
Abstract
Device failure due to undesired biological responses remains a substantial roadblock in the development and translation of new devices into clinical care. Polyethylene glycol (PEG)-based hydrogel coatings can be used to confer antifouling properties to medical devices—enabling minimization of biological responses such as bacterial infection, thrombosis, and foreign body reactions. Application of hydrogel coatings to diverse substrates requires careful consideration of multiple material factors. Herein, we report a systematic investigation of two coating methods: (1) traditional photoinitiated hydrogel coatings; (2) diffusion-mediated, redox-initiated hydrogel coatings. The effects of method, substrate, and compositional variables on the resulting hydrogel coating thickness are presented. To expand the redox-based method to include high molecular weight macromers, a mechanistic investigation of the role of cure rate and macromer viscosity was necessary to balance solution infiltration and gelation. Overall, these structure–property relationships provide users with a toolbox for hydrogel coating design for a broad range of medical devices.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Device failure due to undesired biological responses remains a substantial roadblock in the development and translation of new devices into clinical care. For example, biofouling of urinary catheters results in a high incidence of urinary tract infections (12–25%) upon usage,60 and synthetic vascular grafts require lifelong anticoagulation therapy with a high associated cost and complications.42,53 Soft tissue-like hydrogels are a popular material to better guide the biological response; however, hydrogels lack the durability to withstand handling or physiological loading in many applications as standalone materials.24,51,66 Hydrogel coatings provide the opportunity to decouple biological design criteria from requisite device mechanical properties. This opens the possibility of adding antifouling properties to medical devices—enabling minimization of biological responses such as bacterial infection, thrombosis, and foreign body reactions. Polyethylene glycol (PEG)-based hydrogels are commonly selected for this purpose due to their established structure–property relationships,11,32,35,38,45 history of safety in broad applications,31,44,50,52,55,58,64 and facile incorporation of biological cues.8,9,10,15,16 Despite these desirable properties, methods for forming and characterizing PEG-based hydrogel coatings are under-reported in the literature.
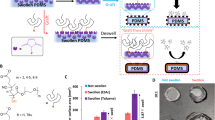
The most common method used to apply PEG-based hydrogel coatings utilize photoinitiated crosslinking. For example, our previous work described photocured hydrogel coatings in multilayered small-diameter vascular grafts8,9,15,43 and heart valves.40,44,47 Protective PEG-dimethacrylate coatings have also been utilized in neural implants and other biomedical applications.19,20,37,50 In the photoinitiation curing process, a hydrogel precursor solution containing a photo-initiator is placed into a mold containing the substrate of choice, and the solution is crosslinked via exposure to UV light (Fig. 1a). Tuning of the hydrogel coating is necessary to meet the requirements of different applications. Modulus is primarily controlled by the precursor solution composition (macromer molecular weight, polymer concentration, crosslinker).11,13,32,45 Although tunable in terms of hydrogel composition, thickness and shape of the construct are determined by the mold. Multiple molds must be employed for each substrate geometry and target coating thickness. It is experimentally difficult to achieve thin, uniform coatings or conformable coatings of complex shapes with this mold-based approach (Fig. 1b and 1c), which limits photoinitiated hydrogel coatings to relatively simple geometries (flat, tubular substrates) with target thicknesses > 100 microns.

Hydrogel coating fabrication methods. (a) Photoinitiated radical crosslinking mechanism with radical generation mediated by UV exposure. (b) Photocured hydrogel coating on a small diameter vascular graft and (c) 3D printed stent. (d) Redox initiated radical crosslinking mechanism with diffusion-based radical generation due to the redox reaction between ammonium persulfate and iron gluconate. (e) Redox-initiated hydrogel coating on a small diameter vascular graft and (f) 3D printed stent.
To achieve hydrogel coatings on materials with more complex geometries, researchers have developed newer methods that do not require molds. Several research groups have accomplished this with surface initiation,36,41,63 surface bridging,34,61,62 or diffusion-mediated crosslinking processes.27,39,46,56 Yu et al. developed an interfacial interpenetration strategy to form “hydrogel skins” of poly(acrylic acid), poly(N,N-dimethylacrylamide), poly(N-vinylpyrrolidone), and poly(hydroxyethyl) methacrylate hydrogels.61 Johnson et al. utilized enzyme-mediated redox chain initiation to generate conformal, micrometer-scale PEG-based hydrogel layers.27,28 Glucose oxidase is used to generate a glucose-dependent source of hydrogen peroxide that reacts with Fe2+ to initiate hydrogel crosslinking. Thus, glucose diffusion from the substrate can be used to provide temporal control and spatial localization.23,28 The Grande-Allen lab utilized this crosslinking platform with PEGDA and PEG-diacrylamide to coat decellularized bovine heart valves with a thin and conformable hydrogel layer.47 Ma et al. developed a similar diffusion-based approach based on leaching of Fe2+ from 3D printed constructs to initiate hydrogel crosslinking at the surface after reaction with a peroxide intiator.39 To expand this approach and remove the requirement for compounding with ferrous iron prior to coating, our lab developed an adsorption-based redox approach. Iron gluconate (IG) adsorption to a substrate and diffusion-mediated desorption from the surface can be used to initiate the reduction of ammonium persulfate (APS) for crosslinking at the substrate surface (Fig. 1d).56 This method enabled conformable hydrogel coatings with tunable thickness at multiple molecular weights and polymer concentrations (Figs. 1e and 1f). Additionally, this method yields tunable thickness over time, eliminating the need for multiple molds. However, due to the increased complexity of the redox crosslinking platform, greater consideration of fabrication conditions is necessary to tune these coatings for individual applications. Concentrations of each reagent (APS, IG, macromer) affect reaction kinetics, and macromer molecular weight affects both acrylate concentration and solution viscosity.12,26,28,54,58
Both the conventional photoinitiated coating method and the newer diffusion-mediated redox coating method have advantages and limitations that require consideration of the intended application and substrate of interest. The goal of this paper is to provide readers with the design considerations for both platforms and the tools necessary to tailor these methods to meet the needs of the target application. Electrospun mesh substrates, a material widely used in biomedical applications, were used in this investigation as an example substrate for consideration.29 First, design parameters of photoinitiated hydrogel coatings are presented and compared to the redox-initiated hydrogel coatings. In photopolymerized systems, initial coating thickness is dependent on mold geometry; whereas, the diffusion-mediated redox method controls coating thickness by IG concentration and macromer immersion time. Both of the methods are dependent on compositional determinants of swelling degree to predict the final hydrogel coating thickness. An added challenge of the diffusion-mediated redox system in regards to higher molecular weight macromer applications is then discussed and a series of experiments used to elucidate key parameters and prevent coating delamination. Finally, individual effects of substrate properties and processing prior to coating is demonstrated. Ultimately, this work provides users with guidelines to adapt these coating methods to a broad range of device applications.
Experimental
Materials
Reagents were purchased from Sigma-Aldrich and used without further purification unless otherwise noted.
Fabrication of Electrospun Meshes
Bionate® 80A (DSM Biomedical Inc., Berkeley, CA, USA) was dissolved in 70:30 dimethylacetamide:tetrahydrofuran at 14 wt% to generate electrospinning solutions. The polymer solution was pumped at a flow rate of 0.5 mL/h from a 20-gauge blunted syringe needle onto a rotating mandrel (50 RPM) positioned 50 cm from the needle tip. Electrospinning was performed under controlled humidity between 40 and 50% with temperatures of 21.0–26.5 °C. Using a high voltage source, the mandrel was charged at − 5 kV and the needle tip voltage range from + 11.5 to 15.5 kV to maintain Taylor cone stability. Meshes were spun for 3.5–4 h to obtain the desired thickness (~ 0.13–0.18 mm, n = 15). Following spinning, the mandrel with attached mesh was annealed at 70 °C overnight. The meshes were characterized via scanning electron microscopy (SEM) by taking three sections of the mesh with five images taken per section for a total of 15 images per mesh, SI Fig. 1. Fiber diameter was obtained from the images in ImageJ by drawing a midline across the SEM image and measuring the first ten fibers that crossed the line for a total of 150 fibers per mesh.
Synthesis of PEGDA
Poly(ethylene glycol) diacrylate (PEGDA) was synthesized as previously described.11 Briefly, a solution of PEG diol in anhydrous dichloromethane was prepared under a nitrogen atmosphere (PEG 3.4 kDa or 10 kDa, 0.1 mmol/mL; PEG 20 kDa, 15 wt%; 1 mol equivalents). Anhydrous triethylamine (2 mol equivalents) was added dropwise, followed by dropwise addition of acryloyl chloride (4 mol equivalents). The reaction was stirred for 24 h after addition for PEGDA 3.4 kDa and 10 kDa and 48 h after addition for PEGDA 20 kDa. The reaction was then neutralized with a potassium bicarbonate wash (8 mol equivalents) and dried with anhydrous sodium sulfate. PEGDA was precipitated in chilled diethyl ether, filtered, and dried at room temperature overnight followed by vacuum drying for 5 min. Product acrylation was determined using proton nuclear magnetic resonance (1H NMR) spectroscopy. Spectra were recorded on a Varian MR-400 400 MHz spectrometer and analyzed using an internal reference of TMS/solvent signal. Polymers with conversions of hydroxyl to acrylate end groups over 85% were used in this work. 1H-NMR (CDCl3): 3.6 ppm (m, –OCH2CH2–), 4.3 ppm (t, –CH2OCO–), 6.1 ppm (dd, –CH=CH2), 5.8 and 6.4 ppm (dd, –CH=CH2).
Hydrogel Composite Fabrication and Characterization
Precursor Preparation
All polymer solutions were prepared by dissolving PEGDA (3.4, 10, or 20 kDa) in deionized water at a concentration of 10 or 20 wt%/vol. For photoinitiated hydrogels, precursor solutions with 0.1 wt% Irgacure 2959 were prepared. For hydrogel coatings, precursor solutions were prepared with ammonium persulfate (0.01, 0.025, 0.05, or 0.1 wt%).
Photoinitiated Hydrogel Composite Fabrication
Photoinitiated bulk hydrogels were fabricated by placing precursor solutions between 1.5 mm spaced glass plates and curing on a UV transilluminator (UVP, 25-W, 365 nm) for 6 min on both sides. Photoinitiated composites were prepared by soaking mesh substrates in precursor solutions for 30 min prior to crosslinking. Mesh samples (0.15–0.16 mm) were then placed between glass slides, held by coverslips to set thickness (two No. 1 coverslips, 0.32–0.36 mm spaced), and precursor solution was pipetted between the glass slides. Samples were cured on a UV transilluminator for 6 min on both sides identically to bulk hydrogels. Thicknesses of mesh and hydrogel coatings at crosslinking and equilibrium swelling was characterized with force-normalized calipers (Mitutoyo, n = 9).
Diffusion-Mediated Redox Hydrogel Composite Fabrication
Redox hydrogel coatings were formed as previously described with minor modifications.56 Electrospun mesh samples (120–190 µm thick) were cut to 5 × 10 mm and passed through a graded ethanol ramp (70, 50, 30, and 0% ethanol in water, 15 min each) prior to use to ensure proper wetting of the substrate with the iron gluconate (IG) solution and homogenous adsorption of the IG to the substrate. Mesh substrates (0.12–0.19 mm thick) were then coated in a solution of IG (3 wt% [Fe2+] as determined with the Ferrozine Assay)25 via adsorption by soaking ramped meshes in IG for 15 min. After soaking, meshes were briefly dipped in methanol then dried under compressed air for 1 min. After drying, meshes were immediately transferred to 3D printed clamps (SI Fig. 2) and immersed in aqueous solutions of PEGDA with APS (detailed above) for 10, 20, or 30 s in a 96 well plate. After fabrication, composites were either immediately immersed in deionized water or allowed to continue crosslinking for 15 min or until dry (~ 2 h). All composites were then washed in deionized water with three exchanges of water at 10 min, 15 min, and overnight to remove the sol fraction.
The minimum concentration of APS necessary to form hydrogels was determined by characterizing the mass of dried hydrogels (3.4 kDa 10 wt%, 20 kDa 10 wt% or 20 wt%) at three APS concentrations (0.01, 0.025, or 0.05 wt% APS). The mass of dry composites after swelling and removal of the sol fraction was weighed and the mass of the uncoated meshes subtracted. If the resultant mass of the dry hydrogel was > 0.1 mg, a gel was considered to have formed (n = 12).
The effect of molecular weight and macromer concentration (3.4 or 20 kDa, 10 or 20 wt%, 0.05 wt% APS), time,13,16,35 and APS concentration (20 kDa 10 wt%, 0.05 wt% or 0.1 wt% APS, 30 s) on hydrogel coating thickness was determined (n = 12). Composites were first trimmed to eliminate edge effects and then thickness was measured with a force-normalized caliper (Mitutoyo). The effect of swelling on dimensional changes were determined by measuring composite thickness immediately after curing and again after swelling overnight (n = 12).
Statistical Analysis
The data for all measurements are displayed as mean ± standard deviation. An analysis of variation (ANOVA) comparison utilizing Tukey’s multiple comparison test was used to analyze the significance of data among multiple compositions. Outliers were removed using a ROUT analysis (Q = 0.1%). All tests were carried out at a 95% confidence interval (p < 0.05).
Results and Discussion
Hydrogel coatings have enabled broad innovation in medical device design through the decoupling of surface properties from bulk mechanical durability. In this study, two fabrication methods for hydrogel coatings are described. To demonstrate the benefits and limitations of photoinitiated and redox-initiated radical crosslinking methods, key design variables of each method were characterized including compositional control, swelling behavior, and substrate effects.
Photoinitiated Hydrogel Coatings
The primary variable used to control hydrogel coating thickness with photointiation is through mold geometry. Hydrogel composites (10 wt% 3.4 kDa) were fabricated between glass slides spaced with a range of thickness (0.3, 0.6, or 0.9 mm), photopolymerized, and swollen (Fig. 2a). As expected, the thickness of swollen composites increased with increasing mold thickness with the final hydrogel thickness dependent on the equilibrium swelling of the hydrogel coating. The effects of macromer molecular weight and concentration of the final coating thickness were then evaluated. As expected, thicknesses of composites at cure were similar (SI Fig. 3); whereas, there were distinct differences in the final hydrogel swelling thickness. Swelling ratios were determined by comparing thickness at the swollen state to the as-cured thickness (n = 9). Average swelling ratios of hydrogel composites were 1.4 ± 0.3, 2.3 ± 0.2, and 2.5 ± 0.2 for 10 wt% 3.4 kDa, 10 wt% 20 kDa, or 20 wt% 20 kDa, respectively. There was a significant difference in swelling based on macromer molecular weight (3.4 kDa vs. 20 kDa, p < 0.0001); whereas, the effect of polymer concentration (10 vs. 20 wt%) of the 20 kDa composites was not stastically different (p = 0.2261).

Photoinitiated hydrogel coating thickness control and swelling effects. (a) Hydrogel coating thickness is controlled with mold geometry and the equilibrium swelling of the hydrogel (PEGDA 3.4 kDa). (b) Composition effects on swelling-induced thickness changes of hydrogel coatings with regards to macromer molecular weight and concentration. * represents a significant difference with p < 0.0001. Scale bar represents 500 µm.
For photopolymerized hydrogels, both molding and swelling effects must be considered to achieve desired thicknesses. Lower macromer molecular weight (3.4 kDa) resulted in minimal dimensional change post-swelling; whereas, larger macromer molecular weights (20 kDa) caused over a two-fold increase in thickness change post-swelling. To attain a target thickness, one must characterize the swelling properties of each hydrogel composition in their coating application to select an appropriate mold. In bulk photopolymerized hydrogels, swelling ratios are affected by both macromer molecular weight and concentration. In this study of hydrogel coatings, only macromer molecular weight had a significant effect on swelling ratio (Fig. 2b). In these studies, swelling ratios were estimated by the ratio of swollen to as-cured thickness. Higher macromer molecular weight compositions had higher swelling ratios, consistent with previous studies11; whereas, higher concentrations did not correspond with lower swelling ratios. In contrast to the method employed here to estimate swelling, bulk hydrogel swelling ratios are calculated with volumetric swelling or mass-based to volumetric swelling methods.45 We expect that substrate effects cause restrictions to dimensional changes, and this is responsible for the observed phenomonen. Photopolymerized coatings on electrospun mesh substrates are adhered via a thread-hole topology as described by Yang et al.59 The interdigitated portion of the hydrogel prevents the surface portion from uniform swelling. We hypothesize nonuniform stresses in the photopolymerized coatings resulted in the unexpected swelling results. To rigorously characterize these swelling effects, model-informed thickness control of substrate and hydrogel coating could be employed.
Additional considerations of the photointiation mold setup for hydrogel coatings are substrate geometry and coating uniformity. Simple geometries such as flat sheets and tubular constructs can be fabricated with molds, but more complex substrates such as trileaflet valves or stents are limited. For non-rigid substrates, uneven coating thickness can result in differential swelling forces that deform the substrate. In addition, it is difficult to form hydrogel coatings of uniform thicknesses on thin substrates with this molding approach.
Redox-Initiated Hydrogel Coatings
There are several variables used to control coating thickness in the redox diffusion-mediated crosslinking system (Fig. 3). To demonstrate key design criteria, growth kinetics of hydrogel coatings as a function of APS concentration and macromer molecular weight and concentration were investigated. Hydrogel coatings successfully formed for all compositions except PEGDA 3.4 kDa at 10 s (Fig. 3a). Each condition resulted in significant hydrogel thickness growth with time (10 wt% PEGDA 20 kDa at 0.05 and 0.1 wt% APS: p < 0.0001; PEGDA 3.4 at 10 wt%: p < 0.0001; PEGDA 20 kDa at 20 wt%: p < 0.0001, Fig. 3a). The thinnest hydrogel compositions were 40 ± 20 µm for PEGDA 3.4 kDa at 20 s (Fig. 3a), and the thickest compositions were 500 ± 40 µm for 20 wt% PEGDA 20 kDa at 30 s (Fig. 3a). These results demonstrate the ability of this system to be tuned by initiator concentration and time regardless of macromer molecular weight.

Redox-initiated hydrogel coating thickness control and swelling effects. (a) Hydrogel coating thickness is tunable with time and composition: initiator concentration (10 wt%, 20 kDa), macromer molecular weight (10 wt% solution, 0.05 wt% APS), and macromer concentration (20 kDa, 0.05 wt% APS). (b) As cured thickness of redox hydrogels compared to swollen thickness with regards to macromer molecular weight and concentration (30 s immersion time). ***Significant difference at p < 0.001, ****significant difference at p < 0.0001, *difference from all groups at a 99.9% CI. The scale bar represents 500 µm.
We previously reported the mechanism for redox diffusion-mediated crosslinking.56 As IG desorbs from the surface and diffuses into the solution, redox reactions between APS and IG form persulfate radicals that facilitate the polymerization of reactive polymer end groups. Hydrogel coating thickness increases over time as IG diffuses further into solution. In the present study, the diffusion-mediated crosslinking system yielded growth kinetics specific to each composition tested. Increased initiator concentration (0.05 to 0.1 wt%) resulted in thicker hydrogel coatings at each time point (p < 0.01, n = 12). This increase in thickness was hypothesized to be a result of faster crosslinking kinetics at higher initiator concentrations. It is well established that higher concentrations of redox reactants result in faster gelation time.12,57 This system can also be tuned by raising or lowering IG concentrations with similar thickness effects (SI Fig. 4). Similar to the photoinitiated hydrogel coatings, the final coating thickness is also dependent on the equilibrium swelling of the hydrogel network. Increased macromer molecular weight (3.4 to 20 kDa) resulted in thicker hydrogel coatings at all time points (p < 0.001, n = 12) with an estimated swelling ratio of PEGDA 3.4 kDa was 1.1 ± 0.1 as compared to 1.5 ± 0.2 for 20 kDa (p < 0.0001, n = 12). Increased polymer concentration (10 to 20 wt% PEGDA 20 kDa) also resulted in thicker as-cured hydrogel coatings at all time points (p < 0.001, n = 12); however, the swelling ratios of PEGDA 20 kDa at 10 and 20 wt% were not significantly different, 1.5 ± 0.2 and 1.7 ± 0.1 for 10 and 20 wt%, respectively (n = 12). As-cured hydrogel thickness for 10 wt% PEGDA 20 kDa was 210 ± 40 µm as compared to 300 ± 20 µm for 20 wt% (30 s, SI p < 0.0001, n = 12).This result was contrary to our previous findings for PEGDA 3.4 kDa, where increased macromer concentration resulted in hydrogel coatings with similar as-cured thicknesses differences but significant differences in swelling ratio.56 In this system, faster crosslinking kinetics led to thicker coatings. It was hypothesized that increased viscosity and number of functional groups at higher macromer concentration additionally led to faster crosslinking kinetics in this diffusion-mediated system. These results demonstrate the high degree of tunability in the redox diffusion-mediated crosslinking system and highlight the number of necessary factors for consideration to successfully implement this methodology.
In comparing the two coating methods, the swelling ratios were lower in the redox diffusion-mediated system for all compositions (10 wt% PEGDA 3.4 kDa: p < 0.0004; 10 wt% PEGDA 20 kDa: p < 0.0001; 20 wt% PEGDA 20 kDa: p < 0.0001; n = 9; SI Fig. 5B). For 10 wt% PEGDA 3.4 kDa compositions, photoinitiated coatings had a swelling ratio of 1.4 ± 0.3 in comparison to 1.1 ± 0.1 for the redox-initiated coatings (SI p < 0.0001, n = 9). Overall, both systems can be used to generate coatings in a range of thicknesses and each require consideration of swelling effects on final composite thickness. The redox-initiated system can be used to achieve a broader range of coating thicknesses and applied as a conformable coating of complex geometries.
Preventing Delamination of Redox-Initiated Hydrogel Coatings
Preliminary studies of the redox-initiated coating system conducted with high molecular weight PEGDA 20 kDa resulted in delamination after swelling. This delamination was not observed with previous studies that used lower macromer molecular weight (3.4 kDa), Fig. 4a. The ability to utilize higher molecular weight macromers is necessary for the fabrication of robust hydrogel coatings.14,65 Hydrogel delamination results when the swelling forces exceed the strength of the hydrogel network. Given that delamination was not observed in photoinitiated coatings despite their higher degree of dimensional change with swelling (SI Fig. 5B) we hypothesized that it was a lack of interdigitation with the substrate in the redox-initiated coatings that caused the failure. To understand interdigitation, surface interactions of the substrate and hydrogel precursor solution in the photopolymerization and redox systems were considered. In preparation for photopolymerization, substrates are soaked in the polymer solution prior to crosslinking to ensure solution infiltration. In the redox-initiated coating system, substrates are exposed to the polymer solution upon crosslinking with gelation occurring in as little as 10 s. The redox system relies on desorption of IG from the mesh as well as diffusion of APS and polymer into the mesh. This substrate has ~ 12 µm sized pores, significantly larger than the hydrodynamic radius of PEGDA even at 20 kDa, 7.4 nm.49 However, high macromer solution viscosities slow diffusion and viscosity is further increased during gelation.57 Because gelation occurs so quickly in the diffusion-mediated redox platform, there is only a short period of time allowed for the precursor solution to infiltrate the mesh. When cure rates are fast and the time to gelation short, the macromer solution does not have time to infiltrate the mesh prior to gelation. This results in poor anchoring that fail upon swelling of the hydrogel coating (Fig. 4a).

Delamination considerations in the design of redox-initiated hydrogel coatings. (a) Representative images displaying the effect of macromer molecular weight on swelling-induced delamination of hydrogel coatings. (b) Schematic of hydrogel interdigitation in redox-initiated hydrogel coatings as a function of precursor solution viscosity and cure rate.
To ameliorate the issue of delamination, we hypothesized that decreasing cure rate and prolonging cure time could improve interdigitation by allowing more time for macromer diffusion into the substrate (Fig. 4a). Increasing APS concentration results in faster onset of gelation and complete network formation in bulk hydrogels.57 To slow the cure rate while still forming a gel, the minimum APS concentration necessary for a hydrogel coating to form was investigated (SI Fig. 7). For both 10 wt% PEGDA 3.4 and 20 kDa, 0.05 wt% APS was the minimum concentration necessary to consistently form a hydrogel at a 20 s immersion time (n = 12). This APS concentration was used to conduct the thickness studies described in the prior section of this work. Interestingly, 20 wt% PEGDA 20 kDa formed a hydrogel with APS concentrations as low as 0.01 wt% (n = 12). Increasing macromer concentration increases both precursor solution viscosity and number of reactive groups. The number of reactive acrylate groups for 10 wt% PEGDA 3.4 kDa is 3× higher than that of 20 wt% PEGDA 20 kDa, yet 10 wt% PEGDA 3.4 kDa does not form gels at 0.025 wt%. The viscosity of 20 wt% PEGDA 20 kDa is ~ 20× higher than that of 10 wt% PEGDA 3.4 kDa. This comparison demonstrates the significant impact viscosity has on hydrogel formation in this system, supported by the increased growth kinetics demonstrated with 20 wt% PEGDA 20 kDa relative to 10 wt% PEGDA 20 kDa (Fig. 3a). Despite the increased diffusional time at the lower APS concentration, delamination of the 10 wt% PEGDA 20 kDa hydrogels was still observed and the coatings were qualitatively softer than coatings fabricated at higher APS concentration. We hypothesized that this was the result of reduced crosslink density at the lower APS concentration, Fig. 5a. In the original redox diffusion-mediated crosslinking method, the redox reaction is quenched immediately after removal of the coating from the soaking solution.56 It was hypothesized that excess IG in the composite could react with any unreacted APS in the as-formed hydrogel to continue building crosslinking density after removal of the composite from the precursor solution (SI Fig. 5 and Fig. 6A). To test this hypothesis, the effect of prolonged crosslinking duration was assessed for 10 wt% PEGDA 20 kDa hydrogels at 0.05 wt% APS (Figs. 5b and 5c). Composites were either quenched immediately after initial crosslinking or allowed to sit overnight. Their thicknesses as-cured and after overnight swelling were determined (Fig. 5b). Swollen thicknesses with prolonged crosslinking via an overnight sit were significantly lower than the no sit condition (p < 0.0001, n = 12). Lower swelling indicates the crosslinking density of the network was increased with an overnight sit. Further, immediate delamination after overnight swelling was eliminated with the introduction of an overnight sit, Fig. 5c. This supports that improved crosslinking density prevented rapid delamination. To enable successful utilization of the redox diffusion-mediated crosslinking platform with high molecular weight macromers, the methodology of the redox-diffusion mediated crosslinking system was then updated, Fig. 5d. Concentrations of IG and APS were both lowered from our initial publication to slow cure rate.56 Additionally, a prolonged crosslinking period after the dip coating was introduced to allow crosslinking to continue in air to facilitate complete network formation. These changes enabled more robust hydrogel coatings.

Impact of crosslinking duration on delamination of redox-initiated hydrogel coatings. (a) Schematic depiction of crosslinking density over time as a function of initial initiator concentration. (b) Effect of crosslinking duration on the thicknesses of hydrogel coatings immediately after cure and after swelling (10 wt% PEGDA 20 kDa, 0.05 wt% APS solutions, n = 12). Representative images of hydrogel coatings as cured and after swelling for 24 h (10 wt% PEGDA 20 kDa, 0.05 wt% APS solutions, n = 12) (c). Schematic representation of updated crosslinking method (d). A coated substrate is immersed in polymer and initiator solution with hydrogel coating thickness increasing with time as the iron diffuses away from the substrate. Hydrogel composites are allowed to continue curing overnight after removal from the crosslinking solution. Hydrogel composites are then soaked in water to remove residual redox reagents and terminate the reaction. * indicates a significant difference at p < 0.0001
Substrate Considerations
Finally, consideration of substrate properties and processing is important in both photopolymerized hydrogel coatings and redox-initiated hydrogel coatings. To this end, the role of substrate microarchitecture and hydrophobicity on resulting hydrogel coatings was investigated. First, photopolymerized and redox-based hydrogels were formed on electrospun meshes with two fiber sizes (1 and 10 µm), Fig. 6a. Both composites successfully formed on these meshes without sensitivity to the microarchitecture. In contrast, uniform coating was highly dependent on solution penetration of the mesh (Fig. 6b). Without pre-treatment, a 10 wt% PEGDA 20 kDa solution displayed limited penetration into the hydrophobic polyurethane mesh. When meshes are coated in hydrophilic IG for use in redox initiation, some wetting of the hydrophobic substrate occurs but full solution penetration is heterogeneous. For photopolymerized hydrogel coatings, hydrophobic substrates can be used after running a graded ethanol ramp to improve wettability. For redox-diffusion mediated crosslinking, a graded ethanol ramp is not recommended as this can change the concentration of IG on the substrate and subsequent crosslinking kinetics. As an alternative method, the researcher can immediately transition from the IG coating to the macromer solution soak without drying, Fig. 6b. The degree of drying following the methanol quench will be dependent on the substrate geometry and hydrophobicity. In our experience, microfibrous structures trap bubbles and have slower and more heterogenous wetting than larger geometries such as 3D printed stents.

Substrate considerations for both crosslinking modalities. (a) Effect of electrospun mesh fiber size on composites (10 wt% PEGDA 20 kDa) for UV and redox crosslinking methods. (b) Hydrophobic substrates prevent effective wetting and hydrogel coating formation. Pre-wetting with an ethanol ramp improves polymer diffusivity into mesh for photoinitiated hydrogel coatings. Redox-initiated hydrogel coatings start with an initial step of coating electrospun mesh substrates with iron gluconate through adsorption. The effect of drying time on wetting of the substrate. Scale bar represents 20 µm.
Conclusion
The studies presented here provide design considerations for implementation of the well-established photopolymerization crosslinking method and the recently developed redox-initiated hydrogel coating method. Photopolymerization gelation kinetics have been rigorously characterized by many researchers, including factors such as reaction kinetics, polymer diffusivity, and complicating factors such as auto acceleration and auto deceleration.1,2,3,6,7,30,48 Redox-based crosslinking methods are diverse and include many redox pairs with unique reaction kinetics.4,5,18,21,22,33,57 Introducing further complexity to a redox-initiated process by taking advantage of diffusion-dependent kinetics requires further characterization for successful implementation.28 Use of the redox-initiated crosslinking system presented here specifically requires careful consideration of the interplay of viscosity and crosslinking rate on the hydrogel system. The mechanisms proposed in this work can be used by other researchers to tune this platform to their application. Macromer viscosities and functional groups can vary widely depending on the system used. To utilize the redox-initiated crosslinking method, a stepwise protocol development is suggested. First, researchers should characterize adsorption/desorption kinetics of their desired redox coupling pair on the substrate. Next, the minimum concentration of the redox pair in solution that reliably forms hydrogel coatings in the desired timeframe should be determined. Finally, characterization of the growth kinetics of their hydrogel coating under these set conditions is needed to develop a robust and tunable hydrogel coating methodology. Overall, this study describes and provides tools for the utilization of two important methods for hydrogel coating fabrication, both with their own benefits and limitations. Importantly, this study enables fabrication of high molecular weight hydrogel coatings for damage resistant applications.
References
Anseth, K. S., K. J. Anderson, and C. N. Bowman. Radical concentrations, environments, and reactivities during crosslinking polymerizations. Macromol. Chem. Phys. 197:833–848, 1996.
Anseth, K. S., and C. N. Bowman. Kinetic gelation model predictions of crosslinked polymer network microstructure. Chem. Eng. Sci. 49:2207–2217, 1994.
Anseth, K. S., L. M. Kline, T. A. Walker, K. J. Anderson, and C. N. Bowman. Reaction kinetics and volume relaxation during polymerizations of multiethylene glycol dimethacrylates. Macromolecules. 28:2491–2499, 1995.
Ariff, M., M. Jainuddin, V. Gopalan, and K. V. Rao. Aqueous polymerization of acrylonitrile by ascorbic acid–peroxodisulfate redox system. J. Polym. Sci. Polym. Chem. Ed. 23:2063–2071, 1985.
Berry, K. L., and J. H. Peterson. Tracer studies of oxidation—Reduction polymerization and molecular weight of “Teflon” tetrafluoroethylene resin. J. Am. Chem. Soc. 73:5195–5197, 1951.
Bowman, C. N., and N. A. Peppas. Coupling of kinetics and volume relaxation during polymerizations of multiacrylates and multimethacrylates. Macromolecules. 24:1914–1920, 1991.
Bowman, C. N., and N. A. Peppas. A kinetic gelation method for the simulation of free-radical polymerizations. Chem. Eng. Sci. 47:1411–1419, 1992.
Browning, M. B., D. Dempsey, V. Guiza, S. Becerra, J. Rivera, et al. Multilayer vascular grafts based on collagen-mimetic proteins. Acta Biomater. 8:1010–1021, 2012.
Browning, M. B., V. Guiza, B. Russell, J. Rivera, S. Cereceres, et al. Endothelial cell response to chemical, biological, and physical cues in bioactive hydrogels. Tissue Eng. Part A. 20:3130–3141, 2014.
Browning, M. B., B. Russell, J. Rivera, M. Höök, and E. M. Cosgriff-Hernandez. Bioactive Hydrogels with enhanced initial and sustained cell interactions. Biomacromolecules. 14:2225–2233, 2013.
Browning, M., T. Wilems, M. Hahn, and E. Cosgriff-Hernandez. Compositional control of poly (ethylene glycol) hydrogel modulus independent of mesh size. J. Biomed. Mater. Res. Part A. 98:268–273, 2011.
Chang, J., Y. Tao, B. Wang, X. T. Yang, H. Xu, et al. Evaluation of a redox-initiated in situ hydrogel as vitreous substitute. Polymer. 55:4627–4633, 2014.
Chapla, R., M. Alhaj Abed, and J. West. Modulating functionalized poly(ethylene glycol) diacrylate hydrogel mechanical properties through competitive crosslinking mechanics for soft tissue applications. Polymers. 12:3000, 2020.
Chen, K., Y. Feng, Y. Zhang, L. Yu, X. Hao, et al. Entanglement-driven adhesion, self-healing, and high stretchability of double-network PEG-based hydrogels. ACS Appl. Mater. Interfaces. 11:36458–36468, 2019.
Cosgriff-Hernandez, E., M. S. Hahn, B. Russell, T. Wilems, D. Munoz-Pinto, et al. Bioactive hydrogels based on designer collagens. Acta Biomater. 6:3969–3977, 2010.
Cosgriff-Hernandez, E., A. Post, P. Dhavalikar, T. Wilems, and Z. Lan, Integrin-targeting materials in regenerative medicine, in Proceedings of Abstracts of Papers of the American Chemical Society, vol. 256. Washington, DC: American Chemical Society, 2018.
Dortdivanlioglu, B., N. E. D. Yilmaz, K. B. Goh, X. Zheng, and C. Linder. Swelling-induced interface crease instabilities at hydrogel bilayers. J. Elasticity. 145:31–47, 2021.
Fordham, J. W. L., and H. L. Williams. The persulfate-iron (II) initiator system for free radical polymerizations1. J. Am. Chem. Soc. 73:4855–4859, 1951.
Fu, M., Y. Liang, X. Lv, C. Li, Y. Y. Yang, et al. Recent advances in hydrogel-based anti-infective coatings. J. Mater. Sci. Technol. 85:169–183, 2021.
Fuchs, S., K. Shariati, and M. L. Ma. Specialty tough hydrogels and their biomedical applications. Adv. Healthc. Mater. 9(2):e1901396, 2020.
Gold, G. T., D. M. Varma, P. J. Taub, and S. B. Nicoll. Development of crosslinked methylcellulose hydrogels for soft tissue augmentation using an ammonium persulfate-ascorbic acid redox system. Carbohydr. Polym. 134:497–507, 2015.
House, D. A. Kinetics and mechanism of oxidations by peroxydisulfate. Chem. Rev. 62:185–203, 1962.
Hume, P. S., C. N. Bowman, and K. S. Anseth. Functionalized PEG hydrogels through reactive dip-coating for the formation of immunoactive barriers. Biomaterials. 32:6204–6212, 2011.
Jana, S. Endothelialization of cardiovascular devices. Acta Biomater. 99:53–71, 2019.
Jeitner, T. M. Optimized ferrozine-based assay for dissolved iron. Anal. Biochem. 454:36–37, 2014.
Jiao, Y., D. Gyawali, J. M. Stark, P. Akcora, P. Nair, et al. A rheological study of biodegradable injectable PEGMC/HA composite scaffolds. Soft Matter. 8:1499–1507, 2012.
Johnson, L. M., C. A. DeForest, A. Pendurti, K. S. Anseth, and C. N. Bowman. Formation of three-dimensional hydrogel multilayers using enzyme-mediated redox chain initiation. ACS Appl. Mater. Interfaces. 2:1963–1972, 2010.
Johnson, L. M., B. D. Fairbanks, K. S. Anseth, and C. N. Bowman. Enzyme-mediated redox initiation for hydrogel generation and cellular encapsulation. Biomacromolecules. 10:3114–3121, 2009.
Kishan, A. P., and E. M. Cosgriff-Hernandez. Recent advancements in electrospinning design for tissue engineering applications: a review. J. Biomed. Mater. Res. Part A. 105:2892–2905, 2017.
Kloosterboer, J. G., Network formation by chain crosslinking photopolymerization and its applications in electronics. In: Electronic Applications. Springer, Berlin, 1988, pp. 1–61.
Krsko, P., and M. Libera. Biointeractive hydrogels. Materials Today. 8:36–44, 2005.
Lee, S., X. Tong, and F. Yang. Effects of the poly(ethylene glycol) hydrogel crosslinking mechanism on protein release. Biomater. Sci. 4:405–411, 2016.
Lenka, S., and A. K. Dhal. Polymerization of acrylonitrile initiated by K2S2O8–Fe (II) redox system. J. Polym. Sci. Polym. Chem. Ed. 19:2115–2118, 1981.
Li, J., A. D. Celiz, J. Yang, Q. Yang, I. Wamala, et al. Tough adhesives for diverse wet surfaces. Science. 357:378–381, 2017.
Lin, C.-C., and K. S. Anseth. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm. Res. 26:631–643, 2009.
Liu, K., F. Zhang, Y. Wei, Q. Hu, Q. Luo, et al. Dressing blood-contacting materials by a stable hydrogel coating with embedded antimicrobial peptides for robust antibacterial and antithrombus properties. ACS Appl. Mater. Interfaces. 13:38947–38958, 2021.
Liu, Y., W. He, Z. Zhang, and B. P. Lee. Recent developments in tough hydrogels for biomedical applications. Gels. 4:46, 2018.
Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: toward new generations of smart biocompatible materials. J. Polym. Sci. Part A Polym. Chem. 46:3459–3470, 2008.
Ma, S., C. Yan, M. Cai, J. Yang, X. Wang, et al. Continuous surface polymerization via Fe (II)-mediated redox reaction for thick hydrogel coatings on versatile substrates. Adv. Mater. 30:1803371, 2018.
Motiwale, S., M. D. Russell, O. Conroy, J. Carruth, M. Wancura, et al. Anisotropic elastic behavior of a hydrogel-coated electrospun polyurethane: suitability for heart valve leaflets. J. Mech. Behav. Biomed. Mater. 125:104877, 2021.
Parada, G., Y. Yu, W. Riley, S. Lojovich, D. Tshikudi, et al. Ultrathin and robust hydrogel coatings on cardiovascular medical devices to mitigate thromboembolic and infectious complications. Adv. Healthc. Mater. 9(20):e2001116, 2020.
Pashneh-Tala, S., S. MacNeil, and F. Claeyssens. The tissue-engineered vascular graft—past, present, and future. Tissue Eng. Part B Rev. 22:68–100, 2016.
Post, A., A. P. Kishan, P. Diaz-Rodriguez, E. Tuzun, M. Hahn, and E. Cosgriff-Hernandez. Introduction of sacrificial bonds to hydrogels to increase defect tolerance during suturing of multilayer vascular grafts. Acta Biomater. 69:313–322, 2018.
Puperi, D. S., A. Kishan, Z. E. Punske, Y. Wu, E. Cosgriff-Hernandez, et al. Electrospun polyurethane and hydrogel composite scaffolds as biomechanical mimics for aortic valve tissue engineering. ACS Biomater. Sci. Eng. 2:1546–1558, 2016.
Richbourg, N. R., M. Wancura, A. E. Gilchrist, S. Toubbeh, B. A. C. Harley, et al. Precise control of synthetic hydrogel network structure via linear, independent synthesis-swelling relationships. Sci. Adv. 7:eabe3245, 2021.
Roseen, M. A., M. M. Fahrenholtz, J. P. Connell, and K. J. Grande-Allen. Interfacial coating method for amine-rich surfaces using poly (ethylene glycol) diacrylate applied to bioprosthetic valve tissue models. ACS Appl. Bio Mater. 3:1321–1330, 2020.
Roseen, M. A., R. Lee, A. D. Post, M. Wancura, J. P. Connell, et al. Poly(ethylene glycol)-based coatings for bioprosthetic valve tissues: toward restoration of physiological behavior. ACS Appl. Bio Mater. 3:8352–8360, 2020.
Scranton, A. B., C. N. Bowman, J. Klier, and N. A. Peppas. Polymerization reaction dynamics of ethylene glycol methacrylates and dimethacrylates by calorimetry. Polymer. 33:1683–1689, 1992.
Singh, Y., D. Gao, Z. Gu, S. Li, K. A. Rivera, et al. Influence of molecular size on the retention of polymeric nanocarrier diagnostic agents in breast ducts. Pharm. Res. 29:2377–2388, 2012.
Spencer, K. C., J. C. Sy, K. B. Ramadi, A. M. Graybiel, R. Langer, and M. J. Cima. Characterization of mechanically matched hydrogel coatings to improve the biocompatibility of neural implants. Sci. Rep. 7:1952, 2017.
Stevens, K. R., J. S. Miller, B. L. Blakely, C. S. Chen, and S. N. Bhatia. Degradable hydrogels derived from PEG-diacrylamide for hepatic tissue engineering. J. Biomed. Mater. Res. Part A. 103:3331–3338, 2015.
Stevens, M. M. Biomaterials for bone tissue engineering. Mater. Today. 11:18–25, 2008.
Tatterton, M., S.-P. Wilshaw, E. Ingham, and S. Homer-Vanniasinkam. The use of antithrombotic therapies in reducing synthetic small-diameter vascular graft thrombosis. Vasc. Endovasc. Surg. 46:212–222, 2012.
Temenoff, J. S., H. Shin, D. E. Conway, P. S. Engel, and A. G. Mikos. In vitro cytotoxicity of redox radical initiators for cross-linking of oligo (poly (ethylene glycol) fumarate) macromers. Biomacromolecules. 4:1605–1613, 2003.
Tseng, H., D. S. Puperi, E. J. Kim, S. Ayoub, J. V. Shah, et al. Anisotropic poly(ethylene glycol)/polycaprolactone hydrogel-fiber composites for heart valve tissue engineering. Tissue Eng. Part A. 20:2634–2645, 2014.
Wancura, M., M. Talanker, S. Toubbeh, A. Bryan, and E. Cosgriff-Hernandez. Bioactive hydrogel coatings of complex substrates using diffusion-mediated redox initiation. J. Mater. Chem. B. 8:4289–4298, 2020.
Whitely, M., S. Cereceres, P. Dhavalikar, K. Salhadar, T. Wilems, et al. Improved in situ seeding of 3D printed scaffolds using cell-releasing hydrogels. Biomaterials. 185:194–204, 2018.
Wilems, T. S., X. Lu, Y. E. Kurosu, Z. Khan, H. J. Lim, and L. A. S. Callahan. Effects of free radical initiators on polyethylene glycol dimethacrylate hydrogel properties and biocompatibility. J. Biomed. Mater. Res. Part A. 105:3059–3068, 2017.
Yang, J., R. Bai, B. Chen, and Z. Suo. Hydrogel adhesion: a supramolecular synergy of chemistry, topology, and mechanics. Adv. Funct. Mater. 30(2):1901693, 2019.
Yong, Y., M. Y. Qiao, A. Chiu, S. Fuchs, Q. S. Liu, et al. Conformal hydrogel coatings on catheters to reduce biofouling. Langmuir. 35:1927–1934, 2019.
Yu, Y., H. Yuk, G. A. Parada, Y. Wu, X. Liu, et al. Multifunctional “hydrogel skins” on diverse polymers with arbitrary shapes. Adv. Mater. 31(7):e1807101, 2018.
Yuk, H., T. Zhang, S. Lin, G. A. Parada, and X. Zhao. Tough bonding of hydrogels to diverse non-porous surfaces. Nat. Mater. 15:190, 2016.
Zhang, F., C. Hu, L. Yang, K. Liu, Y. Ge, et al. A conformally adapted all-in-one hydrogel coating: towards robust hemocompatibility and bactericidal activity. J. Mater. Chem. B. 9:2697–2708, 2021.
Zhang, X., B. Xu, D. S. Puperi, A. L. Yonezawa, Y. Wu, et al. Integrating valve-inspired design features into poly(ethylene glycol) hydrogel scaffolds for heart valve tissue engineering. Acta Biomater. 14:11–21, 2015.
Zhang, Y., D. An, Y. Pardo, A. Chiu, W. Song, et al. High-water-content and resilient PEG-containing hydrogels with low fibrotic response. Acta Biomater. 53:100–108, 2017.
Zhu, J., and R. E. Marchant. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices. 8:607–626, 2011.
Acknowledgments
The work presented here was supported by the National Institutes of Health R21 EB020978. The Bionate® Thermoplastic Polycarbonate-urethane was provided by DSM Biomedical (Berkeley, CA).
Conflict of interest
There are no conflicts to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Emmanuel Opara oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wancura, M., Nkansah, A., Robinson, A. et al. PEG-Based Hydrogel Coatings: Design Tools for Biomedical Applications. Ann Biomed Eng 52, 1804–1815 (2024). https://doi.org/10.1007/s10439-023-03154-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-023-03154-9