
Abstract
Helicobacter presence and viability in waters is not well characterized. The identification of natural reservoirs and infection sources may provide novel insights into its waterborne transmission. The goal of this study was to investigated the occurrence of Helicobacter spp. in natural freshwaters from Roraima Tepui, a little studied and unique ecosystem of the Guayana Shield. Freshwaters collected from two localities at Roraima Tepui were cultured in HP selective broth and agar for Helicobacter pylori and analysed by fluorescent in situ hybridization (FISH), specific PCR assays, 16S rRNA gene sequencing and phylogenetic analysis. The presence of other bacteria in freshwater enrichments was determined using clone library sequencing of the 16S rRNA gene and phylogenetic inferences. Helicobacter spp. were detected by semi-nested PCR and FISH in freshwater enrichments from both sites. Coccoid viable but nonculturable (VBNC) cells were evidenced using 16S rRNA gene Helicobacter species and H. pylori-specific probes. Partial 16S rRNA gene sequences of two HP enrichments showed high similarity to H. pylori and Helicobacter nemestrinae (99–100 %). Other bacteria such as Serratia, Aquitalea, Chromobacterium, Mycobacterium, Acinetobacter, Curvibacter and Dysgonomonas were also detected using complete 16S rRNA gene sequences, with Serratia, Aquitalea and Chromobacterium the most common genera (40.9, 18.2 and 15.2 %, respectively). This is the first time that Helicobacter spp. have been reported in freshwaters of a tepui ecosystem. Our results contribute to the current knowledge of these bacteria in the aquatic environment and expand their known/potential sites outside the human host.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Helicobacter (Goodwin et al. 1989) has been expanding since the discovery of Helicobacter pylori, the first bacterium known to colonize the gastric environment (Marshall and Warren 1984) and the causative agent of gastritis, peptic ulcer disease and gastric carcinoma (van Amsterdam et al. 2006). Over half of the world’s population are carriers of H. pylori, and it represents one of the most common bacterial infections. Studies from developing countries have shown a high prevalence of H. pylori infection (70–90 % of population), mainly attributed to low socioeconomic status, age, and the poor management of drinking water (García et al. 2014). The exact mode of H. pylori transmission in humans remains unclear. Several authors have suggested that fecal-oral transmission occurs through drinking water supplies, groundwater, recreational waters, freshwaters streams, and estuary and marine waters contaminated by sewage (Mazari-Hiriart et al. 2001; Moreno et al. 2003; Cellini et al. 2004; Carbone et al. 2005; Voytek et al. 2005; Fernández et al. 2007; Twing et al. 2011; Cunachi et al. 2015; Santiago et al. 2015). Although many studies have found Helicobacter DNA in the environment, reports of live cells are extremely rare and highly inconclusive (Percival and Thomas 2009). Water may also act as a reservoir for the bacteria where they can remain for periods of time in a viable but nonculturable state (VBNC), before being ingested through drinking water (Adams et al. 2003; Bellack et al 2006; Assadi et al. 2015). As H. pylori is one of the EPA drinking water contaminant candidates (USEPA 2009), the detection of reservoirs and sources of infection is important for the understanding of this bacterial presence and persistence in natural environments.
Tepuis (or Tepuys) are table-top mountains that rise from 400 to 3000 m above the surrounding savannas or forests in the Guayana highlands in northern South America (Gansser 1954; Santos et al. 2003). Tepuis have been characterized as biological islands due to their shape and isolation from each other (Berry et al. 1995). Roraima is the highest (2700 m), best-known tepui and the only one open for tourism in the Canaima National Park, Venezuela, in the common border with Brazil and Guyana (Aubrecht et al. 2011). The tepui is located in one of the world largest tropical rainforest (Berry et al. 1995; Aubrecht et al. 2011). As Roraima Tepui is a preserved habitat of ecological relevance, the goal of this study was to determine whether Helicobacter was present in the freshwaters of this tepui ecosystem using a combination of enrichment cultures, fluorescent in situ hybridization (FISH), specific PCR assays, 16S rRNA gene sequencing and phylogenetic analysis.
Materials and methods
Site description and sample collection
The study was conducted on the summit of Roraima Tepui, Canaima National Park, at the south eastern corner of Bolivar State in southern Venezuela. The Tepui is formed by a layer of sandstone that lies over an igneous basement (the Guayana Shield) and presents particular topographical, lithological, hydrological and structural features that have promoted the development of an extensive groundwater system, which probably might emerged through localized upwelling on the base of tepui outer wall (600–700 m below the level of the summit) (Galan et al. 2004). The regional climate is semi-humid/tropical with high rainfalls (5700 mm/y), being the rainiest months between May and September, and a relatively dry season lasting the first three months of the year (Nimer 1989).
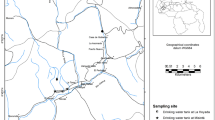
Sampling was authorized with permissions issued by the Environmental Ministery of Venezuela. In October 2007, freshwater samples from two sites were collected at the south central part of the Roraima summit, relatively close to the great wall or south escarpment of the plateau (5°11′52.17″N 60°45′12.45″W): Sampling site 1 named in this study as Laguna de la Puerta (LP), the entrance lagoon of Ojos de Cristal Cave or Roraima Sur Cave System (the world longest cave in quartzite), and sampling site 2 or Los Jacuzzis (LJ), a freshwater well used as a bathing place or pool for visitors (Fig. 1). Both sites are located at the end of a surface sinkhole on Roraima Tepui that takes a permanent stream, which drains through surface vegetation. The water flows are product of surface drainage from precipitation and underground movement (Galan et al. 2004). The adverse environmental conditions and remote geographical location of the tepui difficult the access to the sampling sites and the collection of a large volume and number of water samples to be transported to the laboratory.

Location map of Roraima Tepui and the sampling sites tested. Images were provided by the SPOT satellite, TS-5 (LPAIS, Fundación Instituto de Ingeniería para Investigación y Desarrollo Tecnológico, Caracas, Venezuela) and processed with ArcGIS 10.0 Software (Stanford University)
A volume of 50 mL of freshwater samples were collected with sterile plastic containers (Fisher brand®, USA) in duplicate from each locality (n = 4), about 10 cm below the surface. Particulates in the water were allowed to settle before the water was tested for pH using an Accumet AP61 portable pH meter (Fisher Scientific, USA). Temperature was measured using a RH300 Pshycrometer (Extech instruments, USA).
Bacterial culturing
Bacterial enrichments were established from 1 mL of collected water samples and used to inoculate serum bottles with 9 mL of HP broth containing a mixture of nutritive components (special peptone, beef extract, yeast extract, calf serum with iron, and NaCl), antibiotics (vancomycin, trimethoprim, cefsulodin, amphotericin B, and polymixin B), and a colour indicator system for urease production (urea, phenol red, and HCl) in the presence of a microaerobic gas mixture (5 % CO2, 10 % H2, 85 % N2; Praxair, Inc., Caracas, Venezuela) (Degnan et al. 2003). These enrichments were incubated at 37 °C for 3–5 days in the laboratory. Four freshwater enrichments were obtained from Laguna de la Puerta (LP1 and LP2), and Los Jacuzzis (LJ1 and LJ2) and used for Helicobacter detection by FISH and PCR assays. An aliquot (50 µL) from each enrichment was streaked on HP agar plates (Degnan et al. 2003) and incubated at 37 °C under microaerobic conditions as described above to attempt H. pylori isolation. Eleven urease positive colonies were analysed by biochemical assays and API ATB/Plus system (BioMérieux, France) at Centro Venezolano de Colecciones de Microorganismos (CVCM, Venezuela), cloning and complete 16S rRNA gene sequencing.
Fluorescence in situ hybridization (FISH)
FISH assays were performed by triplicate according to Trebesius et al. (2000) and Contreras et al. (2012) on freshwater enrichments (LP1, LP2, LJ1 and LJ2) obtained in HP broth and clinical strains: H. pylori isolated from a Venezuelan patient with gastritis as positive control and Shigella sp. (ATCC11126) as negative control. Four fluorescent oligonucleotide probes targeting the bacterial 16S rRNA gene were used in this study: two Helicobacter genus-specific probes, HEL274 (5′-GGCCGGATACCCGTCATWGCCT-3′) and HEL717 (5′-AGGTCGCCTTCGCAATGAGTA-3′), both Cy3-5′ labeled (Chan et al. 2005), an H. pylori-specific probe Hpy-1 (5′-CACACCTGACTGACTATCCCG-3′; Cy3-5′ labeled), and a probe for the universal bacteria domain EUB338 (5′-GCTGCCTCCCGT-3′; Alexa Fluor 488 N-5′ labeled) as a positive control to simultaneously visualize the bacteria (Trebesius et al. 2000; Contreras et al. 2012). The hybridization was performed at 54 °C for 90 min and stringent washing at 56 °C for 10 min (Contreras et al. 2012). The slides were air-dried and mounted with anti-fading mounting medium (Immuno-Mount, Thermo Fisher Scientific Inc., USA). The bacteria were visualized with a Nikon Eclipse E600 epifluorescence microscope, which was equipped with filter blocks: G2A for Cy3 (excitation filter: 510–560 nm; beam splitter: 565 nm; barrier filter: 580 nm) and B2A for Alexa Fluor 488 N (excitation filter: 450–490 nm; beam splitter: 500 nm; barrier filter: 515 nm). A Nikon Coolpix 8700 camera digitally acquired the images. Two photographs of the same field were taken with G2A and B2A. The ImageJ 1.34 s (http://rsb.info.nih.gov/Ij) public domain Java image processing package (National Institutes of Health) was used for produces merged images. Positive hybridizations for Helicobacter species were visible as orange labeled-cells, which hybridized with the HEL274 and HEL717 (red) and EUB338 (green) probes, and for H. pylori as red labeled-cells when hybridized with the Hpy-1 (red) probe. Representative images were chosen from about 10 random positions for each sample.
Helicobacter PCR assays and 16S rRNA partial gene sequence analysis
Aliquots of 1 mL from the freshwater enrichments (LP1, LP2, LJ1 and LJ2) were centrifuged at 16,000×g for 1 min. The supernatant was discarded and the DNA was then purified from the pellet using the QIAamp DNA Mini Kit (QIAGEN, USA). Helicobacteriaceae and Helicobacter DNA was amplified using family (Bohr et al. 2002) and genus-specific (Germani et al. 1997) PCR assays for the 16S rRNA gene, respectively. Helicobacteriaceae DNA was amplified by nested PCR; the first with two external universal primers: 8F and 1525R (Contreras et al. 2007), and the second PCR using two internal primers Helicobacteriaceae-specific: BohrF (C97-20) and BohrR (H3A-20), as previously described by Bohr et al. (2002). The initial PCR reaction used a Ready-To-Go PureTaq PCR kit (Amersham Biosciences, USA) and contained 4 µL of extracted DNA and 3 µL of primers (5 µmol L−1) and sterile distilled water to 25 µL. The reaction mixture of the second step (25 µL) contained 3 µL of Helicobacteriaceae-specific primers (5 µmol L−1) and 2 µL of PCR final product from the first step.
Helicobacter DNA was amplified by semi-nested PCR; the first PCR reaction used the Helicobacter genus-specific (HeliF) forward primer and a reverse primer targeting the Epsilon branch of Proteobacteria (EpsilR). The second PCR reaction used the Helicobacter genus-specific internal primers HeliF and HeliR (Germani et al. 1997). The PCR reactions used a Ready-To-Go PureTaq PCR kit and contained 1 µL of extracted DNA and 3 µL of the HeliF/EpsilR primers (5 µmol L−1) and sterile distilled water to 25 µL. The reaction mixture of the second step (25 µL) contained 3 µL of the HeliF/HeliR primers (5 µmol L−1) and 2 µL of the first PCR product. For both nested and semi-nested PCR, the positive control of the first step was prepared by adding 1 µL of H. pylori DNA, the product of which served as the positive control in the subsequent PCR reaction.
The presence of H. pylori DNA was determined using specific PCR assays targeting the glmM (Kansau et al. 1996), ureaA (Peek et al. 1995) and cagA (Rugge et al. 1999) genes. The PCR reactions were performed using the Ready-To-Go PureTaq PCR kit on a GeneAMP PCR System 9700 (Applied Biosystems, USA). Each reaction contained PCR reagents plus 4–6 µL of the extracted DNA, 3 µL of primer mix (5 mol L−1), and sterile distilled water for a final volume of 25 µL. A volume of 1 µL from the positive control was used as a template for each PCR assay. The negative control was a no-template PCR reaction.
Two Helicobacter-specific fragments of the 16S rRNA gene (LP1 and LJ1) were amplified and purified for sequencing using the CONCERT Rapid PCR Purification System kit (GibcoBRL, USA), according to the manufacturer recommendations. Purified amplicons were sequenced at the CeSAAN facility (IVIC, Venezuela) with an ABI PRISM 3130xl Sequencer (Applied Biosystems, USA). The 16S rRNA gene sequences (~292–304 bp) were deposited in the GenBank (Table 1).
Helicobacter sequences together with closest GenBank matches were aligned using the SINA software (Pruesse et al. 2012). The phylogenetic tree was constructed using neighbor-joining method and the Jukes-Cantor model provided in Molecular Evolutionary Genetics Analysis 2.1 software (MEGA, version 5.0) (Tamura et al. 2011), and the stability of grouping was estimated by bootstrap analysis (10,000 replicates).
Cloning, 16S rRNA gene sequencing and phylogenetic inferences
Genomic DNA from eleven urease positive colonies grown on HP agar plates under microaerobic conditions (five colonies obtained from LP and six from LJ) was extracted by ZR Fungal/Bacterial DNA MiniPrep (Zymo Research Corporation, USA). To amplify the 16S rRNA complete gene sequence, a PCR reaction of 40 μL containing 10μL 2X Taq Master Mix (New England Biolabs, USA), ~100 mM of each bacterial primer: 8F (5′-AGAGTTTGATCMTGG CTCAG-3′) and 1391R (5′-GACGGGCGGTGWGTRCA-3′), and 50 ng of DNA template was performed. The PCR amplification was carried out with a hot-start at 94 °C for 8 min, followed by 30 s at 94 °C, 45 s at 58 °C and 1 min at 72 °C for 30 cycles. This was followed by an elongation cycle at 72 °C for 8 min. The PCR products were purified with a ZR DNA Clean & Concentrator-25 Kit (Zymo Research), and cloned into a pTOPO-TA vector and transformed into competent E. coli according to manufacturer’s protocol (Invitrogen, Life Technologies, USA). Clones were picked and screened for unique phylotypes as previously described (Barton et al. 2004). Sequencing was carried out commercially by Agencourt Bioscience and assembled together using DNA Baser software. Assembled sequences were aligned and chimeras removed using the Greengenes NAST algorithm (http://greengenes.lbl.gov). Complete 16S rRNA gene sequences were submitted to the NCBI GenBank database under the accession numbers detailed on Table 1.
A set of sixty-four 16S rRNA sequences (1279–1401 bp) were analysed using QIIME pipeline dependencies (Caporaso et al. 2010). Operational taxonomic units (OTUs) were selected and analysed based on sequence similarity with a 97 % as cut off value. Taxonomic identity was assigned using the RDP classifier (Wang et al. 2007) and taxonomic diversity plots were obtained using QIIME plot generator. Charts were generated at different taxonomic levels.
Results
Environmental parameters
At the collecting sites and during the sampling period the physicochemical parameters tested for freshwater samples were pH 6.9 and temperature 13.3 °C. These parameters were similar to previous reports (Mecchia et al. 2014).
Bacterial culture
At microaerobic conditions described previously (Degnan et al. 2003), growth was observed in four freshwater enrichments in HP broth, two from each site (LP1, LP2, LJ1 and LJ2), and eleven urease positive colonies selected as putative Helicobacter spp. from HP agar (five from LP and six from LJ). Gram staining showed mixed Gram-negative bacteria without the typical spiralled morphology of our target genera. Further attempts to isolate Helicobacter or H. pylori from HP broth enrichments were performed without success.
The eleven urease positive colonies were preliminary identified by API ATB/Plus (BioMérieux, Lyon, France) as Chromobacterium violaceum (99.9 % identity), Photobacterium damsela (86.0 % identity), Pantoea sp. (85.7 % identity), Moraxella lacunata (90.9 % identity) and Serratia liquefaciens/plymuthica (85.9–99.9 % identity), the latter being the most commonly isolated (7/11) from HP enrichments.
Detection of Helicobacter spp. by Fluorescence in situ hybridization (FISH)
While no putative Helicobacter spp. were isolated, the presence of these bacteria was evaluated by FISH on the freshwater enrichments LP1, LP2, LJ1 and LJ2 grown with HP broth. The probes HEL274/HEL717 and Hpy-1 were able to detect Helicobacter and H. pylori in all enrichments, as is shown in Fig. 2. Helicobacter spp. and H. pylori were identified as orange (mixed colour of green and red) (A-B) or red (C-D) whole-cells with different morphological types, predominantly coccoid cells. The presence of other non-Helicobacter bacteria was also detected with the EUB338 (green) probe (A-B).

Detection of Helicobacter spp. and H. pylori by FISH in freshwater enrichments from Roraima Tepui. a, b Helicobacter spp. (orange) and eubacteria (green) cells detected with the mixture of fluorescent probes HEL274+HEL717 (red) and EUB338 (green) in LP and LJ, respectively. c, d H. pylori cells observed with Hpy-1 probe (red) in LP and LJ, respectively. In the images can be seen predominant coccoid cells indicative of the VBNC state. Bar a–d 8 μm
Detection of Helicobacter spp. by PCR and 16S rRNA partial gene sequences
Although H. pylori-specific genes (glmM, cagA and ureA) were not found in any of the enrichments studied, Helicobacteriaceae and Helicobacter DNA were detected by nested and semi-nested PCR in LP (LP1 and LP2) and LJ (LJ1 and LJ2) confirming the presence of members of the genus as observed by FISH.
Helicobacter-specific fragments of the 16S rRNA gene from LP1 and LJ1 enrichments were sequenced and the phylogenetic relationships of these sequences (Helicobacter sp. Roraima enrichment culture LP1Q and J1Q, respectively) with other Helicobacter species based on 16S rRNA sequence data is shown in Fig. 3. Our freshwater enrichments formed a cluster with different primate H. pylori strains, mainly from humans, or H. nemestrinae from pig tailed macaque, both with 99–100 % of homology.

Phylogenetic tree of partial 16S rRNA sequences of Helicobacter obtained from Roraima freshwater enrichments. Tree was constructed using Neighbor-Joining algorithm. Bootstrap values are based on 10,000 replicates and no values are given for groups with Bootstrap values less than 50 %. The scale bar represents 0.03 (3 %) nucleotide sequence difference
Complete 16S rRNA gene sequences of freshwater cultures
In order to identified the presence of other bacterial genera in the eleven cultures obtained from HP agar under microaerobic conditions, we carried out a molecular phylogenetic assessment of the cultures using bacterial clone libraries from 16S rRNA gene sequences and obtained 30 and 34 clones for LP and LJ, respectively. Within each clone library, the OTUs were identified aligning obtained sequences by BLAST using the NCBI database as reference.
These cultures were dominated by two phyla: the Proteobacteria and Actinobacteria. Bacteria from the genera Serratia (40.9 %), Aquitalea (18.2 %), Chromobacterium (15.2 %), Mycobacterium (9.1 %), Acinetobacter (6.1 %), Curvibacter (1.5 %) and Dysgonomonas (1.5 %) were identified on the basis of the complete 16S RNA gene sequences. Phylogenetic inferences show representative sequences grouped mainly in the Gammaproteobacteria, Actinobacteria and Neisseriales (Table 1; Fig. 4).

Phylogenetic composition represented by complete 16S rRNA bacterial sequences of Roraima freshwater cultures grown on HP agar. Pie charts were generated by taxa plotting in QIIME pipeline. The percentage of bacteria from each division was calculated, and results of 1.52 % are shown
Discussion
In this study the presence of Helicobacter spp. was evidenced in freshwaters of a tepui ecosystem. The unsuccessful isolation of these bacteria from HP agar was associated with the large number of coccoid cells observed in the FISH images (Fig. 2), indicating the entrance into a VBNC state. It has been suggested that transformation to a coccoid form enables the bacterium to resist the potentially adverse effects of entering aquatic environments (Bellack et al. 2006). Similarly, several studies have failed to isolate and culture live Helicobacter or H. pylori from environmental samples (Bellack et al. 2006; Delport and van der Merwe 2007). The hostile environment and difficult access to Roraima Tepui determined the low number and volume of water samples and could contribute to the failed isolation of Helicobacter spp.
Good agreement was observed between the detection of Helicobacter by FISH and semi-nested PCR in freshwater enrichments from the two studied sites. Despite our ability to identify H. pylori by FISH and partial sequence analysis of the 16S rRNA, we did not detect the specific genes glmM, ureA and cagA by PCR in the freshwater enrichments. The inability to amplify these typical H. pylori genes could be due to (i) low concentration or fragmented DNA (Fernández et al. 2008) in the freshwater cultures from Roraima, and (ii) the clinical design of primers used in this study which might not be sufficiently robust to provide the accurate detection of H. pylori genes from waters (Voytek et al. 2005). FISH has shown to yield more positive results than PCR for the detection of H. pylori specific genes in environmental samples (Moreno et al. 2003). On the other hand, the discrepancy between the 16S rRNA and specific genes could also be due to variation in primer specificity and sensitivity of the assays (Carbone et al. 2005; Twing et al. 2011). The 16S rRNA PCR assay, while possibly not as specific, is more sensitive than single genes that may vary more in their primer sequence because of their conserved nature within a genome (Twing et al. 2011). The 16S rRNA genes are present in high copy numbers as essential constituents of living cells; therefore, there is no problem with gene loss, as has been previously proposed for H. pylori specific genes (Carbone et al. 2005).
As inferred from the 16S rRNA partial sequencing and phylogenetic analysis, the Helicobacter sequences (LP1 and LJ1) from the two sites examined in the tepui showed a high homology with different H. pylori strains and H. nemestrinae (99–100 %). It is important to note that this part of the analysis was based on a partial 16S rRNA sequencing (292–304 bp). Several reports have probe that 16S rRNA gene analysis are useful for defining prokaryotic relationships from the species to phylum levels. However, cases of very closely related species have highlighted the need to use other molecules to better resolve species identity. For example, H. nemestrinae, once believed to be a novel gastric helicobacter isolated from a pig tailed macaque, was later recognized to be a strain of H. pylori by correction of the 16S rRNA gene sequence and analysis of seven housekeeping and two flagellin genes (Suerbaum et al. 2002). In fact, the 16S rRNA target sequence for H. pylori probe used in our FISH analysis was found to be identical to several H. pylori strains and H. nemestrinae, but in no other species (Trebesius et al. 2000). The presence of these two species of Helicobacter suggests the possibility of anthropogenic or wildlife sources into the freshwaters of the Roraima tepui. These natural aquatic environments might have been secondary contaminated with human waste from tourists or Pemon community. Epidemiological reports of Amerindians groups (Native American peoples) from Venezuela indicate a high prevalence of H. pylori (cagA antigen seropositivity of 95 % in Guajibo-Piaroa) (Contreras et al. 2008). However, a low prevalence of H. pylori (11 %) was found recently in Paraitepuy Pemon community, the aboriginal group established in the basement of Roraima Tepui whom serve as porters and guides for tourists (data not shown). This new information suggests a major influence of tourists in the Helicobacter entrance to Roraima freshwaters. The other possibility is the wildlife origin of helicobacters in Roraima freshwater samples. The high endemism of wildlife described in the Roraima Tepui includes invertebrates (McDiarmid and Gorzula 1989; Macculloch and Lathrop 2007; Brewer-Carias and Audy 2012) and birds (Herrera 2003). Although, there are some reports of mammals from the Canaima National Park represented by lions (Puma concolor), coatíes (Nassua nassua), melero bears (Tamandua tetradactyla), and small howler monkeys (Alouatta) (Brewer-Carias and Audy 2012), no other primates than humans are common visitors at Roraima summit. The limited amount of suitable habitat may not sustain primates or other mammal life on Roraima summit because of the adverse environmental conditions existing in the area (Mecchia et al. 2014). Only a rodent named Roraima mouse (Podoxymys roraimae) has been described as original and collected from the tepui summit, and nowadays is found in a vulnerable condition (Pérez-Zapata et al. 1992). Our evidence placed rodent’s Helicobacter sequences in other clusters far away from Roraima enrichment cultures LP1Q and J1Q (Fig. 3); however, the possibility of Roraima wildlife as Helicobacter carriers is not ruled out.
In addition to Helicobacter detection in these freshwater enrichments, the complete 16S rRNA gene sequencing analysis allowed the identification of other bacteria. The most abundant phylum founded was Proteobacteria (86.37 % of the total sequenced reads), followed by Actinobacteria and Bacteroidetes. These phyla were similar to those reported in water samples (freshwater, seawater, drinking water and wastewater) using high throughput sequencing technology such as 454 pyrosequencing and Illumina (Caporaso et al. 2010; Gilbert et al. 2009; Hong et al. 2010; Kirchman et al. 2010; Ye and Zhang 2013). In our study, Gammaproteobacteria (48.49 %) was the most abundant among the Proteobacteria, followed by Betaproteobacteria (37.88 %); however, Alphaproteobacteria was absent in the freshwater enrichments from Roraima Tepui, while it has been generally dominant in seawater, drinking water and wastewater samples (Gilbert et al. 2009; Hong et al. 2010; Kirchman et al. 2010; Ye and Zhang 2013). The abundance of some members of Gamma and Betaproteobacteria may be favoured by nutrients and selective components present in the HP enrichment broth as well as microaerophilic growth conditions. At the same time, these culture conditions could restrict the growth of genera encompassed within Alphaproteobacteria producing a bias on predominance of certain groups of Proteobacteria. Furthermore, the majority of the sequences of our study were classified to the genus level as Serratia, Aquitalea, Chromobacterium and Mycobacterium (97–99 % of 16S rRNA similarity; Table 1) with the highest percentages of appearance in the two sites (Fig. 4). Serratia, Mycobacterium, Acinetobacter and Dysgonomonas have been reported in abundance in wastewaters (Wiedmann-Al-Ahmad et al. 1994; Wéry et al. 2010; Ye and Zhang 2013), whereas Curvibacter has been only reported in well water (Ding and Yokota 2004). Although the majority of these genera occur in sewage, they are also common in unpolluted water and soil as free-living members (Wiedmann-Al-Ahmad et al. 1994; Ding and Yokota 2004; Wéry et al. 2010; Ye and Zhang 2013).
This represents the first report of Helicobacter in aquatic environments of a little studied and unique ecosystem as the Roraima Tepui. Although further research is necessary to elucidate the human or wildlife Helicobacter origin, our results contribute to the current knowledge of these bacteria in the aquatic environment and expand their known/potential sites outside the human host. Additional attempts should be incorporated to isolate Helicobacter from environmental samples and further research is necessary to assess the bacteriological quality of Roraima freshwaters in order to estimate the possible fecal contamination sources.
References
Adams BL, Bates TC, Oliver JD (2003) Survival of Helicobacter pylori in a natural freshwater environment. Appl Environ Microbiol 69:7462–7466
Assadi M, Chamanrokh P, Whitehouse CA, Huq A (2015) Methods for detecting the environmental coccoid form of Helicobacter pylori. Front Public Health. doi:10.3389/fpubh.2015.00147
Aubrecht R, Lánczos T, Gregor M, Schlögl J, Šmída B, Liščák P, Brewer-Carías C, Vlček L (2011) Sandstone caves on Venezuelan tepuis: return to pseudokarst? Geomorphology 132:351–365
Barton HA, Taylor MR, Pace NR (2004) Molecular phylogenetic analysis of a bacterial community in an oligotrophic cave environment. Geomicrobiol J 21:11–20
Bellack NR, Koehoorn MW, MacNab YC, Morshed MG (2006) A conceptual model of water’s role as a reservoir in Helicobacter pylori transmission: a review of the evidence. Epidemiol Infect 134:439–449
Berry PE, Huber O, Holst BK (1995) Floristic analysis and phytogeography. In: Berry PE, Holst BK, Yatskievych K (eds) Flora of the Venezuelan Guayana, vol 1. Missouri Botanical Garden, St. Louis, pp 161–192
Bohr UR, Primus A, Zagoura AA, Glasbrenner B, Wex T, Malfertheiner P (2002) A group-specific PCR assay for the detection of Helicobacteriaceae in human gut. Helicobacter 7:378–383
Brewer-Carias C, Audy M (2012) Entrañas del Mundo Perdido. Segunda Edicion: Febrero 2012. Caracas: Carlos Capriles de ALTOLITHO C.A
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez A, Goodrich JK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carbone M, Maugeri TL, Gugliandolo C, La Camera E, Biondo C, Fera MT (2005) Occurrence of Helicobacter pylori DNA in the coastal environment of southern Italy (Straits of Messina). J Appl Microbiol 98:768–774
Cellini L, Del Vecchio A, Di Candia M, Di Campli E, Favaro M, Donelli G (2004) Detection of free and plankton-associated Helicobacter pylori in seawater. J Appl Microbiol 97:285–292
Chan V, Crocetti G, Grehan M, Zhang L, Danon S, Lee A, Mitchell H (2005) Visualization of Helicobacter species within the murine cecal mucosa using specific fluorescence in situ hybridization. Helicobacter 10:114–124
Contreras M, Morales A, García-Amado MA, De Vera M, Bermúdez V, Gueneau P (2007) Detection of Helicobacter-like DNA in the gastric mucosa of Thoroughbred horses. Lett Appl Microbiol 45:553–557
Contreras M, Pujol FH, Pérez-Pérez GI, Marini E, Michelangeli FA, Ponce L, Domínguez-Bello MG (2008) Helicobacter pylori seroprevalence in Amerindians from isolated locations. Am J Trop Med Hyg 78:574–576
Contreras M, Salazar V, García-Amado MA, Reyes N, Aparcero M, Silva O, Castro D, Romero R, Gueneau P et al (2012) High frequency of Helicobacter pylori in the esophageal mucosa of dyspeptic patients and its possible association with histopathological alterations. Int J Infect Dis 16:e364–e370
Cunachi AM, Fernández-Delgado M, Suárez P, Contreras M, Michelangeli F, García-Amado MA (2015) Detection of Helicobacter DNA in different water sources and penguin feces from Greenwich, Dee and Barrientos Islands, Antarctica. Polar Biol. doi:10.1007/s00300-015-1879-5
Degnan AJ, Sonzogni WC, Standridge JH (2003) Development of a plating medium for selection of Helicobacter pylori from water samples. Appl Environ Microbiol 69:2914–2918
Delport W, van der Merwe SW (2007) The transmission of Helicobacter pylori: the effects of analysis method and study population on inference. Best Pract Res Clin Gastroenterol 21:215–236
Ding L, Yokota A (2004) Proposals of Curvibacter gracilis gen. nov., sp. nov. and Herbaspirillum putei sp. nov. for bacterial strains isolated from well water and reclassification of [Pseudomonas] huttiensis, [Pseudomonas] lanceolata, [Aquaspirillum] delicatum and [Aquaspirillum] autotrophicum as Herbaspirillum huttiense comb. nov., Curvibacter lanceolatus comb. nov., Curvibacter delicatus comb. nov. and Herbaspirillum autotrophicum comb. nov. Int J Syst Evol Microbiol 54:2223–2230
Fernández M, Contreras M, Suárez P, Gueneau P, García-Amado MA (2007) Use of HP selective medium to detect Helicobacter pylori associated with other enteric bacteria in seawater and marine molluscs. Lett Appl Microbiol 45:213–218
Fernández JL, Cartelle M, Muriel L, Santiso R, Tamayo M, Goyanes V, Gosálvez J, Bou G (2008) DNA fragmentation in microorganisms assessed in situ. Appl Environ Microbiol 74:5925–5933
Galan C, Herrera FF, Carreño R (2004) Geomorfologia e hidrología del Sistema Roraima Sur, Venezuela, la mayor cavidad del mundo en cuarcitas: 10,8 km. Bol Soc Venezolana Espeleol 38:2–16
Gansser H (1954) The Guiana Shield. Ec Geol Helv 47:77–112
García A, Salas-Jara MJ, Herrera C, González C (2014) Biofilm and Helicobacter pylori: from environment to human host. World J Gastroenterol 20:5632
Germani Y, Dauga C, Duval P, Huerre M, Levy M, Pialoux G, Sansonetti P, Grimont PA (1997) Strategy for the detection of Helicobacter species by amplification of 16S rRNA genes and identification of H. felis in a human gastric biopsy. Res Microbiol 148:315–326
Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T, Somerfield PJ, Huse S, Joint I (2009) The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 11:3132–3139
Goodwin CS, Armstrong JA, Chilvers T, Peters M, Colins MD, Sly L, Mcconnell W, Harper W (1989) Transfer of Campylobacter pylori and Campylobacter mustelae to Helicobacter gen. nov. as Helicobacter pylori comb. nov. and Helicobacter mustelae comb. nov., respectively. Int J Syst Bacteriol 39:397–405
Herrera FF (2003) Distribución actualizada de las colonias de guácharos (Steatornis caripensis) en Venezuela. Bol Soc Venezolana Espel 36:6–10
Hong P, Hwang C, Ling F, Andersen GL, LeChevallier MW, Liu WT (2010) Pyrosequencing analysis of bacterial biofilm communities in water meters of a drinking water distribution system. Appl Env Microbiol 76:5631–5635
Kansau I, Raymond J, Bingen E, Courcoux P, Kalach N, Bergeret M, Braimi N, Dupont C, Labigne A (1996) Genotyping of Helicobacter pylori isolates by sequencing of PCR products and comparison with the RAPD technique. Res Microbiol 147:661–669
Kirchman DL, Cottrell MT, Lovejoy C (2010) The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Env Microbiol 12:1132–1143
Macculloch RD, Lathrop A (2007) Herpetofauna of Mount Roraima, Guiana Shield Region, Northeastern South America. Herpetol Rev 38:24–30
Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1:1313–1314
Mazari-Hiriart M, López-Vidal Y, Calva JJ (2001) Helicobacter pylori in water systems for human use in Mexico City. Water Sci Technol 43:93–98
McDiarmid RW, Gorzula S (1989) Aspects of the reproductive ecology and behavior of the Tepui Toads, Genus Oreophrynella (Anura, Bufonidae). Copeia 2:445–451
Mecchia M, Sauro F, Piccini L, Waele J, Sanna L, Tisato N, Lira J, Vergara F (2014) Geochemistry of surface and subsurface waters in quartz-sandstones: significance for the geomorphic evolution of tepui table mountains (Gran Sabana, Venezuela). J Hydrol 511:117–138
Moreno Y, Ferrús MA, Alonso JL, Jimenez A, Hernández J (2003) Use of fluorescent in situ hybridization to evidence the presence of Helicobacter pylori in water. Water Res 37:2251–2256
Nimer E (1989) Climatologia do Brazil. IBGE, Rio de Janeiro
Peek R, Miller G, Tham K, Pérez-Pérez GI, Cover T, Atherton JC, Dunn GD, Blaser MJ (1995) Detection of Helicobacter pylori gene expression in human gastric mucosa. J Clin Microbiol 33:28–32
Percival SL, Thomas JG (2009) Transmission of Helicobacter pylori and the role of water and biofilms. J Water Health 7:469–477
Pérez-Zapata A, Lew D, Aguilera M, Reig OA (1992) New data on the systematics and karyology of Podoxymys roraimae (Rodentia, Cricetidae). Z Saugetierkunde 57:216–224
Pruesse E, Peplies J, Glockner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829
Rugge M, Busatto G, Cassaro M, Shiao YH, Russo V, Leandro G, Avellini C, Fabiano A, Sidoni A et al (1999) Patients younger than 40 years with gastric carcinoma: Helicobacter pylori genotype and associated gastritis phenotype. Cancer 85:2506–2511
Santiago P, Moreno Y, Ferrús MA (2015) Identification of viable Helicobacter pylori in drinking water supplies by cultural and molecular techniques. Helicobacter 20:252–259
Santos JOS, Potter PE, Reis NJ, Hartmann LA, Fletcher IR, Mcnaughton N (2003) Age, source, and regional stratigraphy of the Roraima Supergroup and Roraima-like outliers in northern South america based on U-Pb geochronology. GSA Bull 115:331–348
Suerbaum SC, Kraft C, Dewhirst FE, Fox JG et al (2002) Helicobacter nemestrinae ATCC 49396T is a strain of Helicobacter pylori (Marshall et al. 1985) Goodwin et al. 1989, and Helicobacter nemestrinae Bronsdon et al. 1991 is therefore a junior heterotypic synonym of Helicobacter pylori. Int J Syst Evol Microbiol 52:437–439
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Trebesius K, Panthel K, Strobel S, Vogt K, Faller G, Kirchner T, Kist M, Heesemann J, Haas R (2000) Rapid and specific detection of Helicobacter pylori macrolide resistance gastric tissue by fluorescent in situ hybridisation. Gut 46:608–614
Twing KI, Kirchman DL, Campbell BJ (2011) Temporal study of Helicobacter pylori presence in coastal freshwater, estuary and marine waters. Water Res 45:1897–1905
USEPA. Contaminant Candidate List 3-CCL 2009 [cited 2012 November]
van Amsterdam K, van Vliet AHM, Kusters JG, van der Ende A (2006) Of microbe and man: determinants of Helicobacter pylori-related diseases. FEMS Microbiol Rev 30:131–156
Voytek MA, Ashen JB, Fogarty LR, Kirshtein JD, Landa ER (2005) Detection of Helicobacter pylori and fecal indicator bacteria in five North American rivers. J Water Health 3:405–422
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wéry N, Monteil C, Pourcher A, Godon J (2010) Human-specific fecal bacteria in wastewater treatment plant effluents. Water Res 44:1873–1883
Wiedmann-Al-Ahmad M, Yichy H, Schon G (1994) Characterization of Acinetobacter type strains and isolates obtained from wastewater treatment plants by PCR Fingerprinting. Appl Environ Microbiol 60:4066–4071
Ye L, Zhang T (2013) Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl Microbiol Biotechnol 97:2681–2690
Acknowledgments
This work was partially funded by Grants from Decanato de Investigación y Desarrollo of the Universidad Simón Bolívar and Instituto Venezolano de Investigaciones Científicas. The authors gratefully acknowledge to Grisel Velásquez, UniSIG – IVIC, for her assistance with Fig. 1 (Location map of Roraima Tepui).
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Fernández-Delgado and P. Suárez contributed equally to this paper.
Rights and permissions
About this article

Cite this article
Fernández-Delgado, M., Giarrizzo, J.G., García-Amado, M.A. et al. Evidence of Helicobacter spp. in freshwaters from Roraima Tepui, Guayana Shield, South America. Antonie van Leeuwenhoek 109, 529–542 (2016). https://doi.org/10.1007/s10482-016-0658-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-016-0658-9