
Abstract
The Dll4-Notch-signaling pathway regulates capillary sprouting via the specification of endothelial tip cells. While VEGF is a potent inducer of Dll4 expression, the intracellular mediators that stimulate its expression remain poorly defined. The protein tyrosine phosphatase PTPRJ/DEP-1 is required for angiogenesis in normal or pathological contexts through its modulation of VEGF signaling. Here, we show that in DEP-1 KO mice, retinas at post-natal day 5 show enlarged blood vessels, as well as an increased number of tip cells and vessel branching points at the migrating front of the vascular plexus. Consistent with these observations, the proliferation of endothelial cells is increased in the retinas of DEP-1 KO mice, as revealed by phospho-histone H3 staining, and increased phosphorylation of ERK1/2 in HUVECs transfected with DEP-1 siRNA. The expression of Dll4 was decreased in retinas of DEP-1 KO mice and was associated with decreased Notch activation. Mechanistically, reduced Dll4 expression in the absence of DEP-1 was correlated with the inhibition of the Src/Akt/β-Catenin-signaling pathway in HUVECs. Conversely, overexpression of WT DEP-1 in cultured endothelial cells, but not of mutants unable to activate Src-dependent signaling, promoted Dll4 expression. Inhibition of Src, Akt, and β-catenin transcriptional activity, leading to the inhibition of Dll4 expression, further suggested that their activation through a DEP-1-dependent pathway was required to promote Dll4 expression in VEGF-stimulated endothelial cells. Altogether, these data demonstrate that DEP-1, via Akt and β-catenin, is a significant promoter of the VEGF-induced Dll4-Notch pathway, and can contribute to the regulation of the tip and stalk cell phenotypes of endothelial cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DEP-1 is a receptor-like protein tyrosine phosphatase (PTP) expressed in several cell types. It was initially identified as a negative regulator of cell proliferation, and consistent with its increased expression in confluent epithelial and endothelial cells [1], was suggested to contribute to cell-cell contact inhibition [2]. Many of its identified substrates are growth factor receptors, including VEGFR2, as well as ERK1/2 and cell adhesion proteins [2,3,4]. In vivo, embryonic lethality was reported in a knock-in mutant mouse model, in which a mutant DEP-1 allele was constructed to eliminate its phosphatase activity, which revealed increased endothelial cell proliferation and a disorganized vascular system, suggesting that DEP-1 might regulate neovascularization [5]. Depletion of DEP-1 during the early development of zebrafish caused defects in vasculature, resulting in aberrant blood circulation [6]. In vitro, our group reported that in endothelial cells, DEP-1 binds Src and dephosphorylates its inhibitory tyrosine (Y529), which leads to the auto-phosphorylation of its activating tyrosine (Y418) [4, 7]. We further showed that this regulatory mechanism was involved in the promotion of VEGF-induced capillary formation and vascular permeability in vitro. Surprisingly, no apparent phenotypes were observed in DEP-1 knock-out (KO) mice [8,9,10]. However, further characterization of these mice revealed impaired signaling in subsets of hematopoietic cells, mostly associated with reduced activation of Src family kinases [8, 11,12,13]. Furthermore, we recently showed that VEGF-induced Src activation, capillary formation in Matrigel in vivo and from aortic explants as well as vascular permeability were impaired in DEP-1 KO mice, prohibiting tumor growth and metastasis [14]. All of these results thus indicated that DEP-1 might be involved in the initial steps of capillary sprouting by regulating Src activity.
During sprouting, endothelial cells are activated by VEGF and display specific phenotypes [15]. The highly mobile tip cell leads the extension of the nascent capillary and harbors numerous protrusions that allow the polarized migration of the cell towards the VEGF gradient [16]. Adjacent cells are defined as stalk cells and proliferate to form the growing capillary [16]. Once capillary extension is completed, endothelial cells return to a quiescent-like state associated with the formation of stable cell–cell junctions and vessel maturation, and these are referred to as phalanx cells [17]. Numerous studies have demonstrated the importance of the Notch pathway in regulating these phenotypes [18,19,20,21]. Indeed, VEGF stimulation of endothelial tip cells induces the expression of Dll4, which interacts with the Notch receptor on neighbouring cells and induces its cleavage and the subsequent release of the Notch intracellular domain (NICD) [19, 20]. NICD then acts as a transcription factor co-activator, repressing the tip cell phenotype and VEGF responsiveness [22]. This is accomplished in part by downregulating and upregulating VEGFR2 and VEGFR1, respectively, thus favouring the stalk cell phenotype [20, 21, 23, 24]. Impairment of Notch signaling using antibodies against Dll4 or Notch1, Notch decoys, or gamma secretase inhibitors results in non-productive angiogenesis and oversprouting of blood vessels, without adequate perfusion [21, 25,26,27]. Haploinsufficiency of Dll4 or Notch1 was also shown to be embryonic-lethal, due to highly disorganized vascular structures [28,29,30], further emphasizing the crucial role of Dll4/Notch interactions during vascular development [19, 21, 31, 32].
In the current study, we show that during retinal vascular development, DEP-1 deletion leads to increased vascular density and tip cell formation, correlating with decreased Dll4 expression. At the molecular level, regulation of Dll4 expression is achieved through DEP-1-dependent Src/Akt pathway activation, leading to the phosphorylation (S552) and stabilization of β-catenin, a known promoter of Dll4 transcription [33]. Consequently, we show that DEP-1 acts as an important regulator of Notch signaling and endothelial sprouting during retinal vascular development.
Experimental procedures
Antibodies and reagents
Antibodies against Src, non-pY529Src, cleaved Notch (NICD), Dll4, pS33/S37/T41β-catenin, pS552β-catenin, pY783PLCγ, pS473Akt, Akt, p44/p42 MAPK, pT202/Y204p44/p42MAPK, pS9GSK3β, pY1175VEGFR2 and horseradish peroxidase (HRP)-conjugated secondary IgGs were obtained from Cell Signaling Technology. pY418Src and Alexa Fluor 594-coupled donkey anti-rabbit antibodies were purchased from Invitrogen. Goat anti-DEP-1 and anti-Dll4 antibodies were obtained from R&D Systems. PLCγ antibody was acquired form Millipore. β-catenin and GSK3β antibodies were purchased from BD Pharmingen (BD Biosciences). Actin, ERG1 and Phospho-histone H3 antibodies were acquired from Abcam. Recombinant human VEGF-A was obtained from the Biological Resources Branch Preclinical Repository of the National Cancer Institute—Frederick Cancer Research and Development Center. PP2, DMSO and MG132 were purchased from Sigma. LY294002 and PD98059 were obtained from Cell Signaling Technology. Griffonia simplicifolia FITC-lectin B4 (IsoB4) was purchased from Vectors laboratories. Alexa Fluor 594-labeled IsoB4 was obtained from Life Technologies. PNU74654 and PKF115584 were acquired from Tocris Biosciences.
Mice
DEP-1 KO and WT mice were derived from DEP-1+/− mice in the FVB/N strain background. These were obtained after backcrossing the previously described DEP+/− mice (C57/BL6J background) for at least ten generations with FVB/N mice [9]. All animal experiments were conducted in accordance with the guidelines set out by the CRCHUM Animal Experimentation Ethics Committee.
Fluorescence immunostaining
Eyes of 5-day-old pups were collected and fixed 2 h at room temperature in 4% formalin in PBS, and then enucleated as previously described [34]. For antibody staining, retinas were dissected, permeabilized overnight at 4 °C in PBS (with 1% BSA and 0.5% Triton X-100), rinsed twice and then incubated overnight with the appropriate primary antibodies (1/100). Retinas were next washed in PBS, incubated 2 h at room temperature with secondary antibodies and IsoB4 (1/100) in PBS, rinsed in PBS, and flat mounted. Retinas were analyzed by immunofluorescence microscopy using an Olympus FV1000 microscope. From whole-mount retinas, radial expansion (distance of the migrating front from the center of the retina) was measured using ImageJ. Retinal vessel branch points and vessel length were assessed using the Angiogenesis Analyzer application (ImageJ). Tip cell density at the migrating front was calculated based on the number of tip cells reported by 100 µm of migrating front (tip cell density from the four petals of each retina for at least nine retinas per condition). Retinal endothelial cell nuclear density (ERG1 staining) was calculated using ImageJ (particle analysis) and normalized to vascular (IsoB4) area.
Collection of retinal proteins
The eyes of 5-day-old pups were collected and dissected. Isolated retinas were frozen on dry ice and lysed in RIPA buffer pH 7.4 containing 50 mM Tris-HCl, 150 mM NaCl, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 10 µg/mL aprotinin, 10 µg/mL leupeptin (Roche Applied Science), 1 mM phenylmethylsulfonyl fluoride, 5 mM sodium fluoride, and 1 mM sodium vanadate. Protein concentrations were determined using the BCA protein assay reagent (Pierce).
Cell culture and transfection
Human umbilical vein endothelial cells (HUVECS; Cascade Biologics/Invitrogen) were cultivated between passages 1 and 4 on gelatin (0.2%)-coated tissue culture plates (Corning) in M200 medium (Invitrogen) supplemented with LSGS (Low serum growth supplement; Invitrogen) and gentamycin (50 µg/ml, Wisent). Cells (4 × 104 cells/cm2) were transfected in OptiMEM medium (Invitrogen) 20 h after plating with cDNA constructs and Lipofectamin 2000 (Invitrogen) according to the manufacturer’s instructions. Plasmids encoding WT human DEP-1 and the C/S mutant (in pmT2 vector) were provided by Nicholas Tonks (Cold Spring Harbor Laboratory, NY, USA) [1]. The Y1311F/Y1320F DEP-1 mutant was generated as previously described [7].
Nucleofection of HUVECs
HUVECs (2–5 × 106cells) were nucleofected using the Nucleofector II (program U-001; Lonza) with DEP-1 (Hs_PTPRJ_3_HP) or AllStars control siRNAs (Qiagen, Toronto, ON, Canada) using the Amaxa HUVEC Nucleofector Kit following manufacturer’s recommendations [35]. Cells were then incubated in M200 supplemented with LSGS for 48 h before being processed for in vitro experiments.
Cell lysis and western blotting
For in vitro cell stimulations, nucleofected cells were plated at 3.5 × 105 cells/cm2 on 0.2% gelatin-coated tissue culture plates for 48 h and then starved in M200 with 1% FBS for 3 h prior to an overnight stimulation with VEGF (50 ng/mL). For experiments with chemical inhibitors, HUVEC cells were plated on gelatin (0.2%) at 3.2 × 105 cells/cm2 for 48 h. Cells were then starved and pre-treated with inhibitors or vehicle for 3 h and stimulated with VEGF (50 ng/mL) for 18 h. Transfected cells were similarly starved in M200 with 1% FBS for 3 h and stimulated with VEGF overnight before lysis in a 50 mM HEPES, pH 7.5, buffer containing 0.5% Nonidet P-40, 0.5% Triton X-100, 10% glycerol, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 µg/mL aprotinin, and 10 µg/mL leupeptin. Protein concentration was determined using the Bradford assay. Protein lysates were resolved by SDS-PAGE and transferred on nitrocellulose membranes (Bio-Rad). Western blotting and ECL detection (Amersham Biosciences/GE Healthcare) were then performed according to the manufacturer’s recommendations using the specified antibodies [4, 14].
Data analysis
Densitometric analyses were performed with the Quantity One 4.6.3 software (Bio-Rad, Mississauga, ON, Canada). Levels of phosphorylated proteins were normalized to total proteins. PLCγ levels were used to normalize Dll4 and β-catenin western blots. Statistical significance was evaluated with the unpaired t test using GraphPad Prism software. P values less than 0.05 were considered to be significant.
Results
DEP-1 modulates tip cell formation and vessel branching during retinal vascularization
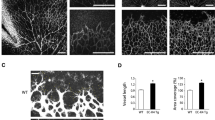
DEP-1 has been shown to regulate the formation of capillaries in vitro and in vivo, prompting us to evaluate its function in vascular sprouting [7, 14]. The retinal model of developmental angiogenesis is widely used to study developmental angiogenesis, where blood vessels emerge from the optic nerve and sprout towards the periphery during the first week of life [16, 36]. Immunostaining of retinas revealed that DEP-1 is expressed in the retinal vasculature (Supplementary Fig. 1). DEP-1 immunostaining was observed in capillaries and the arterial and venous vasculature, and its expression appeared stronger in endothelial cell junctions (Supplementary Fig. 1). The role of DEP-1 in sprouting angiogenesis was investigated in control and DEP-1 KO mice 5 days after birth (P5) during the development of the superficial retinal vascular plexus (Fig. 1). We observed that radial expansion was slightly decreased in DEP-1 KO mice (Fig. 1a, b) while there were no significant differences in radial outgrowth between WT and heterozygote animals. Further analysis of the retinal vascular plexus revealed that DEP-1 KO mice exhibited significantly higher vascular densities at the migrating front, which were characterized by an increased number of branch points (nodes) and a greater length of vascular segments (Fig. 1c, d). Although there was an increase in the vascular surface area (IsoB4-positive) that was accompanied by an increase in endothelial cell numbers in DEP-1 KO mice, analysis of the density of endothelial cells within the retinal vasculature, which was performed by evaluating the number of ERG1-positive EC nuclei in IsoB4-positive blood vessels, revealed no significant difference in the overall size of endothelial cells between WT and DEP-1 KO mice, implying that ECs in DEP-1 KO retinas were able to cover the same surface area as ECs in WT retinas (Fig. 1e, f). However, the venous vascular plexus appeared denser and more branched, and veins were enlarged and showed an increased number of endothelial cells relative to vascular IsoB4-stained area, indicative of increased proliferation (Fig. 1g, h). Since the number of tip cells determines the complexity of the vascular bed, we next investigated the density of tip cells at the leading edge of retinas from control and DEP-1 KO mice. Consistent with the increased number of blood vessel segments and branch points, a greater number of tip cells per length of vascular front was observed in the retinas of DEP-1 KO mice (Fig. 1i). Finally, we analyzed the retinal vasculature in P12 mice to evaluate whether the hypervascularization phenotype persists in DEP-1 KO mice. At this stage of development, we observed no differences in the vasculature of DEP-1 KO mice compared to WT littermates, showing that the hypervascularized phenotype of these mice is transient and has resolved by P12 (Fig. 1j). Together, these data thus suggest that DEP-1 contributes to the regulation of the tip cell phenotype during the developmental stage of retinal sprouting angiogenesis.

Loss of DEP-1 increases sprouting during developmental retinal angiogenesis. a Retinas were isolated from 5-day-old mice pups, fixed and blood vessels were stained using IsoB4 and measurements of radial expansion were performed in WT, heterozygote and DEP-1 KO mice. b Quantification of vascular radial expansion in WT, heterozygote and DEP-1 KO mice. N = 9 WT, 10 heterozygotes and 9 homozygotes per group. c Images of the vascular front of retinas harvested from WT, heterozygote and DEP-1 KO mice. d Quantification of the number of branch points (nodes) and the length of vascular segments in WT, heterozygote and DEP-1 KO mice. N = 9 WT, 10 heterozygotes and 9 homozygotes per group. e ERG1 staining of WT and DEP-1 KO mice. f Quantification of endothelial nuclei, normalized to vascular area (IsoB4 staining). N = 5 animals/group. g, h Images and quantification of the number of endothelial nuclei from veins at the first branching point in WT and DEP-1 KO animals. N = 5 animals/group. i Images and quantification of the number of endothelial tip cells in WT and DEP-1 KO animals. N = 9 animals/group. j Representative images and quantification of vascular nodes in P12 retinas. N = 5 animals/group. Data ± SEM are shown. Student’s t test: *p ≤ 0.05; **p ≤ 0.01.
Increased proliferation of endothelial cells in the retinas of DEP-1 KO mice
The formation of a new vascular plexus involves the proliferation of ECs in response to VEGF. As the retinas of DEP-1 KO mice showed increased vascular density at the leading edge of migrating capillaries, we evaluated whether this phenotype was associated with altered EC proliferation. To investigate the possible contribution of DEP-1 to the proliferation of endothelial cells during vascularization, retinas were stained using the mitosis marker phospho-histone H3 (PH3). Quantification of PH3+/IsoB4 + cells revealed increased numbers of proliferating cells at the vascular front of DEP-1 KO mice at P5, while no differences were observed in the vascular plexus closer to the optic nerve (Fig. 2a). In endothelial cells, VEGF-induced proliferation is mainly mediated by PLCγ-dependent ERK1/2 activation [37, 38]. To assess the status of this signaling pathway in control and DEP-1 KO mice, whole retina lysates were analyzed by Western blot. We observed that ERK1/2 activation was increased in the retinas of DEP-1 KO mice, while the phosphorylation of its upstream regulator, PLCγ, was not affected (Fig. 2b). Collectively, these results demonstrate an anti-proliferative function of DEP-1 in endothelial cells of the retina and suggest that this feature may contribute to the increased neovascularization observed in DEP-1 KO mice.

DEP-1 acts as an inhibitor of endothelial cell proliferation during retinal development. a The retinas of 5-day-old pups were collected, fixed and processed for immunofluorescence using FITC-IsoB4 and PH3 antibody. The number of PH3 and IsoB4-positive cells was determined in the retinal vascular plexus and near the migrating front (5 images/animal; N = 4 animals/group). b Protein extracts from retinas of WT and DEP-1 KO mice were analyzed by Western blotting using the indicated antibodies. Results were quantified by densitometric analyses; N = 5 WT and 5 DEP-1 KO mice. Data ± SEM are shown. Student’s t test: *p ≤ 0.05; **p ≤ 0.01.
Loss of DEP-1 leads to Dll4 down-regulation and decreased Notch cleavage
In addition to increased vessel branching and enlarged veins, DEP-1 KO mice exhibited an increased number of tip cells at the migrating front (Fig. 1). Since these features were reminiscent of the retinal phenotype associated with impaired Notch signaling [19, 21, 23, 31, 32], we evaluated whether DEP-1 regulates Notch activity in the retinal endothelium. We first assessed the expression of the Notch ligand Dll4, which is an important regulator of Notch activity in endothelial cells during vascular development [39]. Interestingly, we observed an important decrease of Dll4 expression in whole-retina lysates from DEP-1 KO mice compared to controls (Fig. 3a). Immunostaining experiments in retinas of DEP-1 KO mice also revealed a significantly decreased expression of Dll4 at the vascular front (Supplementary Fig. 2). Dll4 binding to Notch leads to its cleavage and the generation of the Notch intracellular domain (NICD). Consistent with the decreased expression of Dll4, we also observed a great reduction in the levels of NICD in the retinas of DEP-1 KO mice compared to WT mice (Fig. 3a). As such, these data suggest that DEP is an important mediator of Dll4 expression and Notch activation in the retinal vasculature.

Expression of the Notch ligand Dll4 depends on DEP-1 expression. a Retinas of 5-day-old pups were collected, lysed and analyzed by Western blot using the specified antibodies. Dll4 expression was inhibited in DEP-1 KO animals, which correlates with the decreased cleavage of Notch (NICD). b Activation of the Src-Akt-signaling pathway was analyzed by Western blotting in retinas harvested from P5 pups. c HUVECs nucleofected with siRNAs were starved 3 h in 1% FBS medium, stimulated for 18 h with VEGF (50 ng/mL) and lysed. Western blot analysis was performed using antibodies targeting signaling molecules of the Src/Akt signaling pathway. Quantification of blots from four independent experiments are shown on the right. d Lysates from HUVECs nucleofected with siRNA, starved and stimulated with VEGF as in c were analyzed by Western blot using antibodies targeting pY1175VEGFR2 and total VEGFR2. Quantification of blots from four independent experiments are shown below. e HUVECs were treated with Src or PI3K inhibitors, PP2 (10 µM) and LY290042 (10 µM), then starved and stimulated with VEGF for 18 h, followed by Western blot analysis with the indicated antibodies. Quantification of blots from four independent experiments are shown on the right. f DEP-1 WT, DEP-1 Y1311F/Y1320/F (YY/FF), DEP-1 C/S or empty vector (pmT2) were transfected in HUVECs using Lipofectamin 2000. Cells were then starved, stimulated with VEGF and lysed as described in c. Lysates were analyzed by Western blot using the specified antibodies. Densitometric analyses were performed on at least 4 independent experiments. Data ± SEM are shown. Student’s t test: *p ≤ 0.05; **p ≤ 0.01.
DEP-1 promotes VEGF-dependent Dll4 expression through Src/Akt signaling
Recent reports have suggested that Akt regulates Dll4 expression, Notch cleavage, and sprouting angiogenesis [40, 41]. Furthermore, it was previously demonstrated that in response to VEGF stimulation, DEP-1 promotes Akt activation through a Src/Gab1/PI3K-signaling pathway [4]. In endothelial cells, DEP-1 dephosphorylates the inhibitory tyrosine of Src (Y529), which leads to Src autophosphorylation (Y418) and full activation [4, 7]. Consistent with these earlier findings, Src and Akt (S473) activation levels were impaired in retina lysates of DEP-1 KO mice that showed significant reductions of Dll4 expression and Notch cleavage (Fig. 3a, b). To further investigate this mechanism, DEP-1 was silenced in HUVECs. Similarly, we observed that abrogation of DEP-1 expression in HUVECs cultured in the presence or absence of VEGF resulted in the inhibition of Dll4 expression, which was associated with reduced Src and Akt phosphorylation (Fig. 3c). In contrast, as previously reported [14], no reduction in the relative phosphorylation of VEGFR2, a DEP-1 substrate, was observed in cells transfected with DEP-1 siRNAs (Fig. 3d), indicating that DEP-1 acts downstream of VEGFR2 to promote VEGF-dependent signaling. Additional experiments performed with inhibitors of Src family kinases and PI3K, PP2 and LY294002, respectively, further demonstrated the implication of Src and Akt in the VEGF-dependent expression of Dll4 in endothelial cells (Fig. 3e). To bring further support to this conclusion, the converse experiment was performed and WT DEP-1 was overexpressed in HUVECs. In this case, as expected, increased Dll4 expression was observed (Fig. 3f). However, overexpression of DEP-1 mutants unable to activate Src (YY/FF) and C/S mutants abrogated VEGF-induced expression of Dll4 (Fig. 3f) [7]. Altogether, these results thus establish the importance of the DEP-1/Src/Akt-signaling pathway in the induction of VEGF-dependent Dll4 expression.
DEP-1-mediated β-catenin stabilization is required for Dll4 expression in VEGF-stimulated endothelial cells
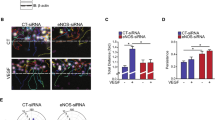
β-catenin/TCF complexes have been shown to bind to the Dll4 promoter and to induce Dll4 expression, demonstrating that β-catenin-transcriptional activation is crucial for proper Notch signaling [33]. Akt signaling has also been shown to regulate β-catenin function via two main mechanisms: (1) directly, by phosphorylating β-catenin on S552, which is associated with β-catenin translocation to the nucleus [42] and thus contributes to its transcriptional activation, (2) indirectly, by inactivating GSK3β, a key component of the β-catenin destruction complex, leading to β-catenin stabilization and increased β-catenin protein levels [42, 43]. Since VEGF is an important inducer of Dll4 and was reported to promote β-catenin stabilization and activation, we postulated that DEP-1 might be involved in the regulation of these processes via Akt activation [4, 7, 19, 44,45,46]. First, we assessed the Akt-mediated phosphorylation of β-catenin on S552 and observed its inhibition in the retinas of DEP-1 KO mice (Fig. 4a), and this was similarly observed in DEP-1-depleted endothelial cell lysates (Fig. 4b), in accordance with reduced Akt activation. These results suggest that, in response to VEGF, DEP-1 promotes β-catenin phosphorylation and translocation to the nucleus through its modulation of Akt signaling. Second, we investigated whether DEP-1 expression had any effect on the stabilization of β-catenin protein levels. Results show that lower levels of β-catenin were present in the lysates of retinas from DEP-1 KO mice (Fig. 4a) or of DEP-1-depleted cells (Fig. 4b). Likewise, the inhibitory phosphorylation of GSK3β (S9), the main regulator of β-catenin degradation, was also impaired in DEP-1 KO mice (Fig. 4a) and in DEP-1-depleted cells (Fig. 4b), suggesting that DEP-1 is involved in the inhibition of GSK3 via the promotion of its phosphorylation by Akt and thus, in the stabilization of β-catenin. To validate this, control and DEP-1-depleted HUVECs were incubated with the proteasome inhibitor MG132 prior to and during their 18 h-stimulation with VEGF to prevent protein degradation and allow the analysis of GSK3-dependent phosphorylation of β-catenin on S33/S37/T41, which signals β-catenin degradation. As expected, the GSK3β-dependent phosphorylation of β-catenin on S33/S37/T41 and β-catenin protein levels were increased in DEP-1-silenced cells compared to controls treated with MG132, supporting the conclusion that DEP-1 depletion promoted degradation of β-catenin (Fig. 4c). Since the results suggested that DEP-1 promotes β-catenin stabilization and translocation to the nucleus (Fig. 4a–c), we next investigated if β-catenin-dependent transcriptional activity was required for Dll4 expression in VEGF-stimulated HUVECs. In the nucleus, activated β-catenin associates with the TCF/LEF transcription factor family to induce gene expression. To test if transcriptionally active β-catenin/TCF complexes were required for VEGF-induced Dll4 expression, their association and activity were disrupted using two inhibitors of the TCF/β-catenin complex, PNU74654 and PKF115584 [47]. Results show that incubation of HUVECs with either inhibitor abrogated VEGF-induced Dll4 expression, suggesting that β-catenin transcriptional activation plays a crucial role in

DEP-1 promotes VEGF-induced β-catenin stabilization and expression of Dll4. a Retinas of 5-day-old pups were collected, lysed and analyzed by Western blot using the specified antibodies. b HUVECs were nucleofected with DEP-1-targeting or control siRNAs. Two days later, cells were starved in M200 medium containing 1% FBS for 3 h and stimulated for 18 h with VEGF (50 ng/mL). Whole cell lysates were resolved by Western blot using the specified antibodies. c HUVECs nucleofected with siRNAs were incubated with MG132 (proteasome inhibitor; 1 µM) and stimulated for 18 h with VEGF, followed by Western blot analyses using the specified antibodies. Quantification of blots from four independent experiments are shown below. d HUVECs were treated with inhibitors of β-catenin/TCF association and activity, PNU74654 (100 µM) and PKF115584 (0.5 µM), during starvation and stimulation with VEGF for 18 h. Expression of Dll4 and PLCγ was evaluated by western blotting. Data ± SEM are shown. Student’s t test: *p ≤ 0.05.
this process in VEGF-stimulated endothelial cells (Fig. 4d). Together, these data suggest that DEP-1 contributes to the regulation of the expression of Dll4 through Akt-dependent phosphorylation and the promotion of β-catenin stabilization and transcriptional activity.
Discussion
Recent evidence demonstrates that the protein tyrosine phosphatase DEP-1 acts as a promoter of angiogenic responses both in vitro and in vivo [4, 7, 14, 35]. In this study, we demonstrate that DEP-1 is also an important regulator of Dll4 expression and of endothelial cell sprouting during retinal vascular development. We propose that this is achieved via Src activation and the Akt-dependent promotion of β-catenin stabilization and transcriptional activity, which has previously been reported to upregulate Dll4 expression [33]. In addition, through its ability to inactivate the proliferative ERK1/2 pathway, DEP-1 also controls the density of the developing retinal vascular network (Fig. 5).

DEP-1 modulates endothelial sprouting via the promotion of Dll4 expression. In response to VEGF, DEP-1 promotes Src activation, leading to Akt activation. Akt regulates β-catenin directly by phosphorylating S552, cueing for its nuclear translocation, and indirectly by inhibiting GSK3β, a major component of the β-catenin destruction complex. In that state, β-catenin degradation is reduced, promoting its translocation to the nucleus. In the nucleus, active β-catenin can then associate with TCF/LEF transcription factor to induce Dll4 expression, which results in Notch activation and the repression of the tip cell phenotype, ensuring proper retinal neovascularization during development.
Previous work demonstrated that DEP-1 modulates VEGF-induced angiogenic responses via its ability to activate the Src kinase [4, 7, 14, 35]. Hence, DEP-1 was shown to regulate VEGF-induced endothelial cell survival, invasion, vascular permeability, and capillary formation in vitro [4, 7, 35]. In vivo, physiological or pathological angiogenesis as well as Src activation were impaired in adult DEP-1 KO mice, further demonstrating the positive role of DEP-1 during these processes [14]. Contrasting with these results, we show here that similar to embryonic vascular development, retinal vascularisation still occurred in these animals (Fig. 1). Indeed, in the absence of DEP-1 expression, an increase in vascular density characterized by a greater number of branching points at the migrating front was found. Consistent with this, a larger number of tip cells was also observed in the retinas of DEP-1 KO mice, showing that DEP-1 controls endothelial sprouting and the proper development of the retinal vascular network. It is noteworthy that the retinal phenotype that we describe in DEP-1 KO mice is much less severe than that previously described in knock-in mice, in which a mutant allele was constructed to eliminate DEP-1 phosphatase activity, which die at midgestation [5]. It is possible that the mutant protein expressed in the knock-in acts in a dominant negative manner, and therefore, strongly interferes with the signaling pathways involved in VEGF-induced angiogenesis. Thus, while the phenotype of the knock-out mice can be rescued during development, expression of a non-functional DEP-1 that still interacts with endogenous protein complexes in the knock-in mice would have fatal consequences.
Endothelial sprouting is regulated by the expression of Dll4, a Notch ligand that limits tip cell formation and favours the stalk cell phenotype [19,20,21, 23, 24]. While it is widely accepted that VEGF stimulation induces Dll4 expression, the molecular pathways implicated remain ill-defined [19]. Here we show that DEP-1 is a promoter of Dll4 expression and, consequently, of Notch activation (Fig. 5). It was previously demonstrated that expression of a dominant-negative Akt mutant or PI3K inhibition impaired Dll4 expression [40, 41]. As DEP-1 expression was shown to be required for Src and Akt signaling in endothelial cells in response to VEGF in vitro and during retinal vascular expansion (Figs. 3, 4a) [4], these results suggest that through its ability to regulate the activity of Src and Akt, DEP-1 can induce Dll4 expression [4, 7]. In support of this, we observed that endothelial cells treated with chemical inhibitors of Src or PI3K, or cells expressing DEP-1 mutants unable to activate Src and Akt, were also deficient in their ability to promote Dll4 expression (Fig. 3e, f).
The β-catenin transcriptional complex was reported to directly bind to the Dll4 promoter, and its transcriptional activity was shown to play a role in Dll4 expression [33]. Accordingly, we observed that the decreased expression of Dll4 in DEP-1 KO mice or DEP-1-silenced endothelial cells correlated with the reduced phosphorylation of β-catenin on S552 and with its decreased protein levels (Fig. 4). In these conditions, GSK3β, an important regulator of β-catenin phosphorylation and degradation by the proteasome, was more active as indicated by its decreased inhibitory phosphorylation on S9 (Fig. 4). Since GSK3β phosphorylation is mainly mediated by Akt and that Akt phosphorylates β-catenin on S552 to allow its nuclear translocation, these results suggest that DEP-1, via the Src-Akt pathway, contributes to the regulation of the extent of β-catenin transcriptional activity in response to VEGF stimulation [42, 48]. The results presented here thus suggest that through these signaling pathways, DEP-1 activity promotes Notch signaling during retinal vascularization. Consistent with our findings, DEP-1 expression in zebrafish was shown to promote arterial specification, which is also dependent on Notch activation [6, 49].
In addition to increased branching, we have also noted the increased proliferation of endothelial cells in DEP-1 KO mice (PH3 staining—Fig. 2). In response to VEGF, DEP-1 is known to negatively regulate proliferation through attenuation of VEGFR2 phosphorylation and the consequent down-modulation of the proliferative PLCγ-ERK1/2-signaling cascade [2, 4]. However, in the retina, the regulation of cell proliferation by DEP-1 seemed to occur downstream of PLCγ as there was no difference in its activation level between WT and DEP-1 KO mice. A possible explanation is that DEP-1 may dephosphorylate ERK1/2 directly [3]. Indeed, several studies have demonstrated that DEP-1 inhibits the VEGF-dependent activation of ERK1/2, leading to decreased proliferation [50,51,52].
On the other hand, it has been shown that Akt activation results in inhibition of Raf, an upstream regulator of ERK1/2, consequently limiting proliferation [53]. As DEP-1 KO mice exhibit impaired Akt activation, it is possible that ERK1/2 is activated through the relieved inhibition of its upstream regulator. Also, the activation of Notch leads to the inhibition of ERK1/2 activation and, consequently, of proliferation [54, 55]. In the absence of DEP-1 and Notch activation, ERK1/2 signalling would therefore increase. Of note, activation of ERK1/2 has also been identified as a promoter of retinal and developmental angiogenesis in vivo [56, 57]. Interestingly, we similarly observed that the inhibition of ERK1/2, using PD98059, resulted in reduced expression of Dll4 in response to VEGF in vitro (Supplementary Fig. 3). However, since DEP-1 KO mice exhibit higher activation levels of ERK1/2 (Fig. 2), but still show reduced Dll4 expression, we conclude that VEGF-induced and DEP-1-dependent-signaling pathways involving Akt and β-catenin contribute to optimal Dll4 expression in endothelial cells. Given that the vascular retinal phenotype observed in DEP-1 KO mice is relatively mild compared to that observed in Dll4 haploinsufficient animals [30], it is likely that other molecular mechanisms are involved in the tight regulation of Dll4 expression. Indeed, the MEK-ERK pathway [58], blood flow [59] and both the RBPJ/Notch intracellular domain and SOX transcription factors [60] have all been shown to regulate Dll4 expression. Here, we describe a novel mechanism by which DEP-1 participates in the precise regulation of Dll4 expression in endothelial cells and maintains the proper response of endothelial cells to VEGF stimulation.
Collectively, these results identify DEP-1 as a novel regulator of the Dll4-Notch signaling pathway controlling the proliferation and sprouting abilities of endothelial cells during retinal vascular development (Fig. 5). These findings could thus provide new avenues for the treatment of vascular pathologies.
References
Ostman A, Yang Q, Tonks NK (1994) Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc Natl Acad Sci USA 91:9680–9684
Lampugnani MG, Zanetti A, Corada M et al (2003) Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol 161:793–804. https://doi.org/10.1083/jcb.200209019
Sacco F, Tinti M, Palma A et al (2009) Tumor suppressor density-enhanced phosphatase-1 (DEP-1) inhibits the RAS pathway by direct dephosphorylation of ERK1/2 kinases. J Biol Chem 284:22048–22058. https://doi.org/10.1074/jbc.M109.002758
Chabot C, Spring K, Gratton J-P et al (2009) New role for the protein tyrosine phosphatase DEP-1 in Akt activation and endothelial cell survival. Mol Cell Biol 29:241–253. https://doi.org/10.1128/MCB.01374-08
Takahashi T, Takahashi K, St John PL et al (2003) A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol 23:1817–1831
Rodriguez F, Vacaru A, Overvoorde J, den Hertog J (2008) The receptor protein-tyrosine phosphatase, Dep1, acts in arterial/venous cell fate decisions in zebrafish development. Dev Biol 324:122–130. https://doi.org/10.1016/j.ydbio.2008.09.011
Spring K, Chabot C, Langlois S et al (2012) Tyrosine phosphorylation of DEP-1/CD148 as a mechanism controlling Src kinase activation, endothelial cell permeability, invasion, and capillary formation. Blood 120:2745–2756. https://doi.org/10.1182/blood-2011-12-398040
Zhu JW, Doan K, Park J et al (2011) Receptor-like tyrosine phosphatases CD45 and CD148 have distinct functions in chemoattractant-mediated neutrophil migration and response to S. aureus. Immunity 35:757–769. https://doi.org/10.1016/j.immuni.2011.09.011
Trapasso F, Drusco A, Costinean S et al (2006) Genetic ablation of Ptprj, a mouse cancer susceptibility gene, results in normal growth and development and does not predispose to spontaneous tumorigenesis. DNA Cell Biol 25:376–382. https://doi.org/10.1089/dna.2006.25.376
Hackbusch D, Dulsner A, Gatzke N et al (2013) Knockout of density-enhanced phosphatase-1 impairs cerebrovascular reserve capacity in an arteriogenesis model in mice. BioMed Res Int 2013:802149. https://doi.org/10.1155/2013/802149
Zhu JW, Brdicka T, Katsumoto TR et al (2008) Structurally distinct phosphatases CD45 and CD148 both regulate B cell and macrophage immunoreceptor signaling. Immunity 28:183–196. https://doi.org/10.1016/j.immuni.2007.11.024
Stepanek O, Kalina T, Draber P et al (2011) Regulation of Src family kinases involved in T cell receptor signaling by protein-tyrosine phosphatase CD148. J Biol Chem 286:22101–22112. https://doi.org/10.1074/jbc.M110.196733
Ellison S, Mori J, Barr AJ, Senis YA (2010) CD148 enhances platelet responsiveness to collagen by maintaining a pool of active Src family kinases. J Thromb Haemost JTH 8:1575–1583. https://doi.org/10.1111/j.1538-7836.2010.03865.x
Fournier P, Dussault S, Fusco A et al (2016) Tyrosine phosphatase PTPRJ/DEP-1 is an essential promoter of vascular permeability, angiogenesis, and tumor progression. Cancer Res 76:5080–5091. https://doi.org/10.1158/0008-5472.CAN-16-1071
Geudens I, Gerhardt H (2011) Coordinating cell behaviour during blood vessel formation. Dev Camb Engl 138:4569–4583. https://doi.org/10.1242/dev.062323
Gerhardt H, Golding M, Fruttiger M et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161:1163–1177. https://doi.org/10.1083/jcb.200302047
Blancas AA, Wong LE, Glaser DE, McCloskey KE (2013) Specialized tip/stalk-like and phalanx-like endothelial cells from embryonic stem cells. Stem Cells Dev 22:1398–1407. https://doi.org/10.1089/scd.2012.0376
Jakobsson L, Franco CA, Bentley K et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953. https://doi.org/10.1038/ncb2103
Lobov IB, Renard RA, Papadopoulos N et al (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104:3219–3224. https://doi.org/10.1073/pnas.0611206104
Suchting S, Freitas C, le Noble F et al (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA 104:3225–3230. https://doi.org/10.1073/pnas.0611177104
Hellstrom M, Phng L-K, Hofmann JJ et al (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445:776–780. https://doi.org/10.1038/nature05571
Borggrefe T, Oswald F (2009) The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci CMLS 66:1631–1646. https://doi.org/10.1007/s00018-009-8668-7
Leslie JD, Ariza-McNaughton L, Bermange AL et al (2007) Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Dev Camb Engl 134:839–844. https://doi.org/10.1242/dev.003244
Williams CK, Li J-L, Murga M et al (2006) Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107:931–939. https://doi.org/10.1182/blood-2005-03-1000
Wu Y, Cain-Hom C, Choy L et al (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464:1052–1057. https://doi.org/10.1038/nature08878
Funahashi Y, Hernandez SL, Das I et al (2008) A Notch1 ectodomain construct inhibits endothelial Notch signaling, tumor growth, and angiogenesis. Cancer Res 68:4727–4735. https://doi.org/10.1158/0008-5472.CAN-07-6499
Thurston G, Noguera-Troise I, Yancopoulos GD (2007) The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer 7:327–331. https://doi.org/10.1038/nrc2130
Limbourg FP, Takeshita K, Radtke F et al (2005) Essential role of endothelial Notch1 in angiogenesis. Circulation 111:1826–1832. https://doi.org/10.1161/01.CIR.0000160870.93058.DD
Krebs LT, Shutter JR, Tanigaki K et al (2004) Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev 18:2469–2473. https://doi.org/10.1101/gad.1239204
Gale NW, Dominguez MG, Noguera I et al (2004) Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA 101:15949–15954. https://doi.org/10.1073/pnas.0407290101
Ridgway J, Zhang G, Wu Y et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444:1083–1087. https://doi.org/10.1038/nature05313
Noguera-Troise I, Daly C, Papadopoulos NJ et al (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444:1032–1037. https://doi.org/10.1038/nature05355
Corada M, Nyqvist D, Orsenigo F et al (2010) The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell 18:938–949. https://doi.org/10.1016/j.devcel.2010.05.006
Larrivee B, Prahst C, Gordon E et al (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 22:489–500. https://doi.org/10.1016/j.devcel.2012.02.005
Spring K, Lapointe L, Caron C et al (2014) Phosphorylation of DEP-1/PTPRJ on threonine 1318 regulates Src activation and endothelial cell permeability induced by vascular endothelial growth factor. Cell Signal 26:1283–1293. https://doi.org/10.1016/j.cellsig.2014.02.008
Nowak-Sliwinska P, Alitalo K, Allen E et al (2018) Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 21:425–532. https://doi.org/10.1007/s10456-018-9613-x
Takahashi T, Yamaguchi S, Chida K, Shibuya M (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20:2768–2778. https://doi.org/10.1093/emboj/20.11.2768
Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17:611–625. https://doi.org/10.1038/nrm.2016.87
Villa N, Walker L, Lindsell CE et al (2001) Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev 108:161–164
Takeshita K, Satoh M, Ii M et al (2007) Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res 100:70–78. https://doi.org/10.1161/01.RES.0000254788.47304.6e
Liu Z-J, Shirakawa T, Li Y et al (2003) Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23:14–25
Fang D, Hawke D, Zheng Y et al (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229. https://doi.org/10.1074/jbc.M611871200
Monick MM, Carter AB, Robeff PK et al (2001) Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J Immunol 1950 166:4713–4720
Stone J, Itin A, Alon T et al (1995) Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci Off J Soc Neurosci 15:4738–4747
Maes C, Goossens S, Bartunkova S et al (2010) Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J 29:424–441. https://doi.org/10.1038/emboj.2009.361
Provis JM, Leech J, Diaz CM et al (1997) Development of the human retinal vasculature: cellular relations and VEGF expression. Exp Eye Res 65:555–568. https://doi.org/10.1006/exer.1997.0365
Sebio A, Kahn M, Lenz H-J (2014) The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin Ther Targets 18:611–615. https://doi.org/10.1517/14728222.2014.906580
Cross DA, Alessi DR, Cohen P et al (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789. https://doi.org/10.1038/378785a0
Trindade A, Kumar SR, Scehnet JS et al (2008) Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112:1720–1729. https://doi.org/10.1182/blood-2007-09-112748
Paduano F, Ortuso F, Campiglia P et al (2012) Isolation and functional characterization of peptide agonists of PTPRJ, a tyrosine phosphatase receptor endowed with tumor suppressor activity. ACS Chem Biol 7:1666–1676. https://doi.org/10.1021/cb300281t
Brunner PM, Heier PC, Mihaly-Bison J et al (2011) Density enhanced phosphatase-1 down-regulates urokinase receptor surface expression in confluent endothelial cells. Blood 117:4154–4161. https://doi.org/10.1182/blood-2010-09-307694
Takahashi T, Takahashi K, Mernaugh RL et al (2006) A monoclonal antibody against CD148, a receptor-like tyrosine phosphatase, inhibits endothelial-cell growth and angiogenesis. Blood 108:1234–1242. https://doi.org/10.1182/blood-2005-10-4296
Ren B, Deng Y, Mukhopadhyay A et al (2010) ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J Clin Invest 120:1217–1228. https://doi.org/10.1172/JCI39837
Sainson RCA, Aoto J, Nakatsu MN et al (2005) Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J Off Publ Fed Am Soc Exp Biol 19:1027–1029. https://doi.org/10.1096/fj.04-3172fje
Liu Z-J, Xiao M, Balint K et al (2006) Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J Off Publ Fed Am Soc Exp Biol 20:1009–1011. https://doi.org/10.1096/fj.05-4880fje
Srinivasan R, Zabuawala T, Huang H et al (2009) Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 4:e8283. https://doi.org/10.1371/journal.pone.0008283
Bullard LE, Qi X, Penn JS (2003) Role for extracellular signal-responsive kinase-1 and -2 in retinal angiogenesis. Invest Ophthalmol Vis Sci 44:1722–1731
Yang C, Guo Y, Jadlowiec CC et al (2013) Vascular endothelial growth factor-A inhibits EphB4 and stimulates delta-like ligand 4 expression in adult endothelial cells. J Surg Res 183:478–486. https://doi.org/10.1016/j.jss.2013.01.009
Watson O, Novodvorsky P, Gray C et al (2013) Blood flow suppresses vascular Notch signalling via dll4 and is required for angiogenesis in response to hypoxic signalling. Cardiovasc Res 100:252–261. https://doi.org/10.1093/cvr/cvt170
Sacilotto N, Monteiro R, Fritzsche M et al (2013) Analysis of Dll4 regulation reveals a combinatorial role for Sox and Notch in arterial development. Proc Natl Acad Sci USA 110:11893–11898. https://doi.org/10.1073/pnas.1300805110
Acknowledgements
We would like to thank A. Fusco for providing DEP-1 KO mice. This work was supported by operating grants from the Cancer Research Society (I.R. and B.L.), an operating grant from the Canadian Institutes of Health Research (363450) (B.L.) and a Grant-in-Aid from the Heart and Stroke Foundation of Canada (B.L.). B.L. is the recipient of a New Investigator Award from the Heart and Stroke Foundation of Canada. P. Fournier was supported by student scholarships from the Canadian Institutes of Health Research (292353) and Institut du cancer de Montréal.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest with the contents of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fournier, P., Viallard, C., Dejda, A. et al. The protein tyrosine phosphatase PTPRJ/DEP-1 contributes to the regulation of the Notch-signaling pathway and sprouting angiogenesis. Angiogenesis 23, 145–157 (2020). https://doi.org/10.1007/s10456-019-09683-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-019-09683-z