
Abstract
The relationships between plants and endophytic bacteria significantly contribute to plant health and yield. However, the microbial diversity in leaves of Eucalyptus spp. is still poorly characterized. Here, we investigated the endophytic diversity in leaves of hybrid Eucalyptus grandis x E. urophylla (Eucalyptus “urograndis”) by using culture-independent and culture-dependent approaches, to better understand their ecology in leaves at different stages of Eucalyptus development, including bacteria with N2 fixation potential. Firmicutes, Proteobacteria (classes alpha-, beta- and gamma-) and Actinobacteria were identified in the Eucalyptus “urograndis” endophytic bacterial community. Within this community, the species Novosphingobium barchaimii, Rhizobium grahamii, Stenotrophomonas panacihumi, Paenibacillus terrigena, P. darwinianus and Terrabacter lapilli represent the first report these bacteria as endophytes. The diversity of the total endophytic bacteria was higher in the leaves from the ‘field’ (the Shannon–Wiener index, 2.99), followed by the indices obtained in the ‘clonal garden’ (2.78), the ‘recently out from under shade (2.68), ‘under shade’ (2.63) and ‘plants for dispatch’ (2.51). In contrast, for diazotrophic bacteria, the highest means of these indices were obtained from the leaves of plants in the ‘under shade’ (2.56), ‘recently out from under shade (2.52)’ and ‘field’ stages (2.54). The distribution of the endophytic bacterial species in Eucalyptus was distinct and specific to the development stages under study, and many of the species had the potential for nitrogen fixation, raising the question of whether these bacteria could contribute to overall nitrogen metabolism of Eucalyptus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eucalyptus is one of the most cultivated plant genera in the world, particularly in tropical and subtropical regions, due to its rapid growth and adaptability and the commercial value of its wood (Brooker 2000). The rapid growth of E. grandis demands a high availability of nutrients, specifically nitrogen (Smethurst et al. 2004). Nitrogen can be obtained by the plant via biological fixation, which can be performed by both free-living and endophytic bacteria (Summan et al. 2005; Reinhold-Hurek and Hurek 2011, Puri et al. 2016a). In this aspect, endophytes improve physical characteristics of plants (height, biomass, and floral bud emergence) in N-limited conditions (Puri et al. 2016b). Studies of the microbial community of E. grandis can improve the knowledge about plant–microorganism interactions and how these interactions can contribute to forest nutrition.
The leaves of plants host a wide variety of microorganisms of ecological importance, particularly bacteria and fungi (Vandenkoornhuyse et al. 2015). This diversity of microorganisms that influences the health and development of different host organs (Brader et al. 2014) as roots, stems and leaves, flowers and seeds (Santoyo and Moreno-Hagelsieb 2016) has been the subject of several studies (Brader et al. 2014). During colonization, endophytic microorganisms adapt to the specific environment of the plant body (Bulgarelli et al. 2013), probably by plant recruitment of microbes. The evolution of obligate endosymbiotic relationship with the host is accompanied by bacterial genome reduction, what means that the evolutionary process among endophytic bacteria and plant is towards an obligate mutualism (Farrar et al. 2014). Endophytic microbes are able to stimulate plant growth through nitrogen fixation (Puri et al. 2016a) as well as the synthesis of auxins, auxin analogs and gibberellins (Waqas et al. 2012). The Eucalyptus–endophytic bacteria interaction is poorly described, and the majority of previous studies have focused on root microorganisms (Bonito et al. 2014; Da Silva et al. 2014).
The molecular analyses of microbial communities via cultivation-dependent (microbial isolation on solid media by classic microbiological methods) (Miguel et al. 2013; Weber et al. 2013; Szilagyi–Zecchin et al. 2014) and cultivation-independent (a metagenomic approach) (West et al. 2010; Jackson et al. 2013; Oliveira et al. 2013) methods have revealed the microbial diversity of a variety of agronomically important plants. One of the cultivation-independent methods used to evaluate the endophytic microbial community is the denaturing gradient gel electrophoresis (DGGE) of 16S rRNA amplicons generated by PCR using universal primers (Wemheuer et al. 2016). Additionally, methods consisting of microbial cultivation followed by DNA extraction, 16S rRNA amplification and phylogeny reconstruction were also performed (Fidalgo et al. 2016).
Despite the existence of studies on the bacterial diversity in Eucalyptus spp. (Ferreira et al. 2008; Procópio et al. 2009; Castellanos et al. 2010), the importance of endophytic bacteria as part of the Eucalyptus microbial community (Brader et al. 2014) and Brazil’s status as the world’s largest producer of Eucalyptus spp., little is known regarding the endophytic microbiota associated with hybrid E. grandis × E. urophylla (“urograndis”). In addition, the variations in the structure of this endophytic community as a function of plant age and stage of development in commercial nurseries are completely unknown. Similarly, the diversity of endophytic bacteria with the potential for nitrogen fixation associated with Eucalyptus “urograndis” remains to be characterized in terms of plant age and stage of development.
Given the above gap, the objective of this study was to evaluate the diversity of the endophytic bacteria, including those with nitrogen fixation potential, associated with Eucalyptus “urograndis” leaves at various stages of development in commercial nurseries and in the field combining dependent and independent cultivation methods.
Materials and methods
Study sites
This study was conducted using seedlings of the hybrid E. grandis × E. urophylla (Eucalyptus “urograndis”) at different stages of development in nurseries and field sites owned by Celulose Nipo-Brasileira (CENIBRA) company in Belo Oriente city, MG, Brazil. Five plant development stages were sampled: ‘clonal garden’ (CG– the production of sprouts from minicuttings), ‘under shade’ (S –six days in the greenhouse receiving 50 % radiation), ‘recently out from under shade’ (OS—first selection with the exclusion of undesirable plants), ‘dispatch’ (DP—the stage prior to planting). Only disease-free plants (determined as those plants with no visual symptom or signal of diseases) with similar standard size and leaf pairs were used) and ‘field’ (18-month-old plants). The average of total radiation in the area was 15.02 MJ/m2. The leaf samples from each stage were collected in quadruplicate.
The ‘under shade’, ‘recently out from under shade’ and ‘dispatch’ plants were grown in a substrate composed primarily of carbonized rice straw and vermiculite. Fertilization of the plants was performed using simple superphosphate and a nutrient solution (Table 1).
The field study site was a CENIBRA forest planted with the hybrid E. grandis × E. urophylla (E. urograndis) at Guanhães city, Minas Gerais, Brazil. The natural vegetation was originally a semi-deciduous forest that was replaced by pastures. The field is currently in its first planting with Eucalyptus. The historical yield of the hybrid is 340 m3 ha−1/6-year rotation. The age of the plants at the time of collection was 18 months.
The site is located at 19° 24′ 54″ S latitude, 42° 25′ 56″ W longitude at an elevation of 318 m. The soil at the study site is highly weathered, with relief conditions ranging from wavy to strongly wavy, with slopes between 20 and 45 % (Scolforo et al. 2008). The most representative soil class is typic acric red latosol with a prominent to moderate clayey texture that is alic, kaolinitic, kaolinitic–oxidic or gibbsitic–oxidic and hypoferric or mesoferric.
The site’s Cwa (“C” indicates temperate climate and warm temperate climate, “w” indicates dry winters or winter with low precipitation, and “a” indicates hot and humid summers with summer rainfalls) climate is mesothermal and has a dry winter and rainy summer, according to the Köppen classification (Köppen 1948). In 2014, the maximum, mean and minimum temperature averages at the site were 23.5, 19.5 and 15.5 °C, respectively, with a mean relative humidity of 74 % and a mean rainfall and water deficit of 56.39 and 8.38 mm, respectively (CENIBRA, Gaspar climatological station).
The Eucalyptus leaf samples were randomly collected in quadruplicate at different stages of plant development: ‘clonal garden’, ‘under shade’, ‘recently out from under shade’, ‘dispatch’ and ‘field’ (18-month old plants). Sampling of plants of ‘clonal garden’, ‘under shade’, ‘recently out from under shade’ and ‘dispatch’ was accomplished by randomly demarcating four blocks in the nursery. Each block represented a repetition. The location of each point was chosen dividing each block into four equal parts throughout the growing area have a more representative sampling procedure. These points were then randomly (by lot) selected and according to the drawing, the leaves were sampled and mixed on composite samples. The sampling of leaves from ‘field’ consisted in the selection of four 18-month-old plants, each plant representing a repetition. The trees were located in a sub-area of 162 m2 (about 16 trees), with a spacing of 3.33 × 3 m. This area was divided into four equal parts and each was chosen by lot a tree. Three regions (upper, middle and lower region) of plant canopy were sampled. Aiming to have a more representative sample from the whole canopy, leafs, were collected from proximal, median and distal parts of the stem for each region. The leaves were sampled and mixed to form composite samples. The leaf samples were stored in sterile plastic bags and immediately transferred to Styrofoam coolers containing ice, transported to the Laboratory of Microbial Ecology and stored at −20 °C for 15 days before processing for diversity analyses.
Surface decontamination of the leaves
Eight healthy leaves from each stages of plant development were surface decontaminated after being washed under tap and distilled water. The material was then immersed twice in distilled water and phosphate buffer (0.05 mmol L−1), pH 7, immersed in 70 % ethanol (v/v) for 1 min, kept in a container containing sodium hypochlorite (5 %) + 0.05 % (v/v) Tween 80 for 5 min, quickly immersed in 70 % ethanol (v/v) and reimmersed in hypochlorite + Tween 80 for 15 min (Miguel et al. 2013; Oliveira et al. 2013). The surface decontamination procedure was performed in laminar flow cabinet at room temperature. After repeating this process, the leaves were individually washed into separate tubes of sterile distilled water and placed in tubes containing 10 mL of R2A culture medium (Reasoner and Gelrdreich 1985) and incubated in bacteriological incubator at 28 °C for 72 h, to verify if the disinfestation protocol was efficient to remove or inactivate all bacteria in the surface of the leaves. Only those leaves with no microbial growth detected in the tubes after incubation were used for metagenomic DNA extraction and isolation of endophytic bacteria.
Extraction of metagenomic DNA from the leaves
After maceration in liquid nitrogen, DNA was extracted from leaves in five different plant development stages (eight leaves per stage). The macerated leaf material was transferred to 2-mL tubes, resuspended and kept in 1000 µL of extraction buffer [(4 % (w/v) cetyl-trimethylammonium bromide (CTAB), 1.4 mol L−1 NaCl, 20 mmol L−1 EDTA, 100 mmol L−1 Tris–HCl, pH 8 and 1 % (w/v) polyvinylpyrrolidone) containing 0.2 % (v/v) β-mercaptoethanol]. The tubes were incubated in a water bath at 37 ºC for 60 min with stirring at constant intervals, followed by the addition of 100 µL of 20 % SDS (sodium dodecyl sulfate) and 30 µg of proteinase K (Sigma), before incubation in a water bath for about 40 min with stirring at constant intervals. The mixture was subjected twice to deproteinization using phenol:chloroform:isoamyl alcohol (25:24:1). After incubation on ice for 30 min and centrifugation at 13,000 × g for 10 min, the aqueous phase was collected. The DNA was precipitated by mixing the supernatant with 50 µL of 7.5 M ammonium acetate at −20 °C for 30 min, and the mixture was centrifuged for 10 min at 7500 × g. The DNA pellet was washed twice in cold 70 % ethanol and absolute ethanol (Sigma) and resuspended overnight in 100 µL of water after drying in a laminar flow cabinet. The concentration and purity of the extracted DNA was measured by its optical density at 260–280 nm (NanoDrop® ND-1000, Thermo Fisher Scientific Inc.). The integrity of the DNA was checked on an agarose gel (0.8 % w/v) stained with Gel Red® 1000X and visualized in the molecular imaging system L-pix Chemi (Loccus Biotechnology, São Paulo, SP, Brazil).
Diversity analysis of the total and diazotrophic bacteria
Nested PCR followed by DGGE was used to assess the diversity of the total bacteria in the Eucalyptus leaves. For the first PCR, total DNA was used as a template for the amplification of the bacterial 16S rRNA gene using the pair of primers P027/1392R (Heuer et al. 1997; Blackwood et al. 2005). The amplicons were used as templates for the second PCR using the pair of primers 984GC/1392R (Heuer et al. 1997; Blackwood et al. 2005).
The PCRs were performed in a final volume of 25 μL containing 5 μL of GoTaq Flex® Reaction Buffer, 200 μM deoxyribonucleotide triphosphates (dNTPs), 2.0 U of GoTaq Flex DNA polymerase, 3.0 mM magnesium chloride, 0.16 μM of each primer, 20 ng of the DNA template and sterile deionized water. The amplification conditions were: initial denaturation at 94 °C for 4 min, 35 cycles of denaturation at 94 °C for 1 min, annealing at 47 °C for 1 min, extension at 72 °C for 2 min and a final extension at 72 °C for 10 min.
The diversity of the potential nitrogen fixing bacteria in the Eucalyptus leaves was assessed using the same method with two different pair of primers, polF/polR (Poly et al. 2001) and polFGC/polR. The reactions were performed in a 50-μL volume containing 1× buffer, 1.5 mM MgCl2, 0.12 mM dNTPs, 0.3 μM of each primer, 1 U of Taq polymerase and 10 ng of the template, and subjected to an initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 0.5 min, 55 °C for 1 min, 72 °C for 2 min and a final extension at 72 °C for 10 min (Mapelli et al. 2013). DNA from pure culture of Agrobacterium tumefaciens was used as template for the positive control in the PCRs targeting the nitrogen-fixing bacteria, and sterile water was used as template for the negative controls.
Amplicons generated after the second round of PCR were analyzed by DGGE (DCode System, Bio-Rad Inc., CA, USA). A DNA mixture of 16S rRNA amplicons from pure cultures of Nocardioides thermolilacinus, Bacillus cereus, Streptomyces setonii, Clavibacter michiganensis, Pectobacterium carotovorum, Pseudomonas putida, P. syringae, Xanthomonas vesicatoria and Ralstonia solanacearum was used as an external marker to normalize the gels using BioNumerics® version 7.1 (Applied Maths, Kortrijk, Belgium). The PCR products were applied to an 8 % polyacrylamide gel (37.5:1 acrylamide—bisacrylamide) (Sigma) using 1× TAE (40 mmol L−1 Tris–HCl, pH 8.0, 20 mmol L−1 acetic acid, 1 mmol L−1 EDTA, pH 8), 0.09 % (v/v) TEMED and 0.07 % (w/v) ammonium persulfate as a buffer for denaturing gradient electrophoresis. The denaturing gradient was optimized at 40–60 % urea/formamide (100 % denaturant containing 7 mol L−1 urea and 40 % (v/v) formamide). DGGE was performed at 60 V for 20 h at a constant temperature of 60 °C in TAE 1× buffer. The DNA fragments in the gel were stained for 20 min in TAE 1× buffer containing SYBR Gold® 1X dye (Invitrogen, Carlsbad, CA, USA) and gel images were obtained using L-pix Chemi (Loccus Biotechnology, São Paulo, SP, Brazil).
Band profiles of DDGE gels were analysed as four repetitions in the BioNumerics® software version 7.1 (Applied Maths, Kortrijk, Belgium) and the gel with better resolution was selected to be shown. The ‘Richness’ variables of the total and nitrogen fixing bacteria were estimated by the program using a binary matrix in which the presence or absence of the band corresponding to each operational taxonomic unit (OTU) was encoded as a one (1) or zero (0), respectively. The structure of these communities was compared using the Dice similarity coefficient and the unweighted pair group method with arithmetic mean. Richness and diversity analyses were performed using the software PAST (Hammer et al. 2001), wherein diversity is estimated using the Shannon–Wiener index, and graphs were constructed using SigmaPlot (Systat Software, Inc., 2008). Statistical analyses were performed in Minitab version 15 (Minitab Inc., State College, PA, USA) (Minitab 2006) using Tukey’s test at 5 % probability.
Isolation of endophytic bacteria
The disinfected leaves in each Eucalyptus development stage were used to establish the density of culturable bacteria per g dry weight of leaf ([colony-forming units (CFU)] g−1). The leaves were homogenized in tubes containing 10 mL of potassium phosphate buffer (0.05 M, pH 7). The suspension was filtered through sterile gauze, and the filtrate was serially diluted in potassium phosphate buffer (0.05 M, pH 7.0). Aliquots (100 μL) were spread onto solid R2A medium and incubated at 28 °C for 72 h.
DNA extraction from pure cultures
Total DNA was extracted from pure cultures grown in liquid R2A medium using a DNA purification kit (Promega™, Madison, USA) following the manufacturer’s instructions. The integrity and quantity of the extracted DNA was visualized on an agarose gel (0.8 % w/v) stained with Gel Red® 1000X, and images were obtained using L-pix Chemi (Loccus Biotechnology, São Paulo, SP, Brazil).
Amplification and sequencing of the 16S rRNA
The universal primers P27F/1392R (Heuer et al. 1997; Blackwood et al. 2005) were used to amplify the 16S rRNA gene from the bacterial isolates from Eucalyptus leaves by BOX-PCR. The reactions contained about 20 ng of DNA, buffer (Promega), 2.25 mmol MgCl2, 210 mmol of each primer, 250 μM dNTPs and 0.02 U of Taq DNA polymerase (Promega). The amplification of the 16S rRNA gene was performed under the following conditions: initial denaturation at 94 °C for 4 min, 35 cycles at 94 °C for 30 s, annealing at 60 °C for 1 min, extension at 72 °C for 1.5 min and a final extension at 72 °C for 7 min. The DNA from P. aeruginosa was used as template for the positive control in the PCRs and sterile water as template for the negative control, respectively. The amplicons were checked by agarose gel electrophoresis (1.2 % w/v), sequenced by Macrogen Inc., Korea, and the obtained sequences were compared with those in the GenBank database (NCBI). For each bacterial sequence, an identity search was performed with the BLASTn algorithm (Basic Local Alignment Search Tool; http://www.ncbi.nlm.nih.gov/BLAST) for nucleotides (Altschul et al. 1990). The sequences reported in this study are available in the GenBank database under the GenBank ID numbers KU180323 to KU180356.
Phylogenetic analysis
The obtained sequences were compared with nucleotide sequences in the GenBank database using the BLAST algorithm (Altschul et al. 1990). The 16S rRNA sequences in the database sharing more than 97 % identity were imported with Mega 6.0 and aligned using ClustalW. The alignments were manually adjusted, and a phylogenetic analysis was performed using the neighbor-joining method (Saitou and Nei 1987). The phylogenetic distance was computed using the p-distance method, and the robustness of the resulting trees and the statistical significance levels of the interior nodes were obtained by bootstrap analysis with 1000 replicates. Bootstrap values greater than 70 % were observed, since nodes supported by values below this number are considered of weak resolution (Schneider 2007).
Results
The diversity and distribution of the endophytic bacterial community in Eucalyptus leaves from the ‘clonal garden’, ‘shade’, ‘recently out from under shade’, ‘dispatch’ and ‘field’ stages were examined by DGGE, plate cultivation and sequencing of the 16S rRNA gene. The nested PCR and DGGE analyses allowed the identification of both large and small populations in the total endophytic and potentially nitrogen fixing bacterial communities. This was accomplished by identifying the largest and smallest members of communities via high or low band intensity, respectively (Figs. 1, 2). The nifH gene was used for rapid differentiation of the populations in the diazotrophic community of Eucalyptus leaves at different stages of plant development (Fig. 2).

DGGE profile of the endophytic bacterial community in Eucalyptus. a Electrophoresis profile based on DGGE with 16S rRNA genes. CG clonal garden, S plants under shade, OS plants recently out from under shade, DP plants for dispatch, F plants in the field, M marker, 1, 2, 3 and 4 repetitions. b Dendrogram constructed from the DGGE of the 16S rRNA gene amplicons from leaf samples in the Eucalyptus development stages. The experiment was conducted with four repetitions

DGGE profile of the potentially nitrogen fixing endophytic bacterial community in Eucalyptus. a Electrophoresis profile based on DGGE with nifH genes. CG clonal garden, S plants under shade, OS plants recently out from under shade, DP plants for dispatch, F plants in the field, M marker; 1, 2, 3 and 4: Repetitions. b Dendrogram constructed from the DGGE of the nifH gene amplicons from leaf samples in the Eucalyptus development stages. The experiment was conducted with four repetitions
The banding profile and cluster analysis showed that the endophytic bacteria community in Eucalyptus leaves was homogeneous (shared high similarity); however, distinct populations were present at the ‘clonal garden’ and ‘field’ stages of development. These two stages formed two groups sharing lower values of similarity than the other groups (Fig. 1). Therefore, the endophytic bacteria community in Eucalyptus leaves was influenced by the stage of development.
Compared with the total endophytic bacterial community, the diazotrophic endophytic bacterial community was more heterogeneous with respect to the Eucalyptus development stages due to its smaller number of distinct groups. Similarity values below 58.3 % indicated that the diazotrophic endophytic community in the field was distinct from that of the other development stages (Fig. 2). Thus, the rare differences seen in the total endophytic community profile (Fig. 1) may be the result of diazotrophs in the ‘field’ and ‘clonal garden’ stages of development (Fig. 2). DGGE was sensitive to detect differences in the endophytic diazotrophic community in Eucalyptus leaves, and the nifH gene was an efficient molecular marker for rapid evaluation of diversity and distribution of the endophytic diazotrophic bacteria in Eucalyptus leaves at different plant development stages.
The distribution of the bands revealed few changes in the community structure of endophytic bacteria in the various Eucalyptus development stages, and most changes were induced by diazotrophic bacteria (Fig. 3). In general, the OTUs were present in the plant development stages of both the total endophytic and diazotrophic endophytic bacterial communities. However, the presence of specific OTUs was evident, reflecting differences in the structure of the community in leaves from the field (Fig. 3). Thus, the development stages of Eucalyptus influenced the endophytic bacteria community under the conditions evaluated, and this influence was most evident for diazotrophs.

Distribution of the OTUs for the total endophytic (a) and potentially diazotrophic bacteria (b). Venn diagram of the leaves of the different Eucalyptus development stages: ‘clonal garden’, ‘shade’, ‘recently out from under shade’, ‘dispatch’ and ‘field’
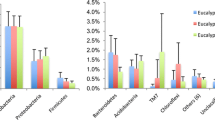
The Shannon diversity and richness indices were calculated for the endophytic bacteria communities in Eucalyptus leaves. The diversity and richness of the total endophytic bacteria were higher in the leaves from the field (Fig. 4). In contrast, for diazotrophic bacteria, the highest mean values of these indices were obtained from eaves of plants in ‘under shade’, ‘recently out from under shade’ and ‘field’ stages (Fig. 5). These results indicate that Eucalyptus development stages had a greater influence on the endophytic diazotrophic bacterial community than on the total bacterial community. Leaves from the ‘clonal garden’ and ‘dispatch’ stages had the lowest mean diversity and richness values among endophytic diazotrophic bacteria (Fig. 5).

Profiles of the richness (a) and diversity (b) of the total endophytic bacteria in the Eucalyptus development stages. CG clonal garden, S plants under shade, OS plants recently out from under shade, DP plants for dispatch, F plants in the field. Means followed by the same letter do not differ significantly according to Tukey’s test at 5 % probability. The experiment was conducted with four repetitions

Profiles of the richness (a) and diversity (b) of the potential nitrogen fixing endophytic bacteria from the Eucalyptus development stages. CG clonal garden, S plants under shade, OS plants recently out from under shade, DP plants for dispatch, F plants in the field. Means followed by the same letter do not differ significantly according to Tukey’s test at 5 % probability
The isolates with distinct BOX–PCR genomic profiles, sharing similarity indices below 90 % (unpublished data), were identified by 16S rRNA sequencing and compared with sequences obtained using the BLAST algorithm (NCBI). The sequencing of the 16S rRNA revealed the presence of three bacterial phyla in the communities: Firmicutes, Proteobacteria (classes alpha-, beta- and gamma-) and Actinobacteria (Fig. 6). These phyla were present in plants at different Eucalyptus development stages, either in the nursery or in the field. The sequences of 16S rRNA obtained from 32 isolates had high identity with sequences deposited in the NCBI database. Among these 32 sequences, only BAC 02 and BAC 31 had identity values below 97 %. Thus, these two isolates were only identified at genus level, whereas the others were identified at species level (Fig. 6) in the present study.

Distribution of the endophytic bacterial species found in the leaves from different Eucalyptus development stages. CG clonal garden, S plants under shade, OS plants recently out from under shade, DP plants for dispatch, F plants in the field; 1, 2, 3 and 4: Replications. Species names are accompanied by their GenBank (NCBI) accession numbers and percent identity values (ID)
The distribution of the endophytic bacterial species in Eucalyptus was largely specific to the plant developmental stage, as only 27 % of the identified species were isolated in more than one stage, such as Bacillus flexus, which was present in leaves from the ‘clonal garden’ to ‘field’ stages of development (Fig. 6). The majority of the endophytic bacteria isolates identified by 16S rRNA sequencing (44 %) were obtained from plants in the ‘under shade’ and ‘recently out from under shade’ (35 %) stages. Endophytic colonization by the genus Bacillus (B. flexus, B. cereus and B. thuringiensis) in Eucalyptus leaves initiates in the ‘clonal garden’ stage and encompasses subsequent stages of development (Fig. 6).
A phylogenetic analysis of the 16S rRNA sequences with greater than 97 % identity confirmed that the endophytic bacteria on leaves from different stages of Eucalyptus development form distinct clades with high bootstrap values and belong to the phyla Firmicutes, Proteobacteria and Actinobacteria (Fig. 7). In general, the bootstrap values exhibited by the groups were strong and support the phylogenetic analysis (Schneider 2007) (Fig. 7).

Phylogenetic tree reconstructed with the neighbor-joining method using sequences of the 16S rRNA gene for the endophytic bacteria found in leaves from different stages of Eucalyptus development. Bootstrap values above 70 % are shown
The phylogenetic analysis of the 16S rRNA sequences also revealed the prevalence of bacteria belonging to the Gammaproteobacteria, including the genera Pantoea and Stenotrophomonas, with high bootstrap values, in leaves from different stages of Eucalyptus development. The Firmicutes and Alphaproteobacteria showed a higher number of distinct genera. Within Proteobacteria, the Betaproteobacteria class was present in lower abundance and the isolate BAC 06 was identified as Massilia timonae and was phylogenetically similar to M. timonae (Fig. 7). Among the Firmicutes, Bacillus was the predominant genus (Fig. 6), with a bootstrap value of 99 % (Fig. 7). Paenibacillus terrigena (BAC 33) and P. darwinianus (BAC 29), exclusive colonizers of the ‘dispatch’ and ‘field’ stages, respectively, have not been previously known to have endophytic interaction with plants. Terrabacter lapilli (BAC 32) is the only member of the phylum Actinobacteria isolated from Eucalyptus leaves that has not been recorded as endophytic. The Alphaproteobacteria class was represented in Eucalyptus leaves by many diazotrophic bacteria, including A. fabrum, Rhizobium grahamii and Rhizobium pusense (Figs. 6, 7). The isolate BAC 16, identified as Rhizobium pusense (Fig. 6), clustered with Agrobacterium fabrum and Rhizobium pusense with high phylogenetic support (Fig. 7). In this phylum, R. grahamii (BAC 25) and Novosphingobium barchaimii (BAC 27) are described for the first time as endophytes (Fig. 6). These two species were only isolated from Eucalyptus seedlings in the ‘recently out from under shade’ stage. N. barchaimii exhibited phylogenetic proximity, with a high bootstrap value, to Sphingobium yanoikuyae and Sphingobium scionense (Fig. 7). For the majority of the isolates, the phylogenetic analysis (Fig. 7) confirmed the identification by BLAST (Fig. 6). An additional endophytic species was Stenotrophomonas panacihumi (Figs. 6, 7) in the Gammaproteobacteria class. This species was isolated from seedlings in the ‘under shade’ and ‘recently out from under shade’ stages (Fig. 6).
Discussion
The amplification of the 16S rRNA gene and separation of the amplicons by DGGE allowed us to assess efficiently the diversity of endophytic bacteria in Eucalyptus leaves. The sequencing and identification of isolates using NCBI resources provided more information on this endophytic bacterial community. DGGE is a solid choice for assessing the diversity of plant-microorganism interactions (Oliveira et al. 2013) because it is a fast semiquantitative method for describing complex microbial communities (Petersen and Dahllöf 2005). The study of plant-microorganism interactions is important because the interior of plants is a habitat for a great diversity of endophytic microorganisms that play important roles in the health and development of plants (Vandenkoornhuyse et al. 2015). To adapt to their plant environment and colonize it, endophytic microorganisms have a different metabolic potential compared with free-living microorganisms (Brader et al. 2014).
Cluster analysis of the DGGE bands of the 16S rRNA fragments indicated clear electrophoretic separation and the formation of distinct groups. These groupings were less evident in the community of diazotrophic bacteria when compared to the total bacteria, with similarity values below 30 % identified across the Eucalyptus development stages. Low similarity percentages (Fig. 1) indicate high variation among the bacterial groups (Altimira et al. 2012) in Eucalyptus leaves. However, the endophytic diazotrophic bacterial community in the ‘field’ seems to recruit its own microbiota, which differs from the other plant development stages, with similarity values of 43 to 58 % (Fig. 2). The gene nifH used in the DGGE, was an efficient molecular marker for detecting differences in the diversity and distribution of the community of potentially diazotrophic endophytic bacteria in Eucalyptus leaves and allowed a rapid diferentiation of populations present in this community (Fig. 2). Our results indicate that these stages modulate the community of a large number of endophytic bacteria having the potential to stimulate plant growth via nitrogen fixation. This ability could improve plant nutrition due to the presence of nitrogenase (Oldroyd and Dixon 2014). The present study may therefore provide contributions to biotechnological solutions for solving the nitrogen problem in plants.
The diversity of endophytic bacteria in trees is studied less frequently than other plants (El-Deeb et al. 2013; Kukla et al. 2014; Miguel et al. 2013). In Eucalyptus, little is known about plant-bacteria interactions, as existing studies have focused on the soil (Da Silva et al. 2014), rhizosphere and roots (Castellanos et al. 2010), stems (Procópio et al. 2009) and seeds (Ferreira et al. 2008).
The importance of Eucalyptus development stages in the modulation of the composition of endophytic bacterial communities is evidenced by higher richness and diversity indices in the leaves from the ‘field’ (Fig. 4). The diversity profile of the endophytic diazotrophic community is distinct (the highest indices means were observed in the leaves obtained from plants in the ‘under shade’, ‘recently out from under shade’ and ‘field’ stages), and the plant development stages also modulate this community (Fig. 5). In addition, Eucalyptus development stages had a greater influence on potentially diazotrophic endophytic bacterial communities than identified for total bacterial communities.
The indices shown in this study mirror those commonly reported in the literature (Fig. 4), which vary between 1.5 and 3.5 (Gazis and Chaverri 2010). The differences in the diversity of endophytic bacteria may be attributed to the distinct sugar concentrations identified in this leaves (unpublished data) and the catabolic variations described in the majority of phylogenetically similar heterotrophic bacteria (Rodionov et al. 2010). Nitrogen fixation requires the use of carbon and energy sources, and the genera with this ability can differ in generation time, growth rate and amount of carbohydrates consumed (Naher et al. 2008). In cyanobacteria, the cleavage of sucrose is essential for carbon metabolism and nitrogen fixation (Figueroa et al. 2013). Thus, in development stages with higher sucrose concentrations, there could be a predominance of diazotrophic bacteria that, like Gluconacetobacter diazotrophicus, use sucrose as an energy source (Velázquez-Hernández et al. 2011).
The endophytic bacterial community in Eucalyptus is composed of the phyla Firmicutes, Proteobacteria (classes alpha-, beta- and gamma-) and Actinobacteria. The endophytic bacteria isolated and identified by 16S rRNA sequencing generally shared identities greater than 97 % with NCBI sequences, and only two isolates, BAC 02 and BAC 31, had identity values lower than 97 % (Fig. 6). In phylogenetic studies in general, identity values exceeding 97 % are the standard for circumscribing organisms of the same species (Al-Batayneh et al. 2011), although some authors recognize that values of 98.5–99 % are more desirable and consistent (Stackebrandt and Ebers 2006). The reliability of the alignments was confirmed using the e-value (data not shown), which indicates the probability of obtaining an alignment with an equivalent or higher score with another random sequence of the same size and base composition (Kerfeld and Scott 2011). After an alignment of the sequences obtained by sequencing the 16S rRNA to the corresponding NCBI sequences, the isolates with identities below 97 % were classified at the genus level (Fig. 6).
The main phylum identified in the ‘under shade’ and ‘recently out from under shade’ plants was Proteobacteria, classes alpha-, beta- and gamma- (Fig. 6). Firmicutes, Actinobacteria and Proteobacteria have the ability to catabolize simple and complex carbohydrates (Li et al. 2014), which facilitate their adaptation to the endophytic habitat. A previous analysis of the bacterial community diversity on leaves using 16S rRNA sequencing via culture-dependent and -independent approaches revealed the dominance of Proteobacteria (Bulgarelli et al. 2013). Many of the microorganisms detected in the present study are potentially diazotrophic (Fig. 6) (Köberl et al. 2013) because they contain the nifH gene, which is associated with nitrogen fixation (Weber et al. 2013). Bacteria in the Firmicutes phylum are typically prevalent in the rhizosphere (Bodenhausen et al. 2013); however, many of its species are well adapted to the endophytic lifestyle (Miguel et al. 2013; Oliveira et al. 2013). This colonization frequently originates from an infection of soil bacteria in the joints of lateral roots followed by rapid dissemination throughout the intercellular spaces of the root system (Chen and Zhu 2013). However, in the present study, the majority of the endophytic bacteria isolated at different stages of Eucalyptus development exhibited stage-specific plant-bacteria interactions, with only a few bacteria present in more than one stage (Fig. 6).
The presence of specific species in the endophytic community of each development stage indicates that a subset of the community cannot be derived from the soil and must instead enter the leaf via the stomata, injury, or other entryways (Romero et al. 2014). Another entry mode of endophytic bacteria is the nutritive solution used in plant fertilization in stages prior to those investigated in this study. In the nutritive solution, Proteobacteria could grow and proliferate most rapidly on the readily available carbon sources (Leff et al. 2015) in the ‘under shade’ and ‘recently out from under shade’ development stages (unpublished data). Thus, the endophytic community can be influenced by the composition and concentration of sugars and amino acids. Both young and shaded plants exhibit higher amino acid content (Chen and Poland 2009; Brader et al. 2014), and the preference of many Proteobacteria genera to utilize organic acids rather than sugars (Portais and Delort 2002) could contribute to the predominance of Proteobacteria in the young and shaded development stages of Eucalyptus (Fig. 6).
The predominance of Firmicutes in the leaves from the field (Fig. 6) can be attributed to the relatively high sugar concentrations (unpublished data) and low amino acid levels (Steinbauer 2013). However, B. flexus was abundant in all the plant development stages (Fig. 6); this could be explained if the colonization by this genus had initiated at the ‘clonal garden’ stage, as evidenced by the occurrence of some species in more than one seedling development stage (Fig. 6). The colonization and permanence of B. flexus, a competent (Hardoim et al. 2008) generalist endophytic bacteria with great competitive ability, suggest the existence of specific genetic machinery for colonization and persistence in Eucalyptus leaves. Compared with the rhizosphere, leaves are more dynamic habitats, and the microbial community interacting with leaves is subject to more variations in temperature, humidity and radiation during the day and night, factors that affect both the microbial community and the physiology of the plant (Turner et al. 2013).
The changes in plant physiology that occur during the stages of Eucalyptus development (Mankessi et al. 2010) may be responsible for altering the endophytic bacterial community. These changes allow endophytic bacteria to multiply rapidly and prevail under favorable conditions and maintain latency under conditions that are less favorable to growth and proliferation. The endophytic bacteria that interact with Eucalyptus may be important for the physiology of the plant, for example, they might provide it with tolerance to biotic and abiotic factors. These characteristics increase plant survival and growth under adverse conditions and when in competition with pathogens. Furthermore, endophytic bacteria produce a wide variety of plant growth regulators such as gibberellins, abscisic acid and auxins (Gaiero et al. 2013).
Our phylogenetic analysis of 16S rRNA sequences confirmed that the endophytic community in Eucalyptus leaves is affiliated the Firmicutes, Proteobacteria (alpha-, beta-, and gamma-) and Actinobacteria phyla as evidenced by the formation of distinct clades (Fig. 7). Bootstrap values above 95 % (Fig. 7) were identified for the majority of the groups and support the phylogenetic analysis (Schneider 2007). Among the Gammaproteobacteria, the genera Pantoea and Stenotrophomonas stand out due to their 99 % bootstrap values. These nitrogen fixing genera are producers and excretors of 3-indoleacetic acid (IAA) and amino acids (Liba et al. 2006). These metabolites released by the endophytes can be used by the plant to increase its growth rate (Kembel et al. 2014). Nitrogen fixation is widespread among bacteria and can be exploited to improve plant nutrition (Oldroyd and Dixon 2014). Among the nitrogen-fixing Proteobacteria, two isolates of Rhizobium (Fig. 6) with high identity and bootstrap values were shown to be genetically related to Agrobacterium, Ochrobactrum rhizosphaerae and Ochrobactrum tritici (Fig. 7).
Ochrobactrum rhizosphaerae was recently reported to have the ability to utilize sucrose and fructose as carbon sources in the soil (Kämpfer et al. 2008), whereas in the present study it was reported to be endophytic in Eucalyptus. Xanthobacter flavus, first reported 36 years ago, is another nitrogen fixer with a system similar to Azotobacter, which utilizes several organic acids. These microorganisms utilized most carbohydrates after prolonged incubation (Malik and Claus 1979). They also form a distinct clade with a high bootstrap value, which validates their phylogenetic proximity (Fig. 7). The presence of nitrogen fixation genes may be one of the characteristics that make them phylogenetically similar.
The majority of the Firmicutes isolated from the various stages of Eucalyptus development belong to the genus Bacillus, and the class Alphaproteobacteria contributed to the largest number of distinct genera (Fig. 6). Bacillus is the most commonly encountered endophytic bacterial genera and is a source of several enzymes with great biotechnological potential such as amylases, proteases, cellulases and lipases (Kannan et al. 2015). These enzymes are critical for the initiation of endophytic colonization (Seo et al. 2010). Bacillus microorganisms produce auxins and gibberellins in addition to fixing nitrogen and also secrete important substances for the biological control of pathogens (Bacon et al. 2015).
The class Betaproteobacteria contained the rarest of the isolates identified in the present study. BAC 06 was identified as M. timonae (Figs. 6, 7), which colonizes the rhizosphere, roots (Ofek et al. 2012) and leaves (Jackson et al. 2013) and is a growth promoter via the production of IAA and siderophores in various plant species. M. timonae is sensitive to competition (Ofek et al. 2012), which could explain it is unable to persist in other stages of Eucalyptus development. This sensitivity to competition may have also occurred in other species in the present study (Fig. 6). The Betaproteobacteria community in Eucalyptus is diverse and development stage-dependent. Many Betaproteobacteria are potentially diazotrophic and can contribute to nitrogen fixation and growth promotion (Fig. 6).
The endophytic bacterial community in Eucalyptus varies according to the organ of isolation, geographical cultivation area and interacting plant species (Ferreira et al. 2008; Procópio et al. 2009; Castellanos et al. 2010). The few existing studies involving Eucalyptus have indicated the presence of Bacillus sp., B. megaterium, Paenibacillus sp., P. humicus, Enterococcus mundtii, Methylobacterium sp., M. variabile, M. gregens, Sphingomonas phyllosphaerae, Paracoccus sp. (Ferreira et al. 2008), Erwinia/Pantoea, Agrobacterium sp., Curtobacterium sp., Brevibacillus sp., Pseudomonas sp., Acinetobacter sp., Burkholderia cepacia, and Lactococcus lactis (Procópio et al. 2009). The genera Azotobacter, Beijerinckia, Derxia, Azospirillum, Herbaspirillum, Gluconacetobacter and Xanthomonas (Castellanos et al. 2010) are also present in Eucalyptus.
The present study has recorded several novel endophytic bacteria in Eucalyptus, including those with nitrogen fixation potential (Figs. 6, 7). The genus Novosphingobium was previously described as endophytic (Castro et al. 2014); however, this is the first record of N. barchaimii (BAC 27) as an endophytic species. This species is phylogenetically related with high bootstrap values to Sphingobium yanoikuyae and Sphingobium scionense (Fig. 7) and has a genetic machinery that allows it to adapt to different habitats such as soils, sediments and aquatic environments (Addison et al. 2007). The interior of the Eucalyptus leaves from the ‘recently out from under shade’ development stage is another important habitat occupied by N. barchaimii (Figs. 6, 7), which utilizes glucose, sucrose and rhamnose as carbon sources and was isolated from soil contaminated with the insecticide hexachlorocyclohexane (Niharika et al. 2013). Rhizobium grahamii, Stenotrophomonas panacihumi, P. terrigena, P. darwinianus and Terrabacter lapilli are also novel endophytic species isolated from Eucalyptus leaves (Figs. 6, 7). Rhizobium is commonly cited as endophytic in many plants; however, the literature does not include Rhizobium grahamii as an endophyte. This bacterium belongs to a new phylogenetic group of Rhizobia, together with Rhizobium mesoamericanum and others (Althabegoiti et al. 2014). The presence of Stenotrophomonas panacihumi in interaction with plants is a novel finding (Fig. 6) because it was previously identified only in soils cultivated with medicinal plants (Yi et al. 2010). Stenotrophomonas panacihumi showed phylogenetic proximity to Stenotrophomonas rhizophila, Stenotrophomonas pavanii and Stenotrophomonas maltophilia (Fig. 7), and the latter two species have been isolated as endophytic nitrogen fixers in sugarcane (Ramos et al. 2014). P. darwinianus and P. terrigena are species included in Firmicutes, the most abundant phylum in environments such as Antarctic soils (Dsouza et al. 2014) and the rhizosphere of bamboo (Han et al. 2009). The ability of Firmicutes bacteria to colonize various environments is attributed to their physiological diversity, to the formation of stress resistant endospores, and secretion of extracellular enzymes and antimicrobial compounds. The presence of these enzymes enables the hydrolysis of a variety of carbohydrates (Priest and Genus 2009). The unprecedented colonization of P. darwinianus and P. terrigena in Eucalyptus occurred in plants from the ‘dispatch’ and ‘field’ stages, respectively (Fig. 6). The phylum Actinobacteria is rarely explored as a source of new endophytic bacteria. However, the number of reports on Actinobacteria in various plants has recently increased (Bodenhausen et al. 2013; Miguel et al. 2013; Romero et al. 2014; Leff et al. 2015). In this study, Terrabacter lapilli, isolated from the leaves of Eucalyptus in the field, has been characterized as an endophytic Actinobacteria (Figs. 6, 7).
The endophytic bacterial community is an important part of the Eucalyptus microbiome. The bacteria that make up this community include endophytic diazotrophs, which can contribute the necessary nitrogen for plant growth. However, new approaches involving nifH genes, responsible for nitrogen fixation, are required. The description of the diversity of these microorganisms in different Eucalyptus development stages enables a better understanding of the dynamics of endophytic colonization in Eucalyptus. The description of the endophytic bacterial diversity in Eucalyptus is an important step towards determining the benefits of the endophytic bacteria–Eucalyptus interaction and the ecological role they play in this interaction.
Conclusions
The nifH gene was used as a marker for the rapid differentiation of the populations in the diazotrophic bacterial community in Eucalyptus leaves at different stages of plant development.
Sequencing of the isolates revealed the presence of three bacterial phyla: Firmicutes, Proteobacteria (classes alpha-, beta- and gamma-) and Actinobacteria. These phyla were present in various Eucalyptus development stages. Among the Gammaproteobacteria, the genera Pantoea and Stenotrophomonas were highlighted, and the majority of the Firmicutes were identified to be Bacillus. The Alphaproteobacteria had the highest number of distinct genera, and the Betaproteobacteria were the less represented in the present study.
The distribution of endophytic bacterial species in Eucalyptus exhibited specificity with respect to the stage of development. The majority of the endophytic bacterial isolates identified by 16S rRNA sequencing were obtained from ‘under shade’ and ‘recently out from under shade’ plants.
This is the first report of P. terrigena, P. darwinianus, Terrabacter lapilli, Rhizobium pusense, Novosphingobium barchaimii and Stenotrophomonas panacihumi as endophytic species.
References
Addison SL, Foote SM, Reid NM, Lloyd-Jones G (2007) Novosphingobium nitrogenifigens sp. nov., a polyhydroxyalkanoate-accumulating diazotroph isolated from a New Zealand pulp and paper wastewater. Int J Syst Evol Microbiol 57:2467–2471
Al-Batayneh KM, Jacob JH, Hussein EI (2011) Isolation and molecular identification of new thermophilic bacterial strain of Geobacillus pallidus and Anoxybacillus flavithermus. Int J Integr Biol 11:39–43
Althabegoiti MJ, Ormeño-Orrillo E, Lozano L, Tejerizo GT, Rogel MA, Mora J, Martínez-Romero E (2014) Characterization of Rhizobium grahamii extrachromosomal replicons and their transfer among rhizobia. BMC Microbiol 14:1–14
Altimira F, Yáñez C, Bravo G, González M, Rojas LA, Seeger M (2012) Characterization of copper-resistant bacteria and bacterial communities from copper-polluted agricultural soils of central Chile. BMC Microbiol 12:1–12
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bacon CW, Palencia ER, Hinton DM (2015) Abiotic and biotic plant stress-tolerant and beneficial secondary metabolites produced by Endophytic Bacillus Species. In: Arora NK (ed) Plant microbes symbiosis: applied facets. Springer, New Delhi, pp 163–177
Blackwood CB, Oaks A, Buyer JS (2005) Phylum- and class-specific PCR primers for general microbial community analysis. Appl Environ Microbiol 71:6193–6198
Bodenhausen N, Horton MW, Bergelson J (2013) Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 8:e56329
Bonito G, Reynolds H, Robeson MS, Nelson J, Hodkinson BP, Tuskan G, Schadt CW, Vilgalys R (2014) Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol Ecol 23:3356–3370
Brader G, Compant S, Mitter B, Trognitz F, Sessitsch A (2014) Metabolic potential of endophytic bacteria. Curr Opin Biotechnol 27:30–37
Brooker MIH (2000) A new classification of the genus Eucalyptus L’Her (Myrtaceae). Aust Syst Bot 13:79–148
Bulgarelli D, Schlaeppi K, Spaepen S, Van Themaat EVL, Schulze-Lefert P (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807–838
Castellanos DMO, Zabala LBB, Botía DIEGOR, Garrido MFR, Baldani VLD, Buitrago RRB (2010) Caracterización de Bacterias diazotróficas asimbióticas asociadas al eucalipto (Eucalyptus sp.) en Codazzi, Cesar (Colombia). Acta Biol Colomb 15:107–120
Castro RA, Quecine MC, Lacava PT, Batista BD, Luvizotto DM, Marcon J, Ferreira A, Melo IS, Azevedo JL (2014) Isolation and enzyme bioprospection of endophytic bacteria associated with plants of Brazilian mangrove ecosystem. SpringerPlus 3:1–10
Chen Y, Poland TM (2009) Interactive influence of leaf age, light intensity, and girdling on green ash foliar chemistry and emerald ash borer development. J Chem Eco 35:806–815
Chen C, Zhu H (2013) Are common symbiosis genes required for endophytic rice–rhizobial interactions? Plant Signal Behav 8:1–4
Da Silva MDCS, De Almeida PT, Moreira BC, Carolino M, Cruz C, Bazzolli DMS, Silva CC, Kasuya MCM (2014) Nitrogen-fixing bacteria in Eucalyptus globulus plantations. PLoS ONE 9:e111313
Dsouza M, Taylor MW, Turner SJ, Aislabie J (2014) Genome-based comparative analyses of Antarctic and temperate species of Paenibacillus. PLoS ONE 9:e108009
El-Deeb B, Fayez K, Gherbawy Y (2013) Isolation and characterization of endophytic bacteria from Plectranthus tenuiflorus medicinal plant in Saudi Arabia desert and their antimicrobial activities. J Plant Interact 8:56–64
Farrar K, Bryant D, Cope-Selby N (2014) Understanding and engineering beneficial plant–microbe interactions: plant growth promotion in energy crops. Plant Biotechnol J 2014(12):1193–1206
Ferreira A, Quecine MC, Lacava PT, Oda S, Azevedo JL, Araújo WL (2008) Diversity of endophytic bacteria from Eucalyptus species seeds and colonization of seedlings by Pantoea agglomerans. FEMS Microbiol Lett 287:8–14
Fidalgo C, Henriques I, Rocha J, Tacão M, Alves A (2016) Culturable endophytic bacteria from the salt marsh plant Halimione portulacoides: phylogenetic diversity, functional characterization, and influence of metal (loid) contamination. Environ Sci and Pollut Res 1:1–15
Figueroa CM, Diez MDA, Kuhn ML, Mcewen S, Salerno GL, Iglesias AA, Ballicora MA (2013) The unique nucleotide specificity of the sucrose synthase from Thermosynechococcus elongatus. FEBS Lett 587:165–169
Gaiero JR, Mccall CA, Thompson KA, Day NJ, Best AS, Dunfield KE (2013) Inside the root microbiome: bacterial root endophytes and plant growth promotion. Am J Bot 2013(100):1738–1750
Gazis R, Chaverri P (2010) Diversity of fungal endophytes in leaves and stems of wild rubber trees (Hevea brasiliensis) in Peru. Fungal Ecol 3:240–254
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:9
Han J, Xia D, Li L, Sun L, Yang K, Zhang L (2009) Diversity of culturable bacteria isolated from root domains of moso bamboo (Phyllostachys edulis). Microb Ecol 58:363–373
Hardoim PR, Overbeek LSV, Elsas KDV (2008) Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol 16:463–471
Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63:3233–3241
Jackson CR, Randolph KC, Osborn SL, Tyler HL (2013) Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol 13:1–12
Kämpfer P, Sessitsch A, Schloter M, Huber B, Busse HJ, Scholz HC (2008) Ochrobactrum rhizosphaerae sp. nov. and Ochrobactrum thiophenivorans sp. nov., isolated from the environment. Int J Syst Evol Micrbiol 58:1426–1431
Kannan R, Damodaran T, Umamaheswari S (2015) Sodicity tolerant polyembryonic mango root stock plants: a putative role of endophytic bacteria. Afr J Biotechnol 14:350–359
Kembel SW, O’connor TK, Arnold HK, Hubbell SP, Wright SJ, Green JL (2014) Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc Natl Acad Sci 111:13715–13720
Kerfeld CA, Scott KM (2011) Using BLAST to Teach ‘‘e-value-evotionary’’ Concepts. PLoS Biol 9:1–4
Köberl M, Schmidt R, Ramadan EM, Bauer R, Berg G (2013) The microbiome of medicinal plants: diversity and importance for plant growth, quality and health. Front Microbiol 4:1–9
Köppen W (1948) Climatologia. Fundo de Cultura Econômica, México
Kukla M, Płociniczak T, Piotrowska-Seget Z (2014) Diversity of endophytic bacteria in Lolium perenne and their potential to degrade petroleum hydrocarbons and promote plant growth. Chemosphere 117:40–46
Leff JW, Del Tredici P, Friedman WE, Fierer N (2015) Spatial structuring of bacterial communities within individual Ginkgo biloba trees. Environ Microbiol 17:1–10
Li X, Rui J, Xiong J, Li J, He Z, Zhou J, Yannarell AC, Roderick I, Mackie RI (2014) Functional potential of soil microbial communities in the maize rhizosphere. PLoS ONE 9:e112609
Liba CM, Ferrara FIS, Manfio GP, Fantinatti-Garboggini F, Albuquerque RC, Pavan C, Ramos PL, Mireira-Filho CA, Barbosa HR (2006) Nitrogen-fixing chemo-organotrophic bacteria isolated from cyanobacteria-deprived lichens and their ability to solubilize phosphate and to release amino acids and phytohormones. J Appl Microbiol 101:1076–1086
Malik KA, Claus D (1979) Xanthobacter flavus, a new species of nitrogen-fixing hydrogen bacteria. Int J Syst Evol Microbiol 29:283–287
Mankessi F, Saya AR, Boudon F, Guédon Y, Montes F, Lartaud M, Verdeil JL, Monteuuis O (2010) Phase change-related variations of dome shape in Eucalyptus urophylla × Eucalyptus grandis shoot apical meristems. Trees 24:743–752
Mapelli F, Marasco R, Rolli E, Barbato M, Cherif H, Guesmi A, Ouzari I, Daffonchio D, Borin S (2013) Potential for plant growth promotion of rhizobacteria associated with Salicornia growing in Tunisian hypersaline soils. Biomed Res Int 2013:1–14
Miguel PSB, Delvaux JC, Oliveira MNV, Monteiro LCP, Freitas FS, Costa MD, Tótola MR, Moraes CA, Borges AC (2013) Diversity of endophytic bacteria in the fruits of Coffee canephora. Afr J Microbiol 7:586–594
Minitab I (2006) MINITAB statistical software. Version: Release, 15
Naher UA, Radziah O, Halimi MS, Shamsuddin ZH, Razi IM (2008) Specific growth rate and carbon sugar consumption of diazotrophs isolated from rice rhizosphere. J Biol Sci 8:1008–1014
Niharika N, Moskalikova H, Kaur J, Sedlackova M, Hampl A, Damborsky J, Prokop Z, Lal R (2013) Novosphingobium barchaimii sp. nov., isolated from a hexachlorocyclohexane (HCH) contaminated soil. Int J Syst Evol Microbiol 63:667–672
Ofek M, Hadar Y, Minz D (2012) Ecology of root colonizing Massilia(Oxalobacteraceae). PLoS ONE 7:e40117
Oldroyd GED, Dixon R (2014) Biotechnological solutions to the nitrogen problem. Curr Opin Biotechnol 26:19–24
Oliveira MNV, Santos TMA, Vale HMM, Delvaux JC, Cordero AP, Ferreira AB, Miguel PSB, Tótola MR, Costa MD, Moraes CA, Borges AC (2013) Endophytic microbial diversity in coffee cherries of Coffea arabica from southeastern Brazil. Can J Microbiol 2013(59):221–230
Petersen DG, Dahllöf I (2005) Improvements for comparative analysis of changes in diversity of microbial communities using internal standards in PCR-DGGE. FEMS Microbiol Ecol 53:339–348
Poly F, Ranjard L, Nazaret S, Gourbière F, Monrozier LJ (2001) Comparison of nifH gene pools in soils and soil microenvironments with contrasting properties. Appl Environ Microbiol 67:2255–2262
Portais JC, Delort AM (2002) Carbohydrate cycling in micro-organisms: what can 13C-NMR tell us? FEMS Microbiol Rev 26:375–402
Priest TFG, Genus I (2009) Paenibacillus. In: De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W et al (eds) Bergey’s manual of systematic bacteriology the firmicutes. Springer, New York
Procópio REL, Araújo WL, Maccheroni JRW, Azevedo JL (2009) Characterization of an endophytic bacterial community associated with Eucalyptus spp. Genet Mol Res 2009(8):1408–1422
Puri A, Padda KP, Chanway CP (2016a) Seedling growth promotion and nitrogen fixation by a bacterial endophyte Paenibacillus polymyxa P2b-2R and its GFP derivative in corn in a long-term trial. Symbiosis 1:1–7
Puri A, Padda KP, Chanway CP (2016b) Evidence of nitrogen fixation and growth promotion in canola (Brassica napus L.) by an endophytic diazotroph Paenibacillus polymyxa P2b-2R. Biol Fert Soils 52:119–125
Ramos PL, Van Trappen S, Thompson FL, Rocha RC, Barbosa HR, De Vos P, Moreira-Filho CA (2014) Screening for endophytic nitrogen-fixing bacteria in Brazilian sugar cane varieties used in organic farming and description of Stenotrophomonas pavanii sp. nov. Int J Syst Evol Microbiol 61:926–931
Reasoner DJ, Geldreich EE (1985) A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49:1–7
Reinhold-Hurek B, Hurek T (2011) Living inside plants: bacterial endophytes. Curr Opin Plant Biol 14:435–443
Rodionov DA, Yang C, Li X, Rodionova IA, Wang Y, Obraztsova AY, Zagnitko OP, Overbeek R, Romine MF, Reed S, Fredrickson JK, Nealson KH, Osterman AL (2010) Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genom 11:494
Romero FM, Marina M, Pieckenstain FL (2014) The communities of tomato (Solanum lycopersicum L.) leaf endophytic bacteria, analyzed by 16S-ribosomal RNA gene pyrosequencing. FEMS Microbiol Lett 351:187–194
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Santoyo G, Moreno-Hagelsieb G, del Carmen Orozco-Mosqueda M, Glick BR (2016) Plant growth-promoting bacterial endophytes. Microbiol Res 183:92–99
Schneider H (2007) Métodos de análise filogenética: um guia prático (Methods for phylogenetic analysis: a practical guide). Holos Editora e Sociedade Brasileira de Genética, Ribeirão Preto
Scolforo JRS, Oliveira AD, Carvalho LMT (eds) (2008) Zoneamento ecológico-econômico do Estado de Minas Gerais: componente socioeconômico. UFLA, Lavras
Seo WT, Lim WJ, Kim EJ, Yun HD, Lee YH, Cho KM (2010) Endophytic bacterial diversity in the young radish and their antimicrobial activity against pathogens. J Korean Soc Appl Biol Chem 53:493–503
Smethurst P, Holz H, Moroni M, Baillie C (2004) Nitrogen management in Eucalyptus nitens plantations. For Ecol Manage 193:63–80
Stackebrandt E, Ebers J (2006) Taxonomic parameters re-visited: tarnished gold standards. Microbiol Today 33:152–155
Steinbauer J (2013) Shoot feeding as a nutrient acquisition strategy in free-living psylloids. PLoS ONE 8:1–9
Summan A, Gaur A, Shrivastava AK, Yadav RL (2005) Improving sugarcane growth and nutrient uptake by inoculating Gluconacetobacter diazotrophicus. Plant Growth Regul 47:155–162
Systat Software, Inc. (2008). SigmaPlot for Windows, version 11.0
Szilagyi-Zecchin VJ, Ikeda AC, Hungria M, Adamoski D, Kava-Cordeiro V, Glienke C, Galli-Terasawa LV (2014) Identification and characterization of endophytic bacteria from corn (Zea mays L.) roots with biotechnological potential in agriculture. AMB Express 4:1–9
Turner TR, James EK, Poole PS (2013) The plant microbiome. Genome Biol 14:1–10
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A (2015) The importance of the microbiome of the plant holobiont. New Phytol 206:1196–1206
Velázquez-Hernández ML, Baizabal-Aguirre VM, Cruz-Vázquez F, Trejo-Contreras MJ, Fuentes-Ramírez LE, Bravo-Patiño A, Cajero-Juárez M, Chávez-Moctezuma MP, Valdez-Alarcón JJ (2011) Gluconacetobacter diazotrophicus levansucrase is involved in tolerance to NaCl, sucrose and desiccation, and in biofilm formation. Arch Microbiol 193:137–149
Waqas M, Khan AL, Kamran M, Hamayun M, Kang SM, Kim YH, Lee IJ (2012) Endophytic fungi produce gibberellins and indoleacetic acid and promotes host-plant growth during stress. Molecules 17:10754–10773
Weber OB, Videira SS, De Araújo JLS (2013) Identification of culturable endophytes in ‘Champaka’ pineapple grown in an organic system. Afr J Agric Res 8:3422–3430
Wemheuer F, Wemheuer B, Kretzschmar D, Pfeiffer B, Herzog S, Daniel R, Vidal S (2016) Impact of grassland management regimes on bacterial endophyte diversity differs with grass species. Lett Appl Microbiol 62:323–329
West ER, Cother EJ, Steel CC, Ash GJ (2010) The characterization and diversity of bacterial endophytes of grapevine. Can J Microbiol 56:209–216
Yi H, Srinivasan S, Kim MK (2010) Stenotrophomonas panacihumi sp. nov., isolated from soil of a ginseng field. J Microbiol 48:30–35
Acknowledgments
The authors thank the Celulose Nipo-Brasileira (CENIBRA) for financial support, Federal Agency for Support and Evaluation of Graduate Education (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES) for financial support and the Minas Gerais State Research Foundation (Fundação de Amparo à Pesquisa do Estado de Minas Gerais—FAPEMIG) for the grant provided to the first author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Miguel, P.S.B., de Oliveira, M.N.V., Delvaux, J.C. et al. Diversity and distribution of the endophytic bacterial community at different stages of Eucalyptus growth. Antonie van Leeuwenhoek 109, 755–771 (2016). https://doi.org/10.1007/s10482-016-0676-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-016-0676-7