study, we used the SOAPaligner/SOAP2 (Li et al. 2009) tool to count the clean reads mapped to each gene. The reads per kb per million reads (RPKM) were used to calculate the expression level for each gene (Mortazavi et al. 2008). Furthermore, hypothesis testing was performed based on the binomial distribution (Volkmann et al. 2017), the pre-set false discovery rate (FDR) cannot exceed 0.05 (Benjamini and Yekutieli 2001), and the DEGs between the two samples were screened out. In our analysis, the DEGs were defined as an FDR ≤ 0.001 and an RPKM ratio of more than 2 times.
GO and KEGG enrichment analysis
The predicted gene sequences were aligned to NR databases through BLAST (version 2.2.23), and the related information was extracted from the Gene Ontology (http://www.geneontology.org/) database using Blast2GO software. GO classification (Ashburner et al. 2000) was achieved using FunRich software (BenitoMartin and Peinado 2015; Pathan et al. 2015). Up- and down-regulation of DEGs involved in the biological processes and signalling pathways, respectively, were analysed, and KEGG significant enrichment analysis of DEGs was performed using Database of Annotation, Visualization, and Integrated Discovery (DAVID) Bioinformatics Resources 6.8 (https://David.ncifcrf.Gov/) (Huang et al. 2008, 2009). We further analyzed the signaling pathways at different stages by using Cytoscape ClueGo (Bindea et al. 2009) software, trying to find a “bridge” that participates in the regulation of two or more signal pathways, which can provide new clues for further research. The P value represents the enrichment degree of the DEGs in GO or KEGG terms. Corrected P < 0.05 were considered significantly enriched (Miao et al. 2017).
Quantitative real time-PCR validation of DEGs
We selected representative genes for the validation of the transcription data by qRT-PCR analysis. These included candidate genes IL-1β, IL-18, CCR10, signal transducer and activator of transcription 4 (STAT4), S100 calcium-binding protein A12 (S100A12), BCL2-related protein A1 (BCL2A1), and were selected based on their differential expression as representatives of inflammation and immunity, cell proliferation and differentiation, cell migration, adhesion, apoptosis and other functional categories. β-actin was used as an endogenous control, designed using the PrimerQuest Tool (http://sg.idtdna.com/Primerquest/Home/Index). The SYBR Green method was used for qRT-PCR amplification. The qRT-PCR conditions were 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s and 60 °C for 45 s. The final melting curve analyses for all primer sets resulted in single product-specific melting curves. The relative expression level of each gene was calculated using the 2−ΔΔCt method.
Statistical analyses
Enrichment for GO terms for individual comparisons was performed by the Storey-Tibshirani method using the FunRich software (BenitoMartin and Peinado 2015; Pathan et al. 2015). Enrichment for KEGG terms for individual comparisons was performed by the Benjamin method using DAVID Bioinformatics Resources 6.8 (https://David.ncifcrf.Gov/) (Huang et al. 2008, 2009).
Results
Establishment of pneumonia model of APP-infected piglets
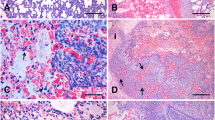
The clinical manifestations of the APP-infected piglets were observed as previously described (Reiner et al. 2014a, b). Piglets began to develop symptoms, such as dyspnoea, loss of appetite and elevated body temperature, at 24 h. Their nasal secretions increased, and their clinical symptoms increased after 120 h. The lungs of the different infection periods were observed using X-ray. X-ray examination showed that the lungs gradually appeared as cloudy shadows, and developed from punctate to flaky. The lungs’ texture gradually thickened and shadows appeared at 24 h. As the infection time increased, the texture gradually became unclear in the lungs, indicating that inflammation had increased. The inflammation of the lungs was maximal at 120 h and then gradually subsided (Fig. 1a). Lung pathology was observed. Inflammatory cell infiltration and necrosis occurred at 6 h. The most severe inflammation occurred at 120 h (Fig. 1b). Lung homogenate (Fig. 1c) and alveolar lavage fluid (Fig. 1d) were collected for bacterial counts, which showed that the largest number of bacteria colonized at 48 h. Based on the above results, 24 and 120 h PBMC samples were processed by RNA-sequencing.

Establishment of the pneumonia model of APP-infected pigs. a Dorsal–ventral (up) and lateral (down) thoracic radiographic projections in APP infected pigs at different infection stages; the lungs gradually appeared cloudy shadows and developed from punctate to flaky. The lungs’ texture gradually thickened and shadows appear at 24 h, and the texture gradually became unclear. b Pathological changes of the lung tissue (HE); Inflammatory cell infiltration and necrosis occurred at 6 h. The most severe inflammation occurred at 120 h. c, d Number of colonized bacteria. The number of bacteria colonized increases with the time of infection, and the largest number of bacteria colonized appear at 48 h
Transcriptomic analysis of PBMCs
RNA-Seq quality assessment
To identify DEGs, PBMCs from the three different infection stages were studied by sequencing the extracted RNA of the different samples. The procedure generated a total of 55, 667, 904; 63, 857, 856, and 46, 927, 328 raw reads for the 0 h-infection group (Pb0), 24 h-infection group (Pb24), and 120 h-infection group (Pb120), respectively. An overview of the sequencing and assembly is shown in Table S1. The key indicators of the clean data, including Q20 and GC%, after the first quality control of the raw data were determined (Table S2), and the reads accounted for more than 80% of the total raw reads (Fig. S1). The number of clean reads that were aligned to the reference gene was determined using the SOAPaligner/SOAP2 tool (Table S3), which provided the general information for further transcriptome analysis. Clean reads in the reference gene distribution were relatively uniform, indicating that the randomness of the RNA-Seq was good (Fig. S2). The sequence data generated in this study were considered suitable for further transcriptome analysis.
Identification of differentially expressed genes (DEGs) between the different groups
As shown in Table 1, a total of 3818 differentially expressed genes were detected among the 24 h-infection group (Pb24) and the uninfected group (Pb0). The expression level of 938 of the 3818 genes were up-regulated, and the 2880 other genes were shown to have down-regulated expression when infected with APP for 24 h. When the 120 h-infection group (Pb120) was compared to the 24 h-infection group (Pb24) and uninfected group (Pb0), we identified 4809 DEGs (3468 up-regulated and 1341 down-regulated), and 2690 DEGs (1402 up-regulated and 1228 down-regulated), respectively.
Gene ontology (GO) analysis of the differentially expressed genes
At the “Pb24-Vs-Pb0” stage, DEGs were predominantly annotated as being in the cytoplasmic, nucleus and platelet membrane compartments, and were mainly enriched in the regulation of nucleic acid metabolism, and signal transduction. However, the amount of DEGs was not significantly enriched (corrected P > 0.05) in the above biological processes at this stage (Fig. 2a). When Pb120 was compared to Pb24 (Pb120-Vs-Pb24), the DEGs were mainly annotated as being in the lysosome, Golgi apparatus, endosome, and exosomes. Unlike the previous stage, DEGs were significantly enriched in protein serine/threonine kinase activity and nucleic acid metabolism (corrected P < 0.05) (Fig. 2b). As expected, the DEGs that encode cellular components of the T cell receptor complex assembly were significantly enriched (2E), as were those in biological processes of the immune response (corrected P < 0.05) when at the “Pb120-Vs-Pb0” stage (Fig. 2c).

Enrichment analysis of Gene Ontology at different infection stage. a Enrichment analysis of Gene Ontology in the “Pb24-Vs-Pb0” stage. b Enrichment analysis of Gene Ontology in the “Pb120-Vs-Pb24” stage. c Enrichment analysis of Gene Ontology in the “Pb120-Vs-Pb0” stage. The results of the three main categories: cellular component (CC), molecular function (MF) and biological process (BP). (Blue indicates CC; Purple indicates MF; Orange indicates BP)(D): Enrichment analysis of the up-regulated and down-regulated DEGs at different infection stage (Blue indicates 0–24 h infection group; Purple indicates 24–120 h infection group; Orange indicates 0–120 h infection group). The X-axis indicates the GO term. The Y-axis indicates the number and the degree of enrichment of the genes in a category. The GO terms presenting a Q-value/Benjamini < 0.05 were considered significantly enriched by the DEGs (TERM with no Q value/Benjamini on the Y axis, indicating no significant enrichment). (Color figure online)
We further analysed the biological processes of each stage, finding that the immune system process mainly involves the immune inflammatory response, cellular migration and chemotaxis, including chemokine-mediated signalling pathways, cellular response to interferon-γ, in the “Pb24-Vs-Pb0” stage (Fig. 2d). At this stage, the majority of up-regulated DEGs, such as CCL4, CXCL8, IL12β, IL18, and IL1 (Fig. S3), were enriched in the above immune process, such as chemokine-mediated signalling pathways (Fig. 2d). The data show that this may be the main stage for the significant expression of cytokines to facilitate the removal of pathogen. As infection time increased, down-regulated DEGs were mainly enriched in platelet aggregation relevant pathways (Fig. 2d). This suggests that APP may facilitate the invasion of the host by destroying platelet formation. Ultimately, these DEGs were enriched in the immune system regulation related to the immune response (Fig. 2c) when the 120 h-infection group was compared to the 0 h-infection group. DEGs associated with inflammation response were also mostly up-regulated (Fig. 2d) after 24 h infection, although some of the cytokines associated with the immune response were down-regulated (Fig. S3). All DEGs involved in the immune response and inflammatory response are summarized in Fig. S3.
KEGG enrichment analysis of DEGs
The DEGs from the “Pb24-Vs-Pb0” stage were mainly enriched in signalling pathways related to cell proliferation, differentiation and renewal and the maintenance of tissue morphology, such as the cytokine–cytokine receptor interactions, hematopoietic cell lineage, and ECM-receptor interactions. In addition, the DEGs are also enriched in the NOD-like receptor signalling pathway, chemokine signalling pathway and other signalling pathways related to the immune response (Fig. 3a). We further analysed this stage and found that the DEGs associated with the above pathways were mostly up-regulated (Fig. S3). The results of the KEGG analysis are consistent with the GO analysis. It is noteworthy that the down-regulation of DEGs related to the T cell receptor signalling pathway also appear to be significantly enriched (Fig. 3b).

Enrichment analysis of KEGG at different infection stage. a Analysis of KEGG at different stages of infection. The X-axis indicates the KEGG term. The Y-axis indicates the number and degree of enrichment of genes in a category (Blue indicates 0–24 h infection group; Purple indicates 24–120 h infection group; Orange indicates 0–120 h infection group) The KEGG terms presenting Benjamini < 0.05 were considered significantly enriched by the DEGs (TERM with no Benjamini on the Y axis, indicating no significant enrichment). b Enrichment analysis of up-regulated and down-regulated DEGs in the “Pb24-Vs-Pb0” stage. c Enrichment analysis of up-regulated and down-regulated DEGs in the “Pb120-Vs-Pb24” stage. d Enrichment analysis of up-regulated and down-regulated DEGs in the “Pb120-Vs-Pb0” stage. The X-axis indicatesthe ratio of the DEGs enriched in this pathway to the total DEGs. The Y-axis indicates the KEGG term. The count and negLog10_qValue indicates the number and degree of enrichment of genes in a category, respectively. *negLog10_qValue > 1.3 were considered significantly enriched by the DEGs. (Color figure online)
With the prolongation of the infection time, it was found that DEGs were more significantly enriched in the NF-kappa B signalling pathway, focal adhesion, the TNF signalling pathway and sugar metabolism (Fig. 3a). DEGs were enriched in the NF-kappa B pathway after 24 h of host infection. The result shows that the NF-κB signalling pathway plays an important role in the process of the immune response to APP. We further found that the expression of DEGs related to the NF-κB signalling pathways was down-regulated (Fig. 3c). When the 120 h-infection group was compared to the 0 h-infection group, it was found that DEGs were more significantly enriched in amoebiasis, rheumatoid arthritis, transcriptional misregulation in cancer and pertussis (Fig. 3a). We further found that the expression of DEGs related to the above pathways were up-regulated (Fig. 3d). From Fig. 3, we found that the down-regulated DEGs are more enriched in the T cell receptor signalling pathway.
We further analyzed the signaling pathways at different stages by using Cytoscape ClueGo (Bindea et al. 2009) software. We found that DEGs were also mainly enriched in cytokine–cytokine receptor interactions, chemokine signalling pathway, and proteoglycans in cancer (Fig. 4a). In addition, IL1, IL8, CCL2, CCL4, CSF1R and other cytokines are involved in the regulation of multiple signalling pathways, such as cytokine–cytokine receptor interactions and chemokine signalling pathway (Fig. 4a). With increasing infection time, the DEGs involved in the NF-ΚB signalling pathway, T cell receptor signalling pathway and focal adhesion, were significantly enriched (Fig. 4b). This result is basically the same as Fig. 3. Moreover, IL8, IL10, IL12, IFN-γ are also associated with two or more pathways (Fig. 4b). These cross-talk genes, which play a bridge role, are shown in Fig. 4.

The relationship between DEGs and signal pathways at different infection stages. a Its represents the infection stage of “Pb24-Vs-Pb0”. b Its represents the infection stage of “Pb120-Vs-Pb24”, respectively. Each cross-node represents a cross-talk gene (Different circles indicate signal pathways where DEGs are enriched and genes on branches indicate DEGs involved in the signaling pathway)
DEG identification by qRT-PCR
To verify the gene expression data obtained by RNA-Seq, qRT-PCR was employed to analyse the DEGs. Primer pairs for the 11 DEGs and the housekeeping gene β-actin are available (Table S4). The result of the qRT-PCR assay was consistent with the RNA-Seq results (Figure S4), suggesting that our sequencing data are accurate and reliable.
Discussion
In this study, a pneumonia model of APP-infected piglets was successfully established, and the PBMCs of the piglets from different infection stages were sequenced by Illumina HiSeq 4000 high-throughput sequencing technology for the first time. The data obtained from each stage were analysed, and then, differentially expressed genes, which were mainly enriched in cell components, molecular functions, biological processes and signalling pathways, were found in each different stage Changes in gene expression of the PBMCs at the different stages of APP infection were systematically summarized, providing new ideas for the study of the immunity and inflammation mechanisms, thus providing an important theoretical basis for the development of diagnostic agents and therapeutic drugs.
This is the first time that transcriptome analysis has been done on PBMCs obtained from different stages of APP-infected piglets. DEGs were found in this study, some of which were related to infection of APP, but the majority are not yet clear.
The effect of differentially expressed cytokines on the host
Our research indicates that cytokine–cytokine receptor interactions and the chemokine signalling pathway play an important role in APP infection. These two pathways were also significantly enriched during APP infection in the study of Li et al. (2017). Collectively, our study and that of Li et al. (2017) show that cytokines play an important role in the APP infection process. Many studies have also shown that piglets naturally or experimentally infected by APP show significantly increased expression of the cytokines IL-1, IL-8, and IL18 (Reiner et al. 2014a, b; Hsu et al. 2016; Wang et al. 2016; Chen et al. 2011). In our study, the expression of IL-1α, IL-1β, and IL-18 was significantly up-regulated in the early stage (Figures S3, S4 and S5), and this result is consistent with the previous studies. From Fig. 4, we can see that these DEGs are also important node molecules. The interleukin-1 family, which contains IL-1α, IL-1β, IL-18, and others, plays a central role in the regulation of immune and inflammatory responses to infections. These cytokines can enable transmigration of immunocompetent cells, such as lymphocytes tosites of infection (Contassot et al. 2012; GomezLaguna et al. 2014; Reed et al. 2015). It can be seen that the piglets’ defence and inflammatory responses were achieved by the expression and migration of inflammatory factors in the early infection of APP.
Interleukin 10 (IL-10) is an anti-inflammatory cytokine with multiple, pleiotropic, effects in immunoregulation and inflammation. The expression of IL-10 was significantly up-regulated in the infection stage (Figures S3 and S4). Previous studies have also reported that the expression of IL10 is significantly increased after APP infection (Li et al. 2017; Reiner et al. 2014a, b). IL10 has been shown to inhibit bacterial product-mediated induction of the pro-inflammatory cytokines IL-1β, IL-12, TNFα, and IFN-γ but also supports the survival of microorganisms in the host via inhibiting the cell-mediated immune response (Redford et al. 2011; Sassu et al. 2017a, b). Furthermore, IL10 is also involved in the regulation of the T cell receptor signaling pathway at 24–120 h infection stage (Fig. 4b). Thus, IL-10, which is significantly elevated, is not only beneficial to the anti-inflammatory response but may be critical to the immune response to APP.
DEGs such as CXCL8 and CCL4 (Figure S3 and Figure S4) induce lymphocyte recruitment to inflammatory sites, affecting the immune response to pathogens. CXCL8 (IL8) is an important chemokine, whose main role is to recruit neutrophils to the site of infection. However, an excessive secretion of IL8 can cause lung damage (Lin et al. 2013; GomezLaguna et al. 2014; Thacker 2006). APP pathogenicity can be enhanced by inducing porcine alveolar macrophages (PAMs) to release IL-8, which results in the recruitment of excessive inflammatory cells to the lungs (GomezLaguna et al. 2014; Wang et al. 2016). At present, most studies have focused on the host stimulation by APP, whose products, such as IL-8, lead to a more severe inflammatory response. However, the mechanism for the up-regulated expression of IL-8 in the early infection stage is not completely clear. The up-regulated genes were significantly enriched in the ECM-receptor interaction pathway at the “Pb0-Vs-Pb24” stage (Fig. 3b). YadA was reported to mediate Yersinia invasion of epithelial cells and the release of high levels of IL-8 by interacting with ECM (Karataev et al. 2008; Uliczka et al. 2009). Recent studies have shown that APP can express multiple adhesins involved in interaction with host ECM components, thereby enhancing adhesion and colonization of the upper respiratory tract of the host (Bujold and MacInnes 2016). The up-regulated expression of IL8 may be associated with changes in the ECM-receptor pathway, but further experimentation is required to determine this.
Chemokine (C–C motif) ligand 4 (CCL4), also known as macrophage inflammatory protein 1β (MIP-1β) (Maurer and von Stebut 2004; Ren et al. 2010), is crucial for infection and inflammation-related immune responses (Ren et al. 2010). CCL4 not only activates neutrophils and induces proinflammatory cytokine, such as IL-1, IL-6 and TNF-α, synthesis and release but also is associated with the process by which B cells and professional APCs recruit regulatory T-cells (Bystry et al. 2001). A recent RNA-seq study of the lungs of APP-infected mice found that the expression of CCL4 was up-regulated (Li et al. 2017). In our study, the expression level of CCL4 was also high at different infection stages (Figures S3 and S4), and which also affected the immune and inflammatory response.
S100A12 is a calcium-, zinc- and copper-binding protein that plays a prominent role in the regulation of the immune inflammatory response. S100A12 is a pro-inflammatory factor that is up-regulated at sites of inflammation. Its pro-inflammatory activity involves the promotion of cytokine production, recruitment of leukocytes, and regulation of leukocyte adhesion and migration (Foell et al. 2004; Yang et al. 2007). The S100A12 gene could also lead to the up-regulation of cell adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM1) and intercellular adhesion molecule-1 (ICAM-1) (Bowman et al. 2010; Kosaki et al. 2004; Li et al. 2014). That these latter two molecules may be involved in APP infection has been found by others (Reiner et al. 2014a, b; Li et al. 2017). S100A12 is considered an important marker for many inflammatory diseases, such as arthritis (Li et al. 2014). The expression of S100A12 was significantly increased in the early infection stage (Figure S4; Figure S5), suggesting that this may be the major expression period of the inflammatory factors.
Some pathways that may affect the host immune response
NF-κB, which controls the transcription of DNA as a protein complex, plays a key role in regulating the immune response to infection. The NF-κB signaling pathway is a key step of these signaling pathways, including the cytokine–cytokine receptor interactions, T cell receptor signaling pathway, focal adhesion, and the TNF signalling pathway (Fig. 4b), and regulated the multiple cytokines. Li et al. also showed the importance of NF-κB in the APP infection process (Li et al. 2017). As can be seen from Fig. 4b, TLR4, IL1B, IL8 and MIP-1β not only participate in the NF-κB signaling pathway, but also participate in the cytokine–cytokine receptor interaction, T cell receptor signaling, and other pathways. The interaction of IL-1β and IL-1R (Doyle and O’Neill 2006), as well as the LY96-TLR4 complex and LPS, had been proven to activate the NF-κB signalling pathway (Doyle and O’Neill 2006; Hayden et al. 2006; Renard et al. 1997), which releases IL-8, MIP-1β, BCL2-related protein A1 (BCL2A1) and VCAM1. In our data, the expression levels of these genes, such as IL8, MIP-1βand BCL2A1, were also significantly elevated (Figure S3 and Figure S4; Figure S5) in early APP infection stages. Some studies have also demonstrated a pivotal role of NF-κB in ApxI-induced IL-1β, IL-8, and TNF-α production (Hsu et al. 2016; Li et al. 2018; Chen et al. 2011). The expression of many pro apoptotic as well as anti-apoptotic genes, such as BCL2L1, was also found to be induced in inflamed lung tissue (Hedegaard et al. 2007). BCL2A1 can reduce the release of mitochondrial apoptotic cytochrome C and block the activation of caspase, which is a direct transcriptional target for NF-κB response to inflammatory mediators (Hind et al. 2015).
It was noted that up-regulated DEGs were significantly enriched in the amoebiasis and pertussis pathways at the “Pb0-Vs-Pb120” stage (Fig. 3a). The amoebiasis pathway can lead to reactive oxygen species (ROS) and other compound production, which influence neutrophil apoptosis and tissue damage (Harbort et al. 2015; Wang et al. 2014). Moreover, the production of IL-10 can inhibit the expression of IFN-γ, thus influencing the immune response (Wang et al. 2014). In a different study, the expression level of INF-γ began to decrease after 6 h and was significantly lower than normal group at 48 h (Brogaard et al. 2015). Lymphocyte antigen 96 (LY96) and TLR4 were all significantly up-regulated and enriched in the pertussis pathway. LY96 can form a complex with the cell surface TLR4 receptor that can recognize LPS and produce an immune response (Re and Strominger 2002). TLR4 has also been confirmed to be associated with APP infection (Brogaard et al. 2015). Toll-like receptor is an important pattern recognition receptor and plays an important role in innate immunity. Li’s study reports that infection by App activated the Toll-like receptor (TLR) and NOD-like receptor (NLR) signaling pathway, and caused up-regulation of TNF, IL1β, IL12, MIP-1β, CCL2 and other cytokines (Li et al. 2017). Our study also showed significant expression of TLR4, TLR7, and TLR10 following APP infection (Figure S3 and Figure S4), and DEGs significant enriched in NOD-like pathways (Fig. 3b). These results suggest that pattern recognition receptors (PRR) play an important role in the anti-APP infection process.
The expression of the key factors of the T-cell receptor signalling pathway, such as the linker for the activation of T-cells (LAT), CD40 ligand (CD40L), appear to be significantly down-regulated (Figure S3 and Figure S4). CD40 ligand (CD40L), also called CD154, is a protein that is primarily expressed on activated T-cells (Schonbeck and Libby 2001). CD40L is essential for germinal centre formation and the maturation of antibody affinity (Gardell and Parker 2017). Activation of endothelial cells by CD40L will lead to ROS, as well as chemokine and cytokine, production and expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) (Szmitko et al. 2003). This inflammatory reaction in endothelial cells promotes the recruitment of leukocytes to lesions (Szmitko et al. 2003). In our study, CD40L was significantly down-regulated after 24 h of APP infection in the host (Figure S3 and Figure S4). Other studies also showed similar changes in CD40 expression levels during APP infection (Li et al. 2017; Reiner et al. 2014a, b). This shows that APP may inhibit the host’s T cell immune response. There are some studies that have shown that CD40L can trigger the inhibition of the Fas signal-mediated apoptosis response by inhibiting caspase-8 cleavage (Meriem et al. 2016). Moreover, it was found that Fas signalling and caspase-8 were critical for APP infection-induced apoptosis and pathogenicity (Wang et al. 2016). These studies suggest that CD40L may play a significant role in the immunological pathogenesis of APP. VCAM-1 mediates the adhesion of immune cells, such as lymphocytes, to the vascular endothelium (Barreiro et al. 2002; Wu 2007). It functions in leukocyte-endothelial cell signal transduction, which plays a role in the development of rheumatoid arthritis (Laura et al. 2016; Toma et al. 2016). DEGs were also significantly enriched in the rheumatoid arthritis (RA) pathway at the 120 h-infection stage (Fig. 3a). This is consistent with the findings of Li (Li et al. 2017). One study has shown that tumour cells can escape T-cell immunity by overexpressing VCAM-1, which normally mediates leukocyte extravasation to the sites of tissue inflammation (Wu 2007). Both of these diseases may affect the host immune response through the same pathway.
Bacterial infection is the result of interaction between the host and bacterial microbial factors. Our data revealed changes in the transcriptional levels of cell proliferation, cell differentiation and other effectors of host PBMCs at different stages of APP infection. Understanding the change in the host’s immune response to APP at different stages of infection can elicit new avenues of research. Future work will explore the role of the signalling pathways and the DEGs related to the pathogenesis of APP infection to clarify the interaction mechanism between APP and the host, as well as provide new targets for the development of new drugs, vaccines, and diagnostic reagents.
Conclusion
Results of this study promote basic knowledge about APP affecting host responses. We revealed a close relationship between the peripheral blood and inflammatory response from the perspective of molecular function, cell composition, biological processes and signalling pathways. With increasing infection time, the host response mechanisms to APP changed, and we speculate that APP may have a mechanism of inhibiting the host T cell immune response. More investigations are necessary to reveal the interaction mechanism between the host and APP, which can provide an important theoretical basis for the development of diagnostic agents and therapeutic drugs.
Abbreviations
- APP:
-
Actinobacillus pleuropneumoniae
- PBMCs:
-
Peripheral blood mononuclear cells
- BALF:
-
Bronchoalveolar lavage fluid
- ELISA:
-
Enzyme linked immunosorbent assay
- DEGs:
-
Differentially expressed genes
- QC:
-
Quality control
- RPKM:
-
Reads per kb per million reads
- FDR:
-
Pre-set false discovery rate
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- DAVID:
-
Database of Annotation, Visualization, and Integrated Discovery
- PAMs:
-
Porcine alveolar macrophages
- qRT-PCR:
-
Quantitative real time-PCR
References
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29
Auger E, Deslandes V, Ramjeet M, Contreras I, Nash JHE, Harel J, Gottschalk M, Olivier M, Jacques M (2009) Host-pathogen Interactions of Actinobacillus pleuropneumoniae with porcine lung and tracheal epithelial cells. Infect Immun 77:1426–1441
Baltes N, Tonpitak W, Gerlach G, Hennig-Pauka I, Hoffmann-Moujahid A, Ganter M, Rothkötter H (2001) Actinobacillus pleuropneumoniae iron transport and urease activity: effects on bacterial virulence and host immune response. Infect Immun 69:472–478
Barreiro O, Yáñez-Mó M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sánchez-Madrid F (2002) Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 157:1233–1245
BenitoMartin A, Peinado H (2015) FunRich proteomics software analysis, let the fun begin! Proteomics 15:2555–2556
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug-into decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093
Brogaard L, Klitgaard K, Heegaard PM, Hansen MS, Jensen TK, Skovgaard K (2015) Concurrent host-pathogen gene expression in the lungs of pigs challenged with Actinobacillus pleuropneumoniae. BMC Genom 16:417
Bujold AR, MacInnes JI (2016) Attachment of Actinobacillus suis H91-0380 and its isogenic adhesin mutants to extracellular matrix components of the tonsils of the soft palate of swine. Infect Immun 84:2944–2952
Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG (2001) B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol 2:1126–1132
Chen ZW, Chien MS, Chang NY, Chen TH, Wu CM, Huang C, Lee WC, Hsuan SL (2011) Mechanisms underlying Actinobacillus pleuropneumoniae exotoxin ApxI induced expression of IL-1b, IL-8 and TNF-a in porcine alveolar macrophages. Vet Res 42:25
Chen XR, Xing YP, Li YP, Tong YH, Xu JY (2013) RNA-Seq reveals infection-related gene expression changes in Phytophthora capsici. PLoS ONE 8:e74588
Contassot E, Beer H, French LE (2012) Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med Wkly 142:w13590
Doyle SL, O’Neill LAJ (2006) Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol 72:1102–1113
Foell D, Frosch M, Sorg C, Roth J (2004) Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta 344:37–51
Gardell JL, Parker DC (2017) CD40L is transferred to antigen-presenting B cells during delivery of T-cell help. Eur J Immunol 47:41–50
GomezLaguna J, Islas A, Muñoz D, Ruiz Á, Villamil A, Carrasco L, Quezada M (2014) Infection dynamics and acute phase response of an Actinobacillus pleuropneumoniae field isolate of moderate virulence in pigs. Vet Microbiol 173:332–339
Halli O, Ala-Kurikka E, Wallgren P, Heinonen M (2014) Actinobacillus pleuropneumoniae seroprevalence in farmed wild boars in Finland. Am Assoc Zoo Vet 45:813–818
Harbort CJ, Soeiropereira PV, Von BH, Kaindl AM, Costacarvalho BT (2015) Neutrophil oxidative burst activates ATM to regulate cytokine production and apoptosis. Blood 126:2842–2851
Hayden M, West A, Ghosh S (2006) NF-kB and the immune response. Oncogene 25:6758–6780
He M, Xu J, He R, Shen N, Gu X, Peng X, Yang G (2016) Preliminary analysis of Psoroptes ovis transcriptome in different developmental stages. Parasite Vector 9:570
Hedegaard J, Skovgaard K, Mortensen S, Sørensen P, Jensen TK, Hornshøj H, Bendixen C, Heegaard PM (2007) Molecular characterisation of the early response in pigs to experimental infection with Actinobacillus pleuropneumoniae using cDNA microarrays. Acta Vet Scand 49:11
Hind CK, Carter MJ, Harris CL, Chan HTC, James S, Cragg MS (2015) Role of the pro-survival molecule Bfl-1 in melanoma. Int J Biochem Cell Biol 59:94–102
Bowman MH, Wilk J, Heydemann A, Kim G, Rehman J, Lodato JA, Raman J, McNally EM (2010) S100A12 mediates aortic wall remodeling and aorticaneurysm. Circ Res 106:145–154
Hsu C, Li S, Chang N, Chen Z, Liao J, Chen T, Wang J, Lin J, Hsuan S (2016) Involvement of NF-κB in regulation of Actinobacillus pleuropneumoniae exotoxin ApxI-induced proinflammatory cytokine production in porcine alveolar macrophages. Vet Microbiol 195:128–135
Huang DW, Sherman BT, Lempicki RA (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Huang Y, Yi Z, Jin Y, Huang M, He K (2017) Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese Nong-flavor liquor starter. Front Microbiol 8:1747
Karataev GI, Markov AR, Sinyashina LN, Miller GG, Klitsunova NV, Titova IV, Semin EG, Goncharova NI, Pokrovskaya MS, Amelina IP, Amoako K, Smirnov GB (2008) Comparative investigation of the role of the yadA, invA, and psaA genes in the pathogenicity of Yersinia pseudotuberculosis. Mol Genet Microbiol Virol 23:168–177
Klitgaard K, Friis C, Jensen TK, Angen Y, Boye M (2012) Transcriptional portrait of Actinobacillus pleuropneumoniae during acute disease—potential strategies for survival and persistence in the host. PLoS ONE 7:e35549
Kosaki A, Hasegawa T, Kimura T, Iida K, Hitomi J, Matsubara H, Mori Y, Okigaki M, Toyoda N, Masaki H, Inoue-Shibata M, Nishikawa M, Iwasaka T (2004) Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. J Clin Endocrinol Metab 89:5423–5428
Laura M, Francesca M, Cristina L, Manuela C, Caterina RM (2016) Th1-Induced CD106 expression mediates leukocytes adhesion on synovial fibroblasts from juvenile idiopathic arthritis Patients. PLoS ONE 11:e154422
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li X, Tang J, Xu J, Zhu M, Cao J (2014) The inflammation related gene S100A12 is positively regulated by C/EBPβ and AP-1 in pigs. Int J Mol Sci 8:13802–13816
Li P, Xu Z, Sun X, Yin Y, Fan Y, Zhao J, Mao X, Huang J, Yang F, Zhu L (2017) Transcript profiling of the immunological interactions between Actinobacillus pleuropneumoniae serotype 7 and the host by dual RNA-seq. BMC Microbiol 17:193
Li B, Fang J, Zuo Z, Yin S, He T, Yang M, Deng J, Shen L, Ma X, Yu S, Wang Y (2018) Activation of porcine alveolar macrophages by Actinobacillus pleuropneumoniae lipopolysaccharide via the Toll-Like receptor 4/NF-κB-mediated pathway. Infect Immun 86:e617–e642
Lin CH, Yu MC, Chiang CC, Bien MY, Chien MH, Chen BC (2013) Thrombin-induced NF-κB activation and IL-8/CXCL8 release is mediated by c-Src-dependent Shc, Raf-1, and ERK pathways in lung epithelial cells. Cell Signal 25:1166–1175
Liu J, Ma Q, Yang F, Zhu R, Gu J, Sun C, Feng X, Du C, Langford PR, Han W, Yang J, Lei L (2017) B cell cross-epitope of Propionibacterium acnes and Actinobacillus pleuropneumonia selected by phage display library can efficiently protect from Actinobacillus pleuropneumonia infection. Vet Microbiol 205:14–21
Maurer M, von Stebut E (2004) Macrophage inflammatory protein-1. Int J Biochem Cell Biol 36:1882–1886
Meriem B, Nadim N, Daniel Y, Suzanne S, Nada A (2016) The interaction of CD154 with the α5β1 integrin inhibits Fas-induced T cell death. PLoS ONE 11:e158987
Miao Y, Shen Y, Xu Y (2017) Effects of inhibitors on the transcriptional profiling of gluconobater oxydans NL71 genes after biooxidation of xylose into cylonate. Front Microbiol 8:716
Mohammadi H, Mohammadnejad J, Yavari K (2014) human peripheral blood derived hematopoietic stem cell: history, the isolation methods and investigation of different parameters effects on their differentiation to the body cells. Int J Stem Cell Res Transplant 2:59–62
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Pathan M, Keerthikumar S, Ang C, Gangoda L, Quek CYJ, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, Bacic A, Hill AF, Stroud DA, Ryan MT, Agbinya JI, Mariadason JM, Burgess AW, Mathivanan S (2015) FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15:2597–2601
Re F, Strominger JL (2002) Monomeric recombinant MD-2 binds Toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem 227:23427–23432
Redford PS, Murray PJ, O’Garra A (2011) The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol 4:261–270
Reed DM, Paschalaki KE, Starke RD, Mohamed NA, Sharp G, Fox B, Eastwood D, Bristow A, Ball C, Vessillier S, Hansel TT, Thorpe SJ, Randi AM, Stebbings R, Mitchell JA (2015) An autologous endothelial cell: peripheral blood mononuclear cell assay that detects cytokine storm responses to biologics. FASEB J 29:2595–2602
Reiner G, Bertsch N, Hoeltig D, Selke M, Willems H, Gerlach GF, Tuemmler B, Probst I, Herwig R, Drungowski M, Waldmann KH (2014a) Identification of QTL affecting resistance/susceptibility to acute Actinobacillus pleuropneumoniae infection in swine. Mamm Genome 25:180–191
Reiner G, Dreher F, Drungowski M, Hoeltig D, Bertsch N, Selke M, Willems H, Gerlach GF, Probst I, Tuemmler B, Waldmann K, Herwig R (2014b) Pathway deregulation and expression QTLs in response to Actinobacillus pleuropneumoniae infection in swine. Mamm Genome 25:600–617
Ren M, Guo Q, Guo L, Lenz M, Qian F, Koenen RR, Xu H, Schilling AB, Weber C, Ye RD, Dinner AR, Tang WJ (2010) Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J 29:3952–3966
Renard P, Zachary MD, Bougelet C, Mirault ME, Haegeman G, Remacle J, Raes M (1997) Effects of antioxidant enzyme modulations on interleukin-1-induced nuclear factor kappa B activation. Biochem Pharmacol 53:149–160
Sassu EL, Bossé JT, Tobias TJ, Gottschalk M, Langford PR, Hennig-Pauka I (2017a) Update on Actinobacillus pleuropneumoniae -knowledge, gaps and challenges. Transbound Emerg Dis 65:72–90
Sassu EL, Ladinig A, Talker SC, Stadler M, Knecht C, Stein H, Frömbling J, Richter B, Spergser J, Ehling-Schulz M, Graage R, Hennig-Pauka I, Gerner W (2017b) Frequency of Th17 cells correlates with the presence of lung lesions in pigs chronically infected with Actinobacillus pleuropneumoniae. Vet Res 48:4
Schonbeck U, Libby P (2001) The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci 58:4–43
Skovgaard K, Mortensen S, Boye M, Poulsen KT, Campbell FM, Eckersall PD, Heegaard PMH (2009) Rapid and widely disseminated acute phase protein response after experimental bacterial infection of pigs. Vet Res 40:23
Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S (2003) New markers of inflammation and endothelial cell activation: part I. Circulation 108:1917–1923
Thacker EL (2006) Lung inflammatory responses. Vet Res 37:469–486
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76:16–32
Toma L, Sanda GM, Deleanu M, Stancu CS, Sima AV (2016) Glycated LDL increase VCAM-1 expression and secretion in endothelial cells and promote monocyte adhesion through mechanisms involving endoplasmic reticulum stress. Mol Cell Biochem 417:169–179
Uliczka F, Kornprobst T, Eitel J, Schneider D, Dersch P (2009) Cell invasion of Yersinia pseudotuberculosis by invasin and YadA requires protein kinase C, phospholipase C-γ1 and Akt kinase. Cell Microbiol 11:1782–1801
Volkmann ER, Hoffmannvold AM, Chang YL, Jacobs JP, Tillisch K (2017) Systemic sclerosis is associated with specific alterations in gastrointestinal microbiota in two independent cohorts. BMJ Open Gastroenterol 4:e134
Wallgren P, Persson M (2000) Relationship between the amounts of antibodies to Actinobacillus pleuropneumoniae serotype 2 detected in blood serum and in fluids collected from muscles of pigs. Zoonoses Public Health 47:727–737
Wang X, Luo F, Zhao H (2014) Paraquat-induced reactive oxygen species inhibit neutrophil apoptosis via a p38 MAPK/NF-kB—IL-6/TNF-a positive-feedback circuit. PLoS ONE 9:e93837
Wang L, Qin W, Zhang J, Bao C, Zhang H, Che Y, Sun C, Gu J, Feng X, Du C, Han W, Richard PL, Lei L (2016) Adh enhances Actinobacillus pleuropneumoniae pathogenicity by binding to OR5M11 and activating p38 which induces apoptosis of PAMs and IL-8 release. Sci Rep 6:24058
Wu T (2007) The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Res 67:6003–6006
Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, Wang HW, Hampartzoumian T, Simpson JL, Gibson PG, Hunt J, Hart P, Hughes JM, Perry MA, Alewood PF, Geczy CL (2007) S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immun 119:106–114
Yu S, Zuo Z, Cui H, Li M, Peng X, Zhu L, Zhang M, Li X, Xu Z, Gan M, Deng J, Fang J, Ma J, Su S, Wang Y, Shen L, Ma X, Ren Z, Wu B, Hu Y (2013) Transcriptionalprofiling of hilar nodes from pigs after experimental infection with Actinobacillus Pleuropneumoniae. Int J Mol Sci 14:23516–23532
Funding
This research was supported by a Grant from the National Natural Science Foundation of China (No. 31520103917).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
All authors approved the final version of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10482_2018_1126_MOESM5_ESM.tif
Fig. S1. Summary of the sequence assembly. After filtering, the percentage of clean data in all samples is more than 80%, indicating that this sequencing machine is stable and has no other operation errors (TIFF 134 kb)
10482_2018_1126_MOESM6_ESM.tif
Fig. S2. The Reads random distribution. On the X-axis, 0–1 represents the relative position of the gene normalized in the 5′ – > 3′ direction, the y-axis represents the number of Reads in the mapping. In the case of no random bias in sequencing, the entire curve should be stable, indicating that reads are evenly distributed in all parts of the gene, and the randomization of this sequencing is qualified (TIFF 126 kb)
10482_2018_1126_MOESM8_ESM.tif
Fig. S4. qRT-PCR validation of ten selected genes that were differentially expressed in the transcriptomic data. Data are representative of three independent experiments and error bars areplotted as mean ± SEM. The height of each bar chart represents the mean average of sample-specifi 2 −ΔΔCt values (*P < 0.05, **P < 0.01) (TIFF 130 kb)
Rights and permissions
About this article

Cite this article
Jiang, H., Zhu, R., Liu, H. et al. Transcriptomic analysis of porcine PBMCs in response to Actinobacillus pleuropneumoniae reveals the dynamic changes of differentially expressed genes related to immuno-inflammatory responses. Antonie van Leeuwenhoek 111, 2371–2384 (2018). https://doi.org/10.1007/s10482-018-1126-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-018-1126-5