
Abstract
The adsorption and separation behaviors of CO2 and CH4 in new siliceous zeolites (IFO, JSR, OKO, SEW) were simulated using the Grand Canonical Monte Carlo method in the paper. The adsorption isotherms for pure components and binary mixtures of CO2 and CH4 in four siliceous zeolites were obtained. The adsorption thermodynamic properties including Gibb’s free energy change, enthalpy change and entropy change were investigated. The results demonstrate that the adsorbed amount of pure components increases with an increase in pressure, and larger pore volume and surface area are beneficial to improve the adsorption capacity. The adsorption amount of CO2 and CH4 in the JSR zeolite is 7.08 and 2.27 mmol g−1 at 1000 kPa, respectively. In view of the thermodynamic results, the new siliceous zeolites show a higher affinity for CO2. The adsorption capacities of CO2 in all zeolites were five times more than those of CH4 in binary mixtures based on the ratios of equilibrium adsorption capacity. Considering the adsorption uptake and selectivity for CO2/CH4, the JSR zeolite is a good candidate for the separation of CO2/CH4 at low pressure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Separation of CH4 and CO2 mixtures is a challenging research topic due to environmental and economic concerns, and the inherent difficulties in conducting experiments. There are several technologies applied for the separation of mixtures of CH4 and CO2 (Singh et al. 2009; Scholes et al. 2012), in which adsorption-based processes are widely used for post-combustion CO2 capture (Ebner and Ritter 2009), the purification of biogas (Alonso-Vicario et al. 2010), and the purification of coal mine methane and coal mine ventilation air (VAM) (Olajossy et al. 2003). The reason is that the separation cost is usually low if the separation is based on the difference of components in adsorption (Ruthven et al. 1994). Furthermore, the adsorbent is a decisive factor of feasibility and separation cost. In a cost-effective manner, zeolites are considered as potential sorbents for the adsorption of CH4 and CO2 gas mixtures (Ackley et al. 2003; Yang et al. 2012; Zhang et al. 2012; Mofarahi and Gholipour 2014).
Zeolites have been intensively investigated in recent decades because of their large surface areas and molecular sized porous structures (Zhang et al. 2014; Dunne et al. 2009; Bakhtyari and Mofarahi 2014). They are an important class of materials in which separations are obtained by selecting the pore size such that in mixtures molecules that are too large are blocked (Smit and Maesen 2008). Therefore, zeolites are widely used in applications such as separation, purification, ion exchange, catalysis and the isolation of biological molecular (Krishna and van Baten 2005; Rouleau et al. 2009). Currently, about 200 types of zeolite have been crystallographically classified and several new types still appear every year (Liu et al. 2013). Thus, studies on the adsorption and separation behaviors for CH4 and CO2 mixtures in new zeolites are very important and attractive from the viewpoints of science and their applications.
However, zeolites are also challenging because it is very difficult to carry out experiments that provide us with information at the molecular level. In this respect, molecular simulations nicely complement experiments (Catlow et al. 2004). With the development of computer technology, molecular simulation methods, such as Grand Canonical Monte Carlo (GCMC), are playing an important role in developing our understanding of the relationship between the microscopic and macroscopic properties of molecular fluids confined in zeolites, and can be used to generate adsorption isotherms for gases on microporous solid adsorbents (Liu et al. 2011; Kim et al. 2013). To date, the adsorption of water/methanol and water/ethanol mixtures in zeolite 4A was simulated using the GCMC simulation (Wu et al. 2009). The Cosoli group (Cosoli et al. 2008b) studied H2S removal from biogas streams using different zeolites such as NaX, NaY, LTA and MFI with the GCMC method. Pham et al. (Pham et al. 2014) studied the adsorption of N2 and CO2 in pure silica zeolites (BEA, CHA, FER, MFI, and STT) experimentally and by GCMC simulation, and the agreement between computed adsorption isotherms and isosteric heats with those found experimentally was excellent. The Calero group investigated the adsorption selectivity of CO2, N2 and CH4 in MFI, MOR, ISV, ITE, CHA and DDR (García-Pérez et al. 2007). The results showed that the selectivity behavior strongly depended on the type of material structure and the bulk composition of the mixture. Therefore, molecular simulations have become a powerful tool to explore both materials science and life science fields (Zeng et al. 2007; Cosoli et al. 2008a).
In this work, the adsorption and separation properties for CO2 and CH4, and the selectivity performance of new siliceous zeolites (IFO, JSR, SEW and OKO) were investigated using the molecular simulation method, which would quickly aid in choosing effective adsorbents for gas adsorption and separation, and could be practically important for gas separation from low-concentration mine gas.
2 Computational methods
2.1 Zeolite structures
New zeolites are discovered and synthesized annually. Their adsorption, separation and selectivity properties are worth investigating and can be a fascinating area of research. Therefore, the separation of CO2 and CH4 in four new siliceous zeolites (IFO, JSR, OKO and SEW) was investigated using the GCMC method. Framework structures of the new siliceous zeolites are shown in Fig. 1, and details of the different zeolite structures used in the calculations are listed in Table 1 (Martinez-Franco et al. 2013; Xu et al. 2013; Verheyen et al. 2012; Xie et al. 2011).

Framework structures of new siliceous zeolites
2.2 Simulation techniques
The Grand Canonical Monte Carlo (GCMC) method was employed in this work (Metropolis et al. 1953), which is widely applied in the study of adsorption problems. The GCMC method uses a grand canonical ensemble, in which the chemical potential of each species, the volume of the system and the temperature are fixed (Agnihotri et al. 2008; Biswas and Cagin 2010). Hence, comparisons between simulation results and experimental data are reasonable because the adsorption conditions (pressure and temperature) are also constant in the experiments (Rahmati and Modarress 2013; Nugent et al. 2013).
In the present work, each zeolite framework was considered to be rigid. All interactions of gas–zeolite and gas–gas were modeled using the PCFF force field (Hill and Sauer 1994; Sun 1995). The PCFF has been proved as a powerful force field supporting for adsorption in many zeolite systems (Song et al. 2002; Yang et al. 2008; Zhao et al. 2012). The cutoff distance for the calculation of the van der Waals potential energy by an atom based technique was taken as 1.3 nm. The partial charges of atoms were estimated by the charge-equilibration method (Rappe and Goddard 1991) and the electrostatic energy was calculated using the Ewald summation method (Karasawa and Goddard 1989) with an accuracy of 0.0001 kcal mol−1. The partial pressure required for describing the chemical potential of the adsorbed molecules was calculated using the ideal gas law. In the GCMC simulation, adsorbate molecules in the zeolite were translated, rotated, created and deleted from the zeolite framework at random with a probability of 0.225, 0.225, 0.275 and 0.275, respectively (Rahmati and Modarress 2009; Rahmati et al. 2012). For every state, 1 × 107 configurations were generated, the first 5 × 106 steps were used to achieve equilibration and the last 5 × 106 steps were used for calculation of the required properties and subsequent data analysis. For the MFI-type zeolite, the supercell consists of 8 unit cells (2 × 2 × 2, a = 2.007 nm, b = 1.992 nm and c = 1.342 nm). Periodic boundary conditions were applied in three-dimensions in order to simulate an infinite system. Further details of the GCMC method can be found in the literature (Dubbeldam et al. 2013).
3 Results and discussion
3.1 Comparisons with experimental data
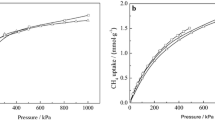
To verify the simulation techniques, the adsorption isotherms of CO2 and CH4 in the MFI zeolite were compared to experimental data (Babarao et al. 2007; Krishna and van Baten 2007; Zhu et al. 2006; Golden and Sircar 1994), as shown in Fig. 2. A good agreement between the simulation results and experimental data was found based on the adsorption isotherms, which demonstrated that the simulation techniques were satisfactory for the simulation of CO2 and CH4 adsorption in purely siliceous zeolites. Therefore, the simulation techniques were applied in the adsorption studies for CO2 and CH4 in the IFO, JSR, OKO and SEW zeolites.

The adsorption isotherms of CO2 and CH4 in purely siliceous zeolite MFI: (a) CO2: open square literature data from Babarao et al. (2007), 300 K; open circle literature data from Krishna and van Baten (2007), 303 K; triangle this work, 298 K. (b) CH4: open square literature data from Zhu et al. (2006), 303 K; open circle literature data from Golden and Sircar (1994), 304 K; triangle this work, 300 K
3.2 Adsorption of CO2 and CH4 in new siliceous zeolites
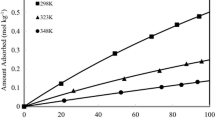
Adsorption isotherms for CO2 and CH4 in the four zeolite structures at 298 K are shown in Fig. 3. The results show that the adsorbed amount of each pure component increases with an increase in pressure, and the sequence for the adsorption amount of CO2 and CH4 in these zeolites is JSR > SEW > OKO > IFO. The adsorption amount of CO2 and CH4 adsorbed in the JSR zeolite is 7.08 and 2.27 mmol g−1 at 1000 kPa, respectively. Comparing with NaX, NaY, MFI, CHA and DDR zeolites, the adsorption amount of CO2 is higher than that in NaX, NaY, CHA or DDR, whereas the adsorption amount of CH4 is smaller than that in these zeolites, and the adsorption amount of CO2 and CH4 in the MFI zeolite is only 2.89 and 2.05 mmol g−1 at 1000 kPa (Zhang et al. 2014; Krishna et al. 2006). On the other hand, for RWY, STW, SOF, PUN, STO, SSF, LTF, IWS, ITR, SVR, BSV and SFS zeolites (Rahmati and Modarress 2013), the maximum adsorption amount of CO2 and the minimum adsorption amount of CH4 in the these zeolites is 3.78 and 1.36 mmol g−1 at 1000 kPa, respectively. Thus, JSR zeolite is a better adsorbent for the separation of CO2 and CH4 mixture among these zeolites because it has higher CO2 adsorption amount and equilibrium adsorption amount ratio between CO2 and CH4. The adsorption capacity for CO2 is higher than that of CH4 in all the zeolites under the same conditions because CO2 is a more slender molecule than CH4 and the energy barrier for inter-cage hopping is lower than that for CH4 (Krishna et al. 2006). Therefore, the steric hindrance in the CH4 molecule is greater than that of CO2 (Krishna and van Baten 2007). Further, CO2 has higher affinity than CH4 because CO2 has quadrupole and the electrostatic potential of the quadrupole term is proportional to the electric charge of ions (Zhang et al. 2014).

The adsorption isotherms of CO2 and CH4 in new siliceous zeolites at 298 K (a) CO2 (b) CH4: diamond IFO zeolite; open square JSR zeolite; triangle OKO zeolite; open circle SEW zeolite
The pore volume and surface area are very important parameters to understand the adsorption and diffusion behavior of penetrated molecules in nanoporous materials. In this work, the pore volume and surface area characterization of the zeolites were analyzed by the Connolly surface method (1985). The probe molecule was selected using hard spheres with a radius of 0.165 nm (the kinetic radius of CO2). The simulated results for the pore volume and surface area were summarized in Table 2. As listed in Table 2, the magnitude of the pore volume and surface area can be ranked as follows: JSR > SEW > OKO > IFO, which is consistent with the adsorption capacity for CO2 and CH4. The result indicates that larger pore volume and surface area are beneficial to improve gas adsorption capacity in zeolites.
3.3 Adsorption thermodynamics
The adsorption process is always accompanied by changes in the system for Gibb’s free energy, enthalpy and entropy, which are favorable to understand the adsorption mechanism and the interactions between adsorbents and adsorbates. The thermodynamic parameters of Gibb’s free energy change, ΔG 0, enthalpy change, ΔH 0, and entropy change, ΔS 0, for the adsorption process were calculated using the following equations:
where K is the equilibrium constant, R is the universal gas constant (8.314 J mol−1 K−1) and T is the temperature in Kelvin. For Eqs. 1 and 2, the temperature dependent K is expressed as follows:
The equilibrium constant at zero coverage limit (Pham et al. 2014; Golden and Sircar 1994) for the pure components (CO2 and CH4) in the new siliceous zeolites over the temperature range from 273 to 348 K are shown in Fig. 4. The results show that the equilibrium constant decreases with an increase in temperature. Moreover, the K values for CO2 are larger than those found for CH4 at the same temperature.

Plot of the logarithm of the equilibrium constants lnK versus 1/T: filled square IFO-CO2; filled circle JSR-CO2; filled triangle OKO-CO2; filled diamond SEW-CO2; open square IFO-CH4; open circle JSR-CH4; open triangle OKO-CH4; open diamond SEW-CH4
The Gibb’s free energy change, ΔG 0, calculated using Eq. 1 is plotted in Fig. 5. The Gibb’s free energy change was found to be linear with temperature. An increase in temperature causes an increase in the ΔG 0 value, which indicates a lower temperature is advantageous for the adsorption process. The negative values of ΔG 0 suggest that the adsorption process is favorable and spontaneous for CO2 and CH4. In addition, the adsorption of CO2 molecules was more favorable than that of CH4 because the ΔG 0 values for CO2 are more negative.

Plot of Gibbs free energy change, ΔG 0, versus temperature T: filled square IFO-CO2; filled circle JSR-CO2; filled triangle OKO-CO2; filled diamond SEW-CO2; open square IFO-CH4; open circle JSR-CH4; open triangle OKO-CH4; open diamond SEW-CH4
The results of the enthalpy change, ΔH 0 and entropy change, ΔS 0 can be estimated from the slope and intercept of the linear relationship in Eq. 3, and the results are presented in Table 3. As listed in Table 3, the new zeolites have a higher affinity to CO2 according to the more negative enthalpy change for CO2. The isosteric heat of adsorption, a commonly reported thermodynamic quantity, is given by q st = −ΔH 0 at the zero occupancy (Zhu et al. 2006; Talu and Myers 2001). Moreover, their isosteric heats of CO2 adsorption are higher than that of CH4, demonstrating the stronger binding capacity between CO2 and zeolites (Colabella et al. 1988).
The negative adsorption entropy ΔS 0 for both CO2 and CH4 suggests that the adsorption of CO2 and CH4 molecules in four zeolites is in an ordered arrangement because the motion of gas molecules is transformed from the three-dimensional space into a two-dimensional space. Further, the less negative entropy of CH4 when compared with CO2 in each new zeolite indicates the increased randomness of CH4 (Zhang et al. 2014). All simulated results for Gibb’s free energy, enthalpy and entropy change, as well as the isosteric heat of adsorption are consistent and show that CO2 is more favorable to be adsorbed than CH4 in these four zeolites.
3.4 Binary mixtures
3.4.1 Adsorption isotherms
The adsorption isotherms for equimolar mixtures of CO2/CH4 in IFO, JSR, OKO and SEW at 298 K are shown in Fig. 6. With an increase in pressure, the amount of CO2 adsorbed initially increases linearly and then flattens, while the amount of CH4 adsorbed changes little. The ratios of equilibrium adsorption capacity for the equimolar mixtures at different pressures are presented in Table 4. It is obvious that with an increase in pressure, the ratio of equilibrium adsorption capacity for CO2/CH4 increases. For a pressure swing adsorption (PSA) processes, the adsorption step generally takes place at moderate pressures (5 atm) in view of energy saving. In this condition, the equilibrium adsorption capacities for CO2 in all zeolites were five times more than those for CH4, which suggests that the new siliceous zeolites have a strong ability to separate a mixture of CO2 and CH4.

The adsorption isotherms of the equimolar mixtures of CO2 and CH4 in new siliceous zeolites at 298 K: (a) IFO, (b) JSR, (c) OKO, (d) SEW; open square CO2; open triangle CH4
3.4.2 Adsorption selectivity
The selectivity coefficient S ij is a good indication of separation ability and is given by the following equation:
where x i and x j are the molar fractions of species i and j in the gas phase, while y i and y j are the molar fraction of species i and j adsorbed in the framework, respectively.
The selectivity coefficients of CO2/CH4 in the four new zeolites as a function of the partial pressure of CO2 at a total pressure 1000 kPa are shown in Fig. 7. The results demonstrate that the selectivity coefficients increase with a decrease in the concentration of CH4 in the mixture. The higher selectivity coefficients for CO2/CH4 at a low concentration of CH4 (<30 mol%) (Zhong et al. 2013) belong to the JSR zeolite. Thus, considering the adsorption uptake and selectivity of CO2/CH4, the JSR zeolite is a good candidate for the separation of CO2/CH4 at low pressure (up to 1000 kPa).

The selectivity coefficients of CO2/CH4 in new siliceous zeolites: open square IFO; open star JSR; open circle OKO; triangle SEW
4 Conclusions
The GCMC method was employed to investigate the adsorption and separation properties for CO2 and CH4 in new siliceous zeolites of IFO, JSR, OKO and SEW. The adsorption isotherms for the pure components and binary mixtures of CO2 and CH4 in these zeolites were obtained. Adsorption thermodynamic parameters were also analyzed.
The adsorbed amount of pure components increases with an increase in pressure and the adsorption amount for both CO2 and CH4 in these zeolites can be ranked as follows: JSR > SEW > OKO > IFO. The adsorption amount of CO2 and CH4 in the JSR zeolite is 7.08 and 2.27 mmol g−1 at 1000 kPa, respectively. The thermodynamic parameters of Gibb’s free energy change and enthalpy change of adsorption show that CO2 displays a higher affinity than that of CH4 in the new siliceous zeolites. A negative adsorption entropy change ΔS 0 indicates the CO2 and CH4 adsorbed are in an ordered arrangement in all the new zeolites. The more negative entropy for CO2 when compared with CH4 in each new zeolite presents a more orderly arrangement of CO2.
The adsorption isotherms for the binary mixtures suggest the new siliceous zeolites can be used to separate a CO2/CH4 mixture. Considering the adsorption uptake and selectivity for CO2/CH4, the JSR zeolite is a good candidate for the separation of CO2/CH4 at low pressure (up to 1000 kPa).
References
Ackley, M.W., Rege, S.U., Saxena, H.: Application of natural zeolites in the purification and separation of gases. Microporous Mesoporous Mater. 61(3), 25–42 (2003)
Agnihotri, S., Kim, P., Zheng, Y., Mota, J.P.B., Yang, L.: Regioselective competitive adsorption of water and organic vapor mixtures on pristine single-walled carbon nanotube bundles. Langmuir 24(11), 5746–5754 (2008)
Alonso-Vicario, A., Ochoa-Gómez, J.R., Gil-Río, S., Gómez-Jiménez-Aberasturi, O., Ramírez-López, C.A., Torrecilla-Soria, J., Domínguez, A.: Purification and upgrading of biogas by pressure swing adsorption on synthetic and natural zeolites. Microporous Mesoporous Mater. 134(1), 100–107 (2010)
Babarao, R., Hu, Z., Jiang, J., Chempath, S., Sandler, S.I.: Storage and separation of CO2 and CH4 in silicalite, C168 schwarzite, and IRMOF-1: a comparative study from Monte Carlo simulation. Langmuir 23(3), 659–666 (2007)
Bakhtyari, A., Mofarahi, M.: Pure and binary adsorption equilibria of methane and nitrogen on zeolite 5A. J. Chem. Eng. Data 59(3), 626–639 (2014)
Biswas, M.M., Cagin, T.: Simulation studies on hydrogen sorption and its thermodynamics in covalently linked carbon nanotube scaffold. J. Phys. Chem. B 114(43), 13752–13763 (2010)
Catlow, C.R.A., Smit, B., van Santen, R.: Computer Modelling of Microporous Materials. Academic Press, New York (2004)
Colabella, J.M., Stall, R.A., Sorenson, C.T.: The adsorption and subsequent oxidation of AsH3 and PH3 on activated carbon. J. Cryst. Growth 92(1–2), 189–195 (1988)
Connolly, M.L.: Computation of molecular volume. J. Am. Chem. Soc. 107(5), 1118–1124 (1985)
Cosoli, P., Ferrone, M., Pricl, S., Fermeglia, M.: Hydrogen sulfide removal from biogas by zeolite adsorption. Part II. MD aimulations. Chem. Eng. J. 145(1), 93–99 (2008a)
Cosoli, P., Ferrone, M., Pricl, S., Fermeglia, M.: Hydrogen sulphide removal from biogas by zeolite adsorption: Part I. GCMC molecular simulations. Chem. Eng. J. 145(1), 86–92 (2008b)
Dubbeldam, D., Torres-Knoop, A., Walton, K.S.: On the inner workings of Monte Carlo codes. Mol. Simul. 39(14–15), 1253–1292 (2013)
Dunne, L.J., Furgani, A., Jalili, S., Manos, G.: Monte-carlo simulations of methane/carbon dioxide and ethane/carbon dioxide mixture adsorption in zeolites and comparison with matrix treatment of statistical mechanical lattice model. Chem. Phys. 359(1–3), 27–30 (2009)
Ebner, A.D., Ritter, J.A.: State-of-the-art adsorption and membrane separation processes for carbon dioxide production from carbon dioxide emitting industries. Sep. Sci. Technol. 44(6), 1273–1421 (2009)
García-Pérez, E., Parra, J.B., Ania, C.O., García-Sánchez, A., van Baten, J.M., Krishna, R., Dubbeldam, D., Calero, S.: A computational study of CO2, N2, and CH4 adsorption in zeolites. Adsorption 13(5–6), 469–476 (2007)
Golden, T.C., Sircar, S.: Gas adsorption on silicalite. J. Colloid Interface Sci. 162(1), 182–188 (1994)
Hill, J.R., Sauer, J.: Molecular mechanics potential for silica and zeolite catalysts based on ab initio calculations. 1. Dense and microporous silica. J. Phys. Chem. 98(4), 1238–1244 (1994)
Karasawa, N., Goddard, W.A.: Acceleration of convergence for lattice sums. J. Phys. Chem. 93(21), 7320–7327 (1989)
Kim, K., Lee, S., Ryu, J.H., Lee, K.S., Lee, W.B.: An improved CO2 adsorption efficiency for the zeolites impregnated with the amino group: a molecular simulation approach. Int. J. Greenhouse Gas Control 19(11), 350–357 (2013)
Krishna, R., van Baten, J.M.: Using molecular simulations for screening of zeolites for separation of CO2/CH4 mixtures. Chem. Eng. J. 133(1–3), 121–131 (2007)
Krishna, R., van Baten, J.M., García-Pérez, E., Calero, S.: Diffusion of CH4 and CO2 in MFI, CHA and DDR zeolites. Chem. Phys. Lett. 429(1), 219–224 (2006)
Krishna, R., van Baten, J.M.: Influence of isotherm inflection on the diffusivities of C5–C8 linear alkanes in MFI zeolite. Chem. Phys. Lett. 407(1), 159–165 (2005)
Liu, B., Wu, Y., Liu, D., Wu, Y., Xi, H., Qian, Y.: Molecular simulation and experimental studies of a mesoporous ZSM-5 type molecular sieve. Phys. Chem. Chem. Phys. 15(8), 2741–2748 (2013)
Liu, J., Keskin, S., Sholl, D.S., Johnson, J.K.: Molecular simulations and theoretical predictions for adsorption and diffusion of CH4/H2 and CO2/CH4 mixtures in ZIFs. J. Phys. Chem. C 115(25), 12560–12566 (2011)
Martinez-Franco, R., Moiner, M., Yun, Y., Sun, J., Wan, W., Zou, X., Corma, A.: Synthesis of an extra-large molecular sieve using proton sponges as organic structure-directing agents. Proc. Natl. Acad. Sci. USA 110(10), 3749–3754 (2013)
Metropolis, N., Rosenbluth, A.W., Rosenbluth, M.N., Teller, A.H., Teller, E.: Equation of state calculations by fast computing machines. J. Chem. Phys. 21(6), 1087–1092 (1953)
Mofarahi, M., Gholipour, F.: Gas adsorption aeparation of CO2/CH4 aystem using zeolite 5A. Microporous Mesoporous Mater. 200, 1–10 (2014)
Nugent, P., Belmabkhout, Y., Burd, S.D., Cairns, A.J., Luebke, R., Forrest, K., Pham, T., Ma, S., Space, B., Wojtas, L., Eddaoudi, M., Zaworotko, M.J.: Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 495, 80–84 (2013)
Olajossy, A., Gawdzik, A., Budner, Z., Dula, J.: Methane separation from coal mine methane gas by vacuum pressure swing adsorption. Chem. Eng. Res. Des. 81(4), 474–482 (2003)
Pham, T.D., Xiong, R., Sandler, S.I., Lobo, R.F.: Experimental and computational studies on the adsorption of CO2 and N2 on pure silica zeolites. Microporous Mesoporous Mater. 185, 157–166 (2014)
Rahmati, M., Modarress, H.: Nitrogen adsorption on nanoporous zeolites studied by grand canonical Monte Carlo simulation. J. Mol. Struct. THEOCHEM 901(1–3), 110–116 (2009)
Rahmati, M., Modarress, H., Gooya, R.: Molecular simulation study of polyurethane membranes. Polymer 53(9), 1939–1950 (2012)
Rahmati, M., Modarress, H.: Selectivity of new siliceous zeolites for separation of methane and carbon dioxide by Monte Carlo simulation. Microporous Mesoporous Mater. 176(8), 168–177 (2013)
Rappe, A.K., Goddard, W.A.: Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 95(8), 3358–3363 (1991)
Rouleau, L., Pirngruber, G., Guillou, F., Barrère-Tricca, C., Omegna, A., Valtchev, V., Pera-Titus, M., Miachon, S., Dalmon, J.A.: Nanocomposite MFI-alumina and FAU-alumina membranes: synthesis, characterization and application to paraffin separation and CO2 capture. Oil Gas Sci. Technol. 64(6), 745–758 (2009)
Ruthven, D.M., Farooq, S., Knaebel, K.S.: Pressure Swing Adsorption. VCH, New York (1994)
Scholes, C.A., Stevens, G.W., Kentish, S.E.: Membrane gas separation applications in natural gas processing. Fuel 96(1), 15–28 (2012)
Singh, P., Niederer, J.P.M., Versteeg, G.F.: Structure and activity relationships for amine-based CO2 absorbents-II. Chem. Eng. Res. Des. 87(1), 135–144 (2009)
Smit, B., Maesen, T.L.M.: Molecular simulations of zeolites: adsorption, diffusion, and shape selectivity. Chem. Rev. 108(10), 4125–4184 (2008)
Song, L., Sun, Z.L., Rees, L.V.C.: Experimental and molecular simulation studies of adsorption and diffusion of cyclic hydrocarbons in silicalite-1. Microporous Mesoporous Mater. 55(2), 31–49 (2002)
Sun, H.: Ab initio calculations and force field development for computer simulation of polysilanes. Macromolecules 28(3), 701–712 (1995)
Talu, O., Myers, A.L.: Molecular simulation of adsorption: Gibbs dividing surface and comparison with experiment. AIChE J. 47(5), 1160–1168 (2001)
Verheyen, E., Joos, L., Van Havebergh, K., Breynaert, E., Kasian, N., Gobechiya, E., Houthoofd, K., Martineau, C., Hinterstein, M., Taulelle, F., Van Speybroeck, V., Waroquier, M., Bals, S., Van Tendeloo, G., Kirschhock, C.E.A., Martens, J.A.: Design of zeolite by inverse sigma transofrmation. Nat. Mater. 11(12), 1059–1064 (2012)
Wu, J.Y., Liu, Q.L., Xiong, Y., Zhu, A.M., Chen, Y.: Molecular simulation of water/alcohol mixtures’ adsorption and diffusion in zeolite 4A membranes. J. Phys. Chem. B 113(13), 4267–4274 (2009)
Xie, D., McCusker, L.B., Baerlocher, C.: Structure of the boroslicate zeolite catalyst SSZ-82 solved using 2D-XPD charge flipping. J. Am. Chem. Soc. 133(50), 20604–20610 (2011)
Xu, Y., Li, Y., Han, Y., Song, X., Yu, J.: A gallogermanate zeolite with eleven-membered-ring channels. Angew. Chem. Int. Ed. 52(21), 5501–5503 (2013)
Yang, J., Zhao, Q., Xu, H., Li, L., Dong, J., Li, J.: Adsorption of CO2, CH4, and N2 on gas diameter grade ion-exchange small pore zeolites. J. Chem. Eng. Data 57(12), 3701–3709 (2012)
Yang, J.Z., Chen, Y., Zhu, A.M., Liu, Q.L., Wu, J.Y.: Analyzing diffusion behaviors of methanol/water through MFI membranes by molecular simulation. J. Membr. Sci. 318(1–2), 327–333 (2008)
Zeng, Y., Ju, S., Xing, W., Chen, C.: Computer simulation of benzene adsorbed in all-silica Y and NaY zeolites. Ind. Eng. Chem. Res. 46(1), 242–248 (2007)
Zhang, J., Burke, N., Zhang, S., Liu, K., Pervukhina, M.: Thermodynamic analysis of molecular simulations of CO2 and CH4 adsorption in FAU zeolites. Chem. Eng. Sci. 113(13), 54–61 (2014)
Zhang, K., Lively, R.P., Noel, J.D., Dose, M.E., McCool, B.A., Chance, R.R., Koros, W.J.: Adsorption of water and ethanol in MFI-type zeolites. Langmuir 28(23), 8664–8673 (2012)
Zhao, L., Zhai, D., Liu, B., Liu, Z., Xu, C., Wei, W., Chen, Y., Gao, J.: Grand canonical Monte Carlo simulations for energy gases on PIM-1 polymer and silicalite-1. Chem. Eng. Sci. 68(1), 101–107 (2012)
Zhong, D.L., Daraboina, N., Englezos, P.: Recovery of CH4 from coal mine model gas mixture (CH4/N2) by hydrate crystallization in the presence of cyclopentane. Fuel 106(4), 425–430 (2013)
Zhu, W., Hrabanek, P., Gora, L., Kapteijn, F., Moulijn, J.A.: Role of adsorption in the permeation of CH4 and CO2 through a silicalite-1 membrane. Ind. Eng. Chem. Res. 45(2), 767–776 (2006)
Acknowledgments
This project was supported by the National Natural Science Foundation of China (21276101 and 21106051), the Jiangsu Program for Production, Teaching, and Research (BY2015052-01), Jiangsu Qing Lan Project and the Six Projects Sponsoring Talent Summits of Jiangsu Province, the Program for Science and Technology Development of Huai’an (HAC2015028, HAG2013073) and the Foundation of Jiangsu Key Laboratory for Chemistry of Low-Dimensional Materials (JSKC15143).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chu, Xz., Liu, Ss., Zhou, Sy. et al. Adsorption and separation of carbon dioxide and methane in new zeolites using the Grand Canonical Monte Carlo method. Adsorption 22, 891–899 (2016). https://doi.org/10.1007/s10450-016-9793-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-016-9793-x