
Abstract
Chitin synthesis inhibitors are successfully used in pest control and an excellent option for integrated pest management programs due to their low non-target effects. However, field-evolved resistance of lepidopteran pests to chitin synthesis inhibitors and the selection of laboratory-resistant strains to these products has been already reported. Therefore, to support efficient resistance management programs it is necessary to expand the knowledge on the resistance mechanisms and potential molecular markers that detect resistant alleles. Teflubenzuron is a chitin synthesis inhibitor used to control the world widely distributed fall armyworm, Spodoptera frugiperda (J.E. Smith, 1797) (Lepidoptera: Noctuidae). Here, we report the selection and inheritance characterization of S. frugiperda strain resistant to teflubenzuron. We also evaluated the cross-resistance to other chitin-synthesis inhibitors and identified single nucleotide polymorphisms (SNPs), which can be used as molecular markers for monitoring the evolution of resistance of S. frugiperda to teflubenzuron. The resistance of the selected strain of S. frugiperda to teflubenzuron was characterized as polygenic, autosomal, and incompletely recessive. The resistance ratio observed was nearly 1,365-fold. Teflubenzuron-resistant strain showed cross-resistance to lufenuron and novaluron, but not to chlorfluazuron. We also detected a set of 72 SNPs that could support monitoring of the resistance frequency to teflubenzuron in field populations. Our data contribute to the understanding of the resistance mechanisms and the inheritance of resistance of S. frugiperda to benzoylureas. We also contribute with candidate markers as tools for monitoring the emergence and spread of teflubenzuron resistance in S. frugiperda.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Key message
-
S. frugiperda resistance to teflubenzuron is polygenic, autosomal, and incompletely recessive.
-
No cross-resistance to chlorfluazuron was observed.
-
Resistance of S. frugiperda to teflubenzuron is not associated to chitin synthase mutation.
-
We detected a set of SNPs that could support monitoring of S. frugiperda resistance to teflubenzuron.
Introduction
Insect resistance evolution to insecticides and Bacillus thuringiensis (Bt)-genetically modified crops is of great concern to biologists, farmers, industry, and government agencies. The strong selection pressure impinged both by numerous insecticides sprays and the wide adoption of Bt-crops increased resistance frequency in many agroecosystems, especially in the successive crop systems used in the central Cerrado savanna region in Brazil (Carvalho et al. 2013; Farias et al. 2014; Nascimento et al. 2016; Okuma et al. 2018; Bolzan et al. 2019; Lira et al. 2020).
The fall armyworm (FAW) Spodoptera frugiperda (J.E. Smith, 1797) (Lepidoptera: Noctuidae) is a polyphagous species native to American tropical regions which gained worldwide distribution after invading Africa a couple of years ago (Goergen et al. 2016). FAW is a serious pest of several economically important crops, such as maize (Cruz 1995), cotton (Santos 2007), and soybean (Bueno et al. 2011). Currently, Bt crops and insecticides are the main tactics in use for FAW management in the world (Assefa 2018).
Insecticides from the benzoylurea group, which were introduced in the early 1970s, act as chitin synthesis inhibitors (van Daalen et al. 1972). These insecticides have been successfully applied to control several pest species in the field. They have high acaricidal and insecticidal activity, exhibiting activity against immatures lepidopterans, coleopterans, hemipterans, and dipterans (Yu 2015). Moreover, the low non-target effects of benzoylureas allow their use in association with other control strategies within well-designed integrated pest management programs (Beeman 1982; Oberlander and Silhacek 1998; Post and Vincent 1973).
Initial studies on the evolution of resistance to chitin synthesis inhibitors under laboratory conditions failed in selecting resistant populations even after 20 generations of directed-selection pressure (Perng et al. 1988). However, their broad use increased the frequency of resistance, leading to evolution of field-evolved resistance of Plutella xylostella (Lepiodptera: Plutellidae) in China and Spodoptera litura (Lepidoptera: Noctuidae) in Pakistan (Ahmad et al. 2008; Lin et al. 1989), and the selection of resistant strains of Spodoptera frugiperda and Cydia pomonella (Lepidoptera: Tortricidae) under laboratory conditions (Sauphanor and Bouvier 1995; Nascimento et al. 2016).
The evolution of resistance in field conditions indicates that the selection pressure and the resistance frequency are high enough to allow the selection of resistant phenotypes, resulting in the complete failure of the management strategies taken in place. The implementation of suitable management plans is required in order to maintain benzoylureas available as a technology for pest control in areas where the resistance frequency is still manageable. The development of reliable and successful resistance management requires the adoption of pro-active strategies and the understanding of the resistance mechanism and its heritability in ways that could contribute to resistance monitoring programs.
Benzoylureas act as chitin synthesis inhibitors by interfering with the synthesis or deposition of chitin in the exoskeleton and other chitinous structures of insects (Merzendorfer and Zimoch 2003; Merzendorfer 2006). The exactly mode of action of benzoylureas has been debated as they were thought to indirectly affect chitin biosynthesis upon binding to sulfonylurea receptors, resulting in vesicle trafficking alterations; however, the role of the ABC transporter sulfonylurea receptor in chitin synthesis was arguable (Abo-Elghar et al. 2004; Meyer et al. 2013). The use of bulk segregants mapping analysis to investigate the resistance mechanism of Tetranychus urticae (Acari: Tetranychidae) to the mite chitin synthesis inhibitor etoxazole led to the characterization of resistance of field populations as monogenic and recessive (Van Leeuwen et al. 2012). The authors of that study also identified a single nonsynonymous mutation (I1017F) in chitin synthase 1 as the resistance mechanism, demonstrating the direct effect of this acaricide on chitin synthase (Van Leeuwen et al. 2012). Their proposition that is benzoylurea with insecticide activity could also target chitin synthase due to the similarities with etoxazole and led to the identification of a mutation at the same position for the isoleucine residue in the chitin synthase 1 in different insect species resistant to chitin synthesis inhibitors (Douris et al. 2016; Fotakis et al. 2020).
Currently, benzoylureas as chlorfluazuron, diflubenzuron, lufenuron, flufenoxuron, novaluron, triflumuron and teflubenzuron are used to control insects in soybean, cotton, and maize crops in Brazil (MAPA 2020). The high selection pressure caused by this group of insecticides has decreased the susceptibility of S. frugiperda to benzoylureas (Schmidt 2002). For instance, FAW populations in central Brazil evolved resistance to lufenuron, carrying a high resistance ratio, and an autosomal and polygenic inheritance of resistance (Nascimento et al. 2016).
Teflubenzuron, 1-(3,5-Dichloro-2,4-difluorophenyl)-3-(2,6-difluorobenzoyl) urea, has been used to control lepidopterans, coleopterans, and dipterans larvae (Yu 2015), since the ovicidal and larvicidal activities of this product were first demonstrated (Ascher and Nemny 1984). Despite the efficacy of this insecticide in controlling insect pests, there are reports of insect resistance to teflubenzuron as early as in the 1980′s, such as for P. xylostella (Iqbal and Wright 1997; Lin et al., 1989; Perng et al. 1988) and Spodoptera littoralis (Lepidoptera: Noctuidae) (Ishaa and Klein 1990). In Brazil, the increased number of control failures with pyrethroids, organophosphates, and benzoylureas (mainly lufenuron) early in 2000s stimulated the use of teflubenzuron to control S. frugiperda in cotton, maize, and soybean crops.
In this study, we characterized the genetic basis of resistance of S. frugiperda to teflubenzuron. By selecting a resistant strain, we (1) characterized the inheritance of resistance, (2) evaluated the cross-resistance to other chitin-synthesis inhibitors, and (3) used a genome scanning approach to identify genomic regions and single nucleotide polymorphisms (SNPs) associated to teflubenzuron resistance for further using them as molecular markers to monitor resistance evolution in field conditions.
Material and methods
Insects
The susceptible S. frugiperda strain (Sf-ss) has been maintained on an artificial diet based on bean, wheat germ and casein (Kasten Junior et al. 1978) in the Arthropod Resistance Laboratory (University of São Paulo, campus ESALQ, Brazil) without insecticide selection for 25 years. The resistant strain (Tef-rr) was selected from field-collected larvae from maize fields from the state of Mato Grosso, Brazil (13°25′35.9″S; 58°38′17.84″W), during the 2014–2015 crop season.
Selection of teflubenzuron-resistant Spodoptera frugiperda strain
Selection for teflubenzuron resistance was carried out using the F2 screen method proposed by Andow and Alstad (1998), since this method increases the likelihood of obtaining a resistant genotype. About 1,000 field-collected larvae were reared under controlled laboratory conditions (25 ± 2 °C; 60 ± 10% RH; 14 h photophase) up to pupation. From the field-collected larvae, we isolated 33 individual couples (families). The progeny obtained for each family was reared under the same controlled conditions described before until adult emergence. Adults originating from these families, which correspond to a F1 generation, were allowed to sib-mate. All eggs laid from each family were collected. Then, 120 larvae from each sib-mated line (i.e., from the F2 generation) were used in insecticide bioassays. We used a common diet-overlay bioassay to select for teflubenzuron resistance at the diagnostic concentration of 10 µg mL−1 of teflubenzuron (Nomolt® 150, teflubenzuron 150 g/L, BASF S.A., São Paulo, Brazil), based on the concentration–response of the susceptible strain (Sf-ss) to this insecticide. The artificial diet (Kasten Junior et al. 1978) was poured into 24-well acrylic plates (Costar®, Corning®), and 30 µL/well of the diagnostic concentration of teflubenzuron in a water solution (v:v) containing 0.1% Triton X-100 was applied on the diet surface. The control diet was treated only with distilled water and 0.1% Triton X-100. Plates were kept under a laminar flow hood for drying the diet surface, and each well was inoculated with one S. frugiperda third instar larva. We allowed larvae to feed for five days under controlled conditions (25 ± 2 °C; 60 ± 10% RH; photophase of 14 h). After five days, we collected and transferred the surviving S. frugiperda larvae to plastic cups (100 mL) containing 50 mL of artificial diet for their rearing until pupation. All surviving individuals from the insecticide treatment were combined and used for successive selection rounds with increasing concentration of teflubenzuron from 10 to 560 µg mL−1 of teflubenzuron for seven generations until the establishment of the teflubenzuron-resistant strain (Tef-rr).
Characterization of Spodoptera frugiperda resistance to teflubenzuron
The susceptible (Sf-ss) and resistant (Tef-rr) strains of S. frugiperda to teflubenzuron were subjected to concentration–response assays with five to 12 logarithmically spaced concentrations between 0.1 and 3,200 μg mL−1 of teflubenzuron. Larval bioassays were conducted using the diet overlay assays earlier described. The lethal concentration 50 (LC50) was estimated with Probit analysis (Finney 1971, 1949) using the POLO software (Robertson et al. 2007). The resistance ratio of Tef-rr was calculated by dividing the LC50 of the Tef-rr by that of the Sf-ss strain.
Estimation of dominance levels
Newly emerged adults from the susceptible (Sf-ss) and resistant (Tef-rr) strains were reciprocally crossed: Tef-rr males × Sf-ss females (RC-1) and Tef-rr females × Sf-ss males (RC-2). Adults (10 couples/cage) were kept in PVC cages (20 cm high × 15 cm diameter) lined with paper to serve as substrate for egg laying. Adults were fed with a 10% honey solution that was replaced every other day. The progenies obtained from each reciprocal cross (F1) were reared on artificial diet until the third instar. Afterwards, third-instars from the reciprocal crosses were exposed to teflubenzuron using the diet-overlay assay explained before.
We estimated the dominance level of resistance from (Bourguet et al. 2000),
where MSS, MRR, and MRS are the mortalities of the Sf-ss, Tef-rr, and heterozygous strains, respectively, exposed to different concentrations of teflubenzuron. Values of D close to zero (D = 0) represent completely recessive inheritance, and values close to 1 (D = 1) represent completely dominant inheritance.
We also estimated dominance level by applying equation [2] (Stone 1968), where D is the degree of dominance and XF, XR, XS are the LC50 values, respectively, for the heterozygote (offspring from reciprocal cross RC1 or RC2), Tef-rr and Sf-ss.
Genetic inheritance associated with teflubenzuron resistance in Spodoptera frugiperda
We used the method proposed by Roush and Daly (1990) and Tsukamoto (1983) to test the hypothesis that a single gene is responsible for teflubenzuron resistance of S. frugiperda. We backcrossed the offspring resulting from the twenty-mating pairs Tef-rr♂ × Sf-ss♀ (heterozygous) with individuals from the resistant strain Tef-rr. We performed diet-overlay bioassays, using eight concentrations of teflubenzuron as earlier described.
The possibility of monogenic inheritance was calculated by using the Chi-square test (Eq. 3) (Sokal and Rohlf 1969), where Ni is the mortality observed in backcrossed larvae at each concentration and ni is the number of individuals tested; q is the expected survival, and p is the expected mortality calculated from the Mendelian model (Eq. 4) (Georghiou, 1969), where \(a\) is the mortality obtained for the parental strain Tef-rr, and \(b\) is the mortality of the heterozygote derived from the reciprocal crosses (Tef-rr♂ x Sf-ss♀). The hypothesis of monogenic inheritance is rejected when the calculated Chi-square is equal or higher than the tabulated Chi-square value, with 1 degree of freedom.
Cross-resistance to other benzoylurea insecticides
Cross-resistance assays of the Tef-rr strain with three other benzoylureas were carried out using the diet-overlay bioassay earlier described utilizing commercial formulations of lufenuron (Match®, 50 g/L, Syngenta, Basel, Switzerland), novaluron (Rimon Supra®, 100 g/L, Syngenta), and chlorfluazuron (Atabron, 50 g/L, ISK Biosciences). For each insecticide, we performed concentration–response bioassays for Tef-rr and Sf-ss as already described. Larval mortality was assessed five days after treatment, and larval mortality was characterized by larval unresponsiveness to stimulation with a fine brush or the occurrence of body malformations. LC50 values were estimated using the POLO software (Robertson et al. 2007).
Genetic crossings and sample selection for gDNA pool sequencing
The pool sequencing was designed to highlight potential markers associated with resistance. We established backcrosses with the resistant strain (Tef-rr) and the offspring from the reciprocal crosses described above (RC-1 and RC-2), e.g., BC1 = (RC-1♂ x Tef-rr♀), BC2 = (RC-1♀ x Tef-rr♂), BC3 = (RC-2♂ x Tef-rr♀), BC4 = (RC-2♀ x Tef-rr♂). Each backcross was split in two groups of insects: (1) individuals randomly collected (BC-random) and (2) individuals that survived the selection pressure by exposure to the diagnostic concentration of 10 µg teflubenzuron/mL (BC-selected) (Fig. 1).

Experimental design, pool sequences, and snp-calling pileline
DNA extraction and sequencing
DNA was extracted from nine larvae from each parental line, Tef-rr (resistant), and Sf-ss (susceptible), and from both groups of each backcross BC1, BC2, BC3, and BC4. Larval genomic DNA was obtained with the modified CTAB method (Doyle 1991). Briefly, 50 mg of tissue from each individual larva was macerated in 650 μL of extraction buffer containing 2% cetyltrimethyl ammonium bromide (CTAB), 1.4 M NaCl, 100 mM tris(hydroxymethyl)aminomethane (Tris–HCl) at pH 8.0, 20 mM ethylenediaminetetraacetic acid (EDTA) at pH 8.0, 1% polyvinylpyrrolidone, 0.2% β-mercaptoethanol, and 20 µL of proteinase K (0.1 μg·mL−1). Samples were incubated at 55 °C for 1 h, added with 650 μL of chloroform: isoamyl alcohol (24:1), and mixed until emulsion. Samples were centrifuged (14,000 g × 5 min × 4 °C), and then the supernatant was collected and transferred to new tubes, where 200 μL of the same extraction buffer (minus the β-mercaptoethanol and proteinase K) was added followed by the addition of the same volume of chloroform: isoamyl alcohol (24:1). The emulsion was thoroughly vortexed, centrifuged (14,000 g × 5 min × 4 °C), and the supernatant collected; we repeated this process 3 times. Samples were combined with 650 μL of cold isopropanol and incubated at −20 °C overnight before centrifugation (14,000 g × 5 min × 4 °C). The pelleted DNA was washed twice with 1 mL of 70% ethanol. The pellet was dried at room temperature, resuspended in 40 µL TE and Rnase A (10 μg mL−1), and stored at −20 °C until further analyses. Genomic DNA was evaluated quantitatively with a Qubit fluorometer (Thermo Fisher Scientific, USA) and checked for degradation with agarose gel electrophoresis. Finally, 7 ng of DNA from each one of nine larvae were combined into a single tube for each treatment.
Briefly, we sheared total pooled DNA into ~ 300–400 bp fragments in an ultrasonicator and used it to build sequencing libraries with the NEBNext Ultra DNA library prep kit (New England Biolabs), according to the manufacturer’s instructions. The whole genome (WGS) for each pool was sequenced in a Miseq platform (Illumina, Inc., San Diego, CA, USA) at the Molecular and Cellular Imaging Center at the Ohio State University.
Sequencing data processing
The quality of raw paired-end reads was assessed using FastQC (Andrews et al. 2015), and reads were filtered using BBmap (http://jgi.doe.gov/data-and-tools/bbtools) by excluding nucleotides with a Phred quality score < 30 from subsequent analyses. Afterwards, the filtered reads were mapped against the S. frugiperda pseudo-genome assembly available at BIPAA—Bioinformatics Platform for Agroecosystem Arthropods (https://bipaa.genouest.org/sp/spodoptera_frugiperda_pub/), using the BWA-MEM (Li and Durbin 2010). Alignment files were converted to SAM/BAM files using SAMtools (Li 2011). Alignment in BAM format from the BC1-random, BC2-random, BC3-random, and BC4-random were combined on a single BAM file (BC-random), whereas BC1-selected, BC2-selected, BC3-selected, and BC4-selected were combined on a single BAM file (BC-treated). Read alignments with PCR duplicates were removed using the MarkDuplicates from Picard software (https://broadinstitute.github.io/picard/), and SNP calling was performed using freebayes (Garrison and Marth 2012). SNPs called were subject to quality filters (quality score > 20 and depth > 10) using the programs Vcftools (Danecek et al. 2011) and Vcffilter (Müller et al. 2017).
Analyses
The vcfR package was used to visualize and manipulate the vcf format. The global FST was calculated for all SNPs using the R package PoolFstat. Tajima’s π and D were calculated for each pooled DNA sample in a 5 kb sliding window with a 5 kb step size for each comparison group using Popoolation v.1.2.2 (Kofler et al. 2011).
Candidate SNPs associated with the resistance of S. frugiperda to teflubenzuron were identified using a population genomics-based approach, which uses the genetic differentiation between the pools to identify genomic regions potentially targeted by selection (Pool-GWAS).
For the population genomics-based approach, SNP count data were analyzed using two different implementations of the bayesian hierarchical models available in the Baypass version 2.2 (Gautier 2015). First, we applied the core model (Coop et al. 2010; Nicholson et al. 2002) to identify loci with significant allele frequency differences. This method is equivalent to the methods that search for loci with higher intra-locus FST. However, in this model a variance–covariance matrix of population allele frequencies (Ω matrix) that works as a kinship matrix is used to control for population structure. Controlling for population structure reduces the likelihood of spurious association between the marker and the phenotype. This method is covariate-free and was expanded to include the calibration of the XtX statistics as proposed by Günther and Coop (2013). Second, we employed the STD model representing an extension of core model, which allows the evaluation of the association of SNP allele frequency with covariates (Gautier 2015). For the covariate model, we conducted two independent analysis: (1) using the LC50 as covariate, and (2) using the mortality obtained with the diagnostic concentration for each bulk (susceptible, resistant, and the BC-random and BC-treated) as a covariate. Because we performed the analysis with the two backcrosses-derived and the parental pools, both genome scans (the FST-like method and the covariate association method) were insensitive to the identification of false positive associations between the markers and the phenotype. These methods are prone to the identification of the strongest signal that might highlight higher differences mostly associated with demography and drift (because the resistant and the susceptible populations share a common ancestor many generations ago), not with selection. The identification of true positives can be done with the identification of SNPs in linkage disequilibrium with the causal gene in the pool that was subjected to selection (BC-treated), and in loci that present allele frequency differences between the BC before and after selection.
Functional annotation and identification of putative markers associated with the resistant phenotype
Annotation of loci associated with SNPs was proposed using the gff file available for the S. frugiperda genome (https://bipaa.genouest.org/sp/spodoptera_frugiperda_pub/) using SnpEff (Cingolani et al. 2012). Genes with no functional annotation in the available genome were annotated after heuristic search using the BLASTx algorithm against the non-redundant protein database available at the NCBI.
Results
Characterization of teflubenzuron resistance in Spodoptera frugiperda
Eleven out of the 33 lines of the F2 generation subjected to selection yielded survivors and were considered to carry resistance traits. The high resistance traits selected in the teflubenzuron-resistant strains were observed by comparing the concentration response curves of the Tef-rr against the Sf-ss and their backcrosses (Fig. 2), and the obtained values of LC50. The LC50 for the resistant Tef-rr (641.47 μg mL−1; IC = 213.05–2,748.81 μg mL−1) was nearly 1,365-fold the LC50 for the susceptible Sf-ss strain (0.47 μg mL−1; IC = 0.35–0.63 μg mL−1). Both Sf-ss (P = 0.02) and Tef-rr (P = 0.03) showed no evidence of distortion at χ2 > 0.01, indicating a good fit to the probit inheritance model of resistance (Table 1).

Log concentration–probit of susceptible (Sf-ss) and resistant (Tef-rr) S. frugiperda strains and progenies of reciprocal crosses between susceptible and resistant strains
Bioassays with progenies of the two reciprocal crosses showed no significant differences, since there was an overlap of 95% CI of the LC50 values (Table 1). Therefore, the hypothesis of parallelism was not rejected (P = 0.247, df = 1). The overlap of the confidence intervals indicated that inheritance of teflubenzuron resistance of S. frugiperda is autosomal, and not related to maternal effects or sex linked.
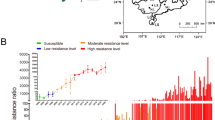
The dominance values for the reciprocal crosses of the offspring estimated following Stone (1968) were 0.32 (Tef-rr♂ × Sf-ss♀) and 0.29 (Tef-rr♀ × Sf-ss♂). The dominance level estimated using the Bourguet-Genissel-Raymond method showed decreased dominance with increased teflubenzuron concentrations (Fig. 3). In both cases, teflubenzuron resistance of S. frugiperda was shown to have an incompletely recessive inheritance.

Level of dominance of S. frugiperda resistance as a function of teflubenzuron concentration
The direct hypothesis test for monogenic inheritance of the teflubenzuron resistance of S. frugiperda based on larval mortality of the F1 × Tef-rr backcross was significant (P < 0.01) for concentrations between 1 and 10 μg mL−1. This result allows the rejection of the hypothesis that teflubenzuron resistance in the selected strain is monogenic (Table 2).
Cross-resistance
Both susceptible and teflubenzuron-resistant strains were tested against chlorfluazuron, lufenuron, and novaluron (Table 3). Teflubenzuron-resistant strain showed some cross-resistance to lufenuron (121.7-fold) and novaluron (75.8-fold), but no cross-resistance of the Tef-rr strain to chlorfluazuron (fourfold) was detected (Table 3).
Genome-scan of Tef-rr and Sf-ss strains of Spodoptera frugiperda
The sequencing of the pooled WGS libraries generated 220 million high-quality paired-end reads after adapter removal and quality trimming. The read mean length was 229 bp and the maximum length was 300 bp, resulting in paired-end fragments with an average of 429 bp in length, with an estimated coverage of 108-fold (Table S1). After mapping and filtering the reads against the S. frugiperda reference genome, 890,209 SNPs were called.
The global genetic differentiation among the resistant, the susceptible and the two backcrosses samples were moderate and significant (FST = 0.10169 with 95% CI of 0.10138–0.10205). The pairwise measurements between BC-random and BC-treated showed that both backcross pools were virtually identical (FST = − 0.01454 95% CI of − 0.0146–0.01442). However, the pairwise FST between Sf-ss and Tef-rr was high (FST = 0.37581 with 95% CI of 0.37489–0.37684). Genetic differentiation was also high between Sf-ss and BC-random (FST = 0.20461 with 95% CI of 0.20394–0.20532), and Sf-ss and BC-treated (FST = 0.21186 with 95% CI of 0.21116–0.21253) (Figure S2).
The variation within the sliding window estimates for π resulted in nucleotide diversities of 0.0192, 0.0285, 0.0336, and 0.0337, respectively, for the Sf-ss and Tef-rr strains and their backcrosses. We identified genomic regions with reduced diversity when comparing the Sf-ss and Tef-rr strains. Large genomic regions with reduced diversity were observed when comparing the resistant and the susceptible strains (Tajima’s π < 0.002), including 600 scaffolds from S. frugiperda genome. Regions of low nucleotide diversity can be observed in Table S3.
The total number of SNPs called was narrowed to 9,161 after the application of XtX statistics (XtX P-value < 0.001) with evidence of divergent selection. We also identified 4,120 SNPs with eBF > 3 db, supporting a moderate evidence of association. Four SNPs exceeded the threshold for strong evidence of association with LC50 values according to the Jeffreys’ rule, whereas 38 SNPs supported moderate evidence of association with mortality using the diagnostic concentration as variable. Finally, 537 SNPs overlapped with both the neutrality model and the association with environmental variables model (Figure S4), distributed in 232 scaffolds.
Scaffold annotation indicated that most of the variants under selection were located in intergenic (47%) and regulatory regions (downstream regions—18%; upstream regions—18%), with 2% presenting missense effect (Figure S5). GO terms distribution demonstrated an impressive number of variants on processes related to primary metabolism, metabolism of organic compounds, and components of membrane (Figure S4). We identified 19 SNPs with non-synonymous effects (Table 4), distributed in 19 scaffolds. Most of the variants are in regulatory regions (Table 5).
Discussion
In the present study, we selected a strain highly resistant (≈ 1,365-fold) to teflubenzuron from a field-collected population of S. frugiperda in the state of Mato Grosso, Brazil. Teflubenzuron resistance was found to be polygenic, incompletely recessive with an autosomal mode of inheritance. Spodoptera frugiperda resistant to teflubenzuron showed evidence of cross-resistance to lufenuron and novaluron, but not to chlorfluazuron.
The pattern of genetic inheritance of S. frugiperda resistance to teflubenzuron is similar and common to lepidopteran species resistant to several insecticides and Bt toxins, e.g., Dipel resistance and Cry1Ab resistance in Ostrinia nubilalis (Lepidoptera: Crambidae) (Huang et al. 1999), and in S. frugiperda resistance to lufenuron (Nascimento et al. 2016), spinosad (Okuma et al. 2018), and spinetoram (Lira et al. 2020).
The inheritance mechanism of teflubenzuron resistance was influenced by insecticide concentration. At lower concentrations, resistance inheritance assumes incompletely dominant features, but at higher concentrations it becomes incompletely recessive. The higher concentration is close to the recommended concentration currently used in field applications for S. frugiperda control. In resistance management, the level of dominance is a variable feature, resulting not only from the genetic background, but also from the interaction between phenotypes and environmental conditions (Bourguet et al. 2000). The level of dominance is one of the most important features for successful IRM (Lenormand and Raymond 1998), since the frequency of resistant insects can be related to the level of dominance. But if resistance inheritance is recessive, the evolution of resistance is delayed because the resistant phenotype is present only in homozygotes, and the alleles that confer resistance are rare (Ffrench-Constant 2013). However, the use of lower concentrations than the level recommended for field application helps to maintain heterozygous individuals in the system and increases the frequency of the resistant alleles within the population. Thus, the continued exposure to the selection pressure (teflubenzuron applications) favors the rapid increase of individual resistance, leading to a concomitant increase in the likelihood of heterozygous mating, and an ultimate production of resistant homozygotes.
The significant deviation between the observed and expected mortalities for the offspring of the resistant–susceptible backcrosses in three concentrations of teflubenzuron indicates that resistance is anchored on more than one gene. Multiple genes with additive and quantitative effects can also lead to the generation of resistance features, and it has been difficult to identify a specific gene or genetic marker associated with such evolutionary process in these cases. The polygenic nature of resistance of S. frugiperda to teflubenzuron agrees with the resistance characterized for another strain of this insect to the benzoylurea lufenuron (Nascimento et al. 2016).
The cross-resistance of teflubenzuron-resistant insects to lufenuron and novaluron may be related to strong selection of insects with overexpression of the detoxification genes, such as cytochrome P450 (CYP), glutathione S-transferases (GSTs), UDP-glucosyltransferases (UGTs), and esterases (CCEs) (Nascimento et al. 2015). These genes are largely associated with detoxification of xenobiotics in several lepidopteran species. Therefore, selection of these genes within these superfamilies may be responsible for the evolution of resistance to different insecticide compounds within the same IRAC group.
The lack of cross-resistance detected to chlorfluazuron as compared to the high cross-resistance levels observed toward lufenuron and novaluron agrees with data available on cross-resistance among different benzoylurea compounds. Cross-resistance to benzoylureas with other chemical compounds was also reported to C. pomonella (Sauphanor and Bouvier 1995; Sauphanor et al. 1998, 2000; Stará and Kocourek 2007). Chlorfluazuron cross-resistance to other benzoylureas, when detected, is very low (cross-resistance to teflubenzuron = 9.9-fold) (Fahmy et al. 1991). The lack of cross-resistance of other benzoylureas to chlorfluazuron is likely linked to the higher toxicity and delayed excretion of chlorfluazuron (Gazit et al. 1989; Guyer and Neumann 1988), but mostly due to the structural nature of this compound. Insect resistance to benzoylureas has been reported to occur due to the overexpression of cytochrome P450 monooxygenases (P450) (Bogwitz et al. 2005a, b) or site mutations in the chitin synthase gene (Douris et al. 2016; Fotakis et al. 2020; Suzuki et al. 2017). In one particular case, resistance of a natural population of Drosophila melanogaster to the benzoylurea lufenuron due to the overexpression of a P450 (Cyp6g1) has evolved as a result of cross-resistance to chemical compounds this fly commonly encounters in nature (Daborn et al. 2002; Wilson and Cain 1997). But chlorfluazuron resistance of a highly resistant (resistance ratio > 2,000-fold) strain of P. xylostella was not affected when using the synergist piperonyl butoxide (PBO), a P450 inhibitor (QingJun et al. 1997). Although PBO had been previously shown to reduce the resistance ratio of a chlorfluazuron selected strain of P. xylostella (resistance ratio = 50-fold) to 4.3-fold, it was suggested the microsomal enzymes acting on this pesticide might be different from other benzoylurea compounds (Ismail and Wright, 1992). In fact, glutathione-S-transferases were later suggested to play a role in P. xylostella resistance to chlorfluazuron (Sonoda and Tsumuki 2005). However, other evolutionary events, such as gene duplication, could be alternative or additional adaptations routes, resulting in the overexpression and/or neofunctionalization of cytochrome P450s (Zimmer et al. 2018).
Several candidate SNPs showed signals of strong positive selection, supporting the polygenic nature of the resistance of S. frugiperda to teflubenzuron. Although resistance of insects to benzoylureas has been associated with site mutations in chitin synthase (Douris et al. 2016; Fotakis et al. 2020; Suzuki et al. 2017), the resistant population of S. frugiperda analyzed did not display any missense variants in this gene. Missense variants were identified in two other genes. One is up-regulated in a chlorantraniliprole-resistant strain of S. exigua, the cytochrome P450 monooxygenase Cyp340AB1 (Wang et al. 2018). The other is the cuticular protein CPG4855, a gene that participates in the formation of the larval and pupal endocuticle in S. exigua (Jan et al. 2017). Although P450 enzymes have been implicated in the metabolization of several pesticides, including benzoylureas, as earlier discussed, we do not believe the resistance mechanism of S. frugiperda to teflubenzuron would be associated with a point mutation in the Cyp340AB1 gene, a P450 belonging to the CYP4 clan. The Cyp genes mostly commonly involved in the metabolization of xenobiotics, including insecticides, belongs to the family Cyp6 of the CYP3 Clan of P450 enzymes (Feyereisen 2012), as the Cyp6g1 gene previously reported in lufenuron-resistant D. melanogaster (Daborn et al. 2002; Wilson and Cain 1997). On the other hand, the site mutation in the cuticular protein CPG4855 could have a close association with the resistance mechanism observed in S. frugiperda to teflubenzuron, once RNAi experiments targeting cuticular proteins demonstrated association of these genes and insects resistant to insecticides (Fang et al. 2015; Huang et al. 2018).
The resistance of S. frugiperda to teflubenzuron was found to be polygenic, and as such much more likely to involve mechanisms of regulation of gene expression, as reported to other benzoylurea-resistant insects. In fact, several polymorphic SNPs were detected upstream (within 5 Kb of the start codon) and downstream (within 5 Kb of the stop codon) of gene regions, including intronic regions of the genome. These SNP variations might be responsible for modifications leading to regulation of gene expression and protein function. In humans, GWAS analysis demonstrated that more than 90% disease-associated SNPs were located up and downstream (promoter and enhancer regions) gene regions or even in non-coding regions of the genome (Hindorff et al., 2009; Hrdlickova et al. 2014; Ricaño-Ponce and Wijmenga 2013).
Annotation allowed the identification of several upstream, downstream, and intron variants in genes associated with several biological processes besides biological regulation and regulation of cellular processes. These annotation results point to multiple gene interactions and regulation playing decisive roles in insecticide resistance.
Thus, the high number of SNPs in genomic regions involved in mechanisms that interfere with the regulation of gene expression of a number of genes involved in processes of metabolization and excretion of xenobiotics are thought to serve as candidate molecular markers for monitoring the polygenic teflubenzuron-resistant phenotypes in the field. These candidates involve the multidrug resistance-associated protein 4 or ATP-binding cassette subfamily C member 4 (ABCC4), a transmembrane protein involved in the efflux of organic compounds from cells (Hardy et al. 2019), including xenobiotics in insects (Labbé et al. 2011). ABCCs can be functionally diverse, but they are capable to translocate a range of organic xenobiotics including insecticides, and have been involved in insecticide resistance mechanisms in insects and drug resistance in humans (Dermauw and Van Leeuwen 2014). Moreover, ABCC transporters act in synergy with glutathione-S-transferases (GSTs) and UDP-glucosyltransferases (UGTs) which are enzymes acting in phase II of detoxification. In humans, the synergy of ABCCs, GSTs, and UGTs confer resistance to drugs and carcinogens (Dermauw and Van Leeuwen 2014).
G-protein-coupled receptors (GPCRs) play a central role in cell signaling as receptors of neuromodulators, neurotransmitters, hormones and neuropeptides. GWAS analysis for the identification of SNPs in the teflubenzuron-resistant strain of S. frugiperda identified variants in genomic regions that can interfere with gene expression of a GPCR belonging to the methuselah (mth) subfamily of the secretin family. Methuselah is a secretin receptor reported to be insect-specific. Mth play a role in several biological processes in insects, such as stress response, regulation of fluid and ion secretion, and longevity, among others (Araújo et al. 2013). An exploration of GPCRs in Tribolium castaneum indicated mth is also important for larval molt and metamorphosis (Bai et al. 2011). Moreover, mth gene mutation led D. melanogaster to become tolerant to dichlorvos (Pandey et al., 2015), but insect response to insecticide exposure was also altered with changes in the levels of mth expression (Cao et al. 2019; Li et al. 2015; Lucas et al. 2019; Ma et al. 2020). Similarly to the early discussed synergism of ABCCs and phase II conjugation enzymes in insect response to insecticide exposure, mth is co-expressed with the phase I detoxification P450 enzymes, contributing to insect resistance to insecticides (Cao et al. 2019; Li et al. 2015).
The selection of a strain of S. frugiperda highly resistant to teflubenzuron, the identification of cross-resistance to lufenuron and novaluron, and the use of genome wide association analysis led us to identify several candidate molecular markers for monitoring resistance evolution to benzoylureas. Several SNPs identified in association with the teflubenzuron-resistant strain indicates that the polygenic mechanism of resistance selected in the resistant strain of S. frugiperda is based on a dense and intricate network of co-expressing genes, of which many are important regulatory genes. The variation in the levels of cross-resistance observed for the different benzoylureas assayed against the teflubenzuron-resistant strain of S. frugiperda provides us with additional tools to investigate and understand the particular differences in the target sites of the many structural compounds sharing chitin synthesis inhibition as a mode of action. The well-defined mutation in the chitin synthase gene related to target site mutation resistance to benzoylureas in arthropods (Van Leeuwen et al. 2012) and the several molecular marker candidates we added as sources of metabolic resistance to benzoylureas can be strategically used for monitoring the resistance of S. frugiperda and for future implementation of successful insecticide resistance management strategies.
Data Availability Statement
Illumina data will be publicly available at the NCBI BioProject PRJNA678657.
References
Abo-Elghar GE, Fujiyoshi P, Matsumura F (2004) Significance of the sulfonylurea receptor (SUR) as the target of diflubenzuron in chitin synthesis inhibition in Drosophila melanogaster and Blattella germanica. Insect Biochem Mol Biol 34:743–752. https://doi.org/10.1016/j.ibmb.2004.03.009
Ahmad M, Sayyed AH, Saleem MA, Ahmad M (2008) Evidence for field evolved resistance to newer insecticides in Spodoptera litura (Lepidoptera: Noctuidae) from Pakistan. Crop Prot 27:1367–1372. https://doi.org/10.1016/j.cropro.2008.05.003
Andow DA, Alstad DN (1998) F2 screen for rare resistance alleles. J Econ Entomol 91:572. https://doi.org/10.1093/jee/91.3.572
Andrews S, Krueger F, Seconds-Pichon A, et al (2015) FastQC. A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 26 June 2018
Araújo AR, Reis M, Rocha H, et al (2013) The Drosophila melanogaster methuselah Gene: a novel gene with ancient functions. PLoS One 8. https://doi.org/https://doi.org/10.1371/journal.pone.0063747
Ascher KRS, Nemny NE (1984) The effect of CME 134 on Spodoptera littoralis eggs and larvae. Phytoparasitica 12:13–27. https://doi.org/10.1007/BF02980794
Assefa F (2018) Status of fall armyworm (Spodoptera frugiperda), biology and control measures on maize crop in Ethiopia: a review. Int J Entomol Res 6:75–85. https://doi.org/https://doi.org/10.33687/entomol.006.02.2498
Bai H, Zhu F, Shah K, Palli SR (2011) Large-scale RNAi screen of G protein-coupled receptors involved in larval growth, molting and metamorphosis in the red flour beetle. BMC Genomics 12. https://doi.org/https://doi.org/10.1186/1471-2164-12-388
Beeman RW (1982) Recent advances in mode of action of insecticides. Annu Rev Entomol 27:253–281. https://doi.org/10.1146/annurev.en.27.010182.001345
Bogwitz MR, Chung H, Magoc L et al (2005a) Cyp12a4 confers lufenuron resistance in a natural population of Drosophila melanogaster. Proc Natl Acad Sci USA 102:12807–12812. https://doi.org/10.1073/pnas.0503709102
Bogwitz MR, Chung H, Magoc L et al (2005b) Cyp12a4 confers lufenuron resistance in a natural population of Drosophila melanogaster. Proc Natl Acad Sci USA 102:12807–12812. https://doi.org/10.1073/pnas.0503709102
Bolzan A, Padovez FEO, Nascimento ARB et al (2019) Selection and characterization of the inheritance of resistance of Spodoptera frugiperda (Lepidoptera: Noctuidae) to chlorantraniliprole and cross-resistance to other diamide insecticides. Pest Manag Sci 75:2682–2689. https://doi.org/10.1002/ps.5376
Bourguet D, Genissel A, Raymond M (2000) Insecticide resistance and dominance levels. J Econ Entomol 93:1588–1595. https://doi.org/10.1603/0022-0493-93.6.1588
Bueno RCOF, De Freitas BA, Moscardi F et al (2011) Lepidopteran larva consumption of soybean foliage: Basis for developing multiple-species economic thresholds for pest management decisions. Pest Manag Sci 67:170–174. https://doi.org/10.1002/ps.2047
Cao C, Sun L, Du H et al (2019) Physiological functions of a methuselah-like G protein coupled receptor in Lymantria dispar Linnaeus. Pestic Biochem Physiol 160:1–10. https://doi.org/10.1016/j.pestbp.2019.07.002
Carvalho RA, Omoto C, Field LM et al (2013) Investigating the molecular mechanisms of organophosphate and pyrethroid resistance in the fall armyworm Spodoptera frugiperda. PLoS ONE 8:e62268. https://doi.org/10.1371/journal.pone.0062268
Cingolani P, Platts A, Wang LL et al (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. https://doi.org/10.4161/fly.19695
Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423. https://doi.org/10.1534/genetics.110.114819
Cruz I (1995) Manejo Integrado de pragas de milho com ênfase para o controle biológico. http://ainfo.cnptia.embrapa.br/digital/bitstream/item/43838/1/Manejo-integrado-2.pdf. Accessed 11 November 2020
Daborn PJ, Yen JL, Bogwitz MR et al (2002) A single P450 allele associated with insecticide resistance in Drosophila. Science 297:2253–2256. https://doi.org/10.1126/science.1074170
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Dermauw W, Van Leeuwen T (2014) The ABC gene family in arthropods: comparative genomics and role ininsecticide transport and resistance. Insect Biochem Mol Biol 45:89–110. https://doi.org/10.1016/j.ibmb.2013.11.001
Douris V, Steinbach D, Panteleri R et al (2016) Resistance mutation conserved between insects and mites unravels the benzoylurea insecticide mode of action on chitin biosynthesis. Proc Natl Acad Sci USA 113:14692–14697. https://doi.org/10.1073/pnas.1618258113
Doyle J (1991) DNA Protocols for Plants. In: Hewitt GM, Johnston AWB, Young JPW (eds) Molecular Techniques in Taxonomy. NATO ASI Series (Series H: Cell Biology), vol 57. Springer, Berlin, pp 283–285. https://doi.org/10.1007/978-3-642-83962-7_18
Fahmy AR, Sinchaisri N, Miyata T (1991) Development of chlorfluazuron resistance and pattern of cross-resistance in the diamondback Moth, Plutella xylostella. J Pestic Sci 16:665–672. https://doi.org/10.1584/jpestics.16.665
Fang F, Wang W, Zhang D, et al (2015) The cuticle proteins: a putative role for deltamethrin resistance in Culex pipiens pallens. Parasitol Res 114. https://doi.org/https://doi.org/10.1007/s00436-015-4683-9
Farias JR, Horikoshi RJ, Santos AC, Omoto C (2014) Geographical and temporal variability in susceptibility to Cry1F toxin from Bacillus thuringiensis in Spodoptera frugiperda (Lepidoptera: Noctuidae) populations in Brazil. J Econ Entomol 107:2182–2189. https://doi.org/10.1603/EC14190
Feyereisen R (2012) Insect CYP Genes and P450 Enzymes. Insect Mol Biol Biochem 236–316. https://doi.org/https://doi.org/10.1016/B978-0-12-384747-8.10008-X
Ffrench-Constant RH (2013) The molecular genetics of insecticide resistance. Genetics 194:807–815. https://doi.org/10.1534/genetics.112.141895
Finney DJ (1949) the Adjustment for a natural response rate in Probit analysis. Ann Appl Biol 36:187–195. https://doi.org/10.1111/j.1744-7348.1949.tb06408.x
Finney DJ (1971) Probit analysis, 3rd edn. Cambridge University Press, London
Fotakis EA, Mastrantonio V, Grigoraki L et al (2020) Identification and detection of a novel point mutation in the chitin synthase gene of Culex pipiens associated with diflubenzuron resistance. PLoS Negl Trop Dis 14:1–10. https://doi.org/10.1371/journal.pntd.0008284
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv Prepr arXiv12073907
Gautier M (2015) Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics 201:1555–1579. https://doi.org/10.1534/genetics.115.181453
Gazit Y, Ishaaya I, Perry AS (1989) Detoxification and synergism of diflubenzuron and chlorfluazuron in the red flour beetle Tribolium castaneum. Pestic Biochem Physiol 34:103–110. https://doi.org/10.1016/0048-3575(89)90147-8
Georghiou GP (1969) Genetics of resistance to insecticides in houseflies and mosquitoes. Exp Parasitol 26:224–255. https://doi.org/10.1016/0014-4894(69)90116-7
Goergen G, Kumar PL, Sankung SB, et al (2016) First report of outbreaks of the fall armyworm Spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidae), a new alien invasive pest in West and Central Africa. PLoS One 11. https://doi.org/https://doi.org/10.1371/journal.pone.0165632
Günther T, Coop G (2013) Robust identification of local adaptation from allele frequencies. Genetics 195:205–220. https://doi.org/10.1534/genetics.113.152462
Guyer W, Neumann R (1988) Activity and fate of chlorfluazuron and diflubenzuron in the larvae of Spodoptera littoralis and Heliothis virescens. Pestic Biochem Physiol 30:166–177. https://doi.org/10.1016/0048-3575(88)90050-8
Hardy D, Bill RM, Jawhari A, Rothnie AJ (2019) Functional expression of multidrug resistance protein 4 MRP4/ABCC4. SLAS Discov 24:1000–1008. https://doi.org/10.1177/2472555219867070
Hindorff LA, Sethupathy P, Junkins HA et al (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106:9362–9367. https://doi.org/10.1073/pnas.0903103106
Hrdlickova B, de Almeida RC, Borek Z, Withoff S (2014) Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim Biophys Acta Mol Basis Dis 1842:1910–1922. https://doi.org/10.1016/j.bbadis.2014.03.011
Huang F, Buschman LL, Higgins RA, McGaughey WH (1999) Inheritance of resistance to Bacillus thuringiensis toxin (dipel ES) in the European corn borer. Science 284(80):965–967. https://doi.org/https://doi.org/10.1126/science.284.5416.965
Huang Y, Guo Q, Sun X, et al (2018) Culex pipiens pallens cuticular protein CPLCG5 participates in pyrethroid resistance by forming a rigid matrix. Parasit Vectors 11. https://doi.org/https://doi.org/10.1186/s13071-017-2567-9
Iqbal M, Wright DJ (1997) Evaluation of resistance, cross-resistance and synergism of abamectin and teflubenzuron in a multi-resistant field population of Plutella xylostella (Lepidoptera: Plutellidae). Bull Entomol Res 87:481–486. https://doi.org/10.1017/s0007485300041341
Ishaa Ya I, Klein M (1990) Response of susceptible laboratory and resistant field strains of Spodoptera littoralis (Lepidoptera: Noctuidae) to teflubenzuron. J Econ Entomol 83:59–62. https://doi.org/10.1093/jee/83.1.59
Ismail F, Wright DJ (1992) Synergism of teflubenzuron and chlorfluazuron in an acylurea-resistant field strain of Plutella xylostella L. (lepidoptera: Yponomeutidae). Pestic Sci 34:221–226. https://doi.org/10.1002/ps.2780340306
Jan S, Liu S, Hafeez M, et al (2017) Isolation and functional identification of three cuticle protein genes during metamorphosis of the beet armyworm, Spodoptera exigua. Sci Rep 7. https://doi.org/https://doi.org/10.1038/s41598-017-16435-w
Kasten P Jr, Precetti AACM, Parra JRP (1978) Dados biológicos comparativos de Spodoptera frugiperda em duas dietas artificiais e substrato natural. Rev Agric 53:68–78. https://doi.org/10.37856/bja.v53i1-2.3948
Kofler R, Orozco-terWengel P, de Maio N et al (2011) Popoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE 6:e15925. https://doi.org/10.1371/journal.pone.0015925
Labbé R, Caveney S, Donly C (2011) Genetic analysis of the xenobiotic resistance-associated ABC gene subfamilies of the Lepidoptera. Insect Mol Biol 20:243–256. https://doi.org/10.1111/j.1365-2583.2010.01064.x
Lenormand T, Raymond M (1998) Resistance management: the stable zone strategy. Proc R Soc B Biol Sci 265:1985–1990. https://doi.org/10.1098/rspb.1998.0529
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. https://doi.org/10.1093/bioinformatics/btp698
Li T, Cao C, Yang T et al (2015) A G-protein-coupled receptor regulation pathway in cytochrome P450-mediated permethrin-resistance in mosquitoes, Culex quinquefasciatus. Sci Rep 5:1–13. https://doi.org/10.1038/srep17772
Lin JG, Hung CF, Sun CN (1989) Teflubenzuron resistance and microsomal monooxygenases in larvae of the diamondback moth. Pestic Biochem Physiol 35:20–25. https://doi.org/10.1016/0048-3575(89)90098-9
Lira EC, Bolzan A, Nascimento ARB et al (2020) Resistance of Spodoptera frugiperda (Lepidoptera: Noctuidae) to spinetoram: inheritance and cross-resistance to spinosad. Pest Manag Sci 76:2674–2680. https://doi.org/10.1002/ps.5812
Lucas ER, Rockett KA, Lynd A, et al (2019) A high throughput multi-locus insecticide resistance marker panel for tracking resistance emergence and spread in Anopheles gambiae. Sci Rep. https://doi.org/https://doi.org/10.1038/s41598-019-49892-6
Ma Z, Zhang Y, You C, et al (2020) The role of G protein‐coupled receptor‐related genes in cytochrome P450‐mediated resistance of the house fly, Musca domestica (Diptera: Muscidae), to imidacloprid. Insect Mol Biol 29. https://doi.org/https://doi.org/10.1111/imb.12615
MAPA (2020) Sistema de Agrotóxicos Fitossanitários. Ministério da Agricultura, Pecuária e Abastecimento. http://agrofit.agricultura.gov.br/agrofit_cons/principal_agrofit_cons. Accessed 11 November 2020
Merzendorfer H (2006) Insect chitin synthases: a review. J Comp Physiol B Biochem Syst Environ Physiol 176:1–15. https://doi.org/10.1007/s00360-005-0005-3
Merzendorfer H, Zimoch L (2003) Chitin metabolism in insects: structure, function and regulation of chitin synthases and chitinases. J Exp Biol 206:4393–4412
Meyer F, Flötenmeyer M, Moussian B (2013) The sulfonylurea receptor Sur is dispensable for chitin synthesis in Drosophila melanogaster embryos. Pest Manag Sci 69:1136–1140. https://doi.org/10.1002/ps.3476
Müller H, Jimenez-Heredia R, Krolo A et al (2017) VCF.Filter: interactive prioritization of disease-linked genetic variants from sequencing data. Nucleic Acids Res 45:W567–W572. https://doi.org/10.1093/nar/gkx425
Nascimento ARB, Farias JR, Bernardi D et al (2016) Genetic basis of Spodoptera frugiperda (Lepidoptera: Noctuidae) resistance to the chitin synthesis inhibitor lufenuron. Pest Manag Sci 72:810–815. https://doi.org/10.1002/ps.4057
Nascimento ARB, Fresia P, Cônsoli FL, Omoto C (2015) Comparative transcriptome analysis of lufenuron-resistant and susceptible strains of Spodoptera frugiperda (Lepidoptera: Noctuidae). BMC Genomics 16. https://doi.org/10.1186/s12864-015-2183-z
Nicholson G, Smith AV, Jónsson F et al (2002) Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Stat Soc Ser B Stat Methodol 64:695–715. https://doi.org/10.1111/1467-9868.00357
Oberlander H, Silhacek DL (1998) Mode of action of insect growth regulators in lepidopteran tissue culture. Pestic Sci 54:300–302. https://doi.org/10.1002/(sici)1096-9063(1998110)54:3%3c300::aid-ps830%3e3.0.co;2-8
Okuma DM, Bernardi D, Horikoshi RJ et al (2018) Inheritance and fitness costs of Spodoptera frugiperda (Lepidoptera: Noctuidae) resistance to spinosad in Brazil. Pest Manag Sci 74:1441–1448. https://doi.org/10.1002/ps.4829
Pandey A, Khatoon R, Saini S et al (2015) Efficacy of methuselah gene mutation toward tolerance of dichlorvos exposure in Drosophila melanogaster. Free Radic Biol Med 83:54–65. https://doi.org/10.1016/j.freeradbiomed.2015.02.025
Perng FS, Yao MC, Hung CF, Sun CN (1988) Teflubenzuron resistance in diamondback moth (Lepidoptera: Plutellidae). J Econ Entomol 81:1277–1282. https://doi.org/10.1093/jee/81.5.1277
Post LC, Vincent WR (1973) A new insecticide inhibits chitin synthesis. Naturwissenschaften 60:431–432. https://doi.org/10.1007/BF00623561
QingJun W, GuoRen Z, JianZhou Z, Xing Z, Xiwu G (1997) Resistance selection of Plutella xylostella (L.) by chlorfluazuron and patterns of cross-resistance. Acta Entomol Sin 41:34–41
Ricaño-Ponce I, Wijmenga C (2013) Mapping of immune-mediated disease genes. Annu Rev Genomics Hum Genet 14:325–353. https://doi.org/10.1146/annurev-genom-091212-153450
Robertson JL, Savin NE, Savin NE, Preisler HK (2007) Bioassays with arthropods, 2nd ed. CRC Press, Boca Raton. https://doi.org/10.1201/9781420004045
Roush RT, Daly JC (1990) The role of population genetics in resistance research and management. Pestic Resist Arthropods 97–152. https://doi.org/https://doi.org/10.1007/978-1-4684-6429-0_5
Santos WJ (2007) Manejo das pragas do algodão com destaque para o cerrado brasileiro. In: Freire EC (ed) Algodão no cerrado do Brasil. ABAPA, Brasilia, p 403–478
Sauphanor B, Bouvier JC (1995) Cross‐resistance between benzoylureas and benzoylhydrazines in the codling moth, Cydia pomonella L. Pestic Sci. https://doi.org/https://doi.org/10.1002/ps.2780450412
Sauphanor B, Bouvier JC, Brosse V (1998) Spectrum of insecticide resistance in Cydia pomonella (Lepidoptera: Tortricidae) in Southeastern France. J Econ Entomol. https://doi.org/10.1093/jee/91.6.1225
Sauphanor B, Brosse V, Bouvier JC, et al (2000) Monitoring resistance to diflubenzuron and deltamethrin in French codling moth populations (Cydia pomonella). Pest Manag Sci. https://doi.org/https://doi.org/10.1002/(SICI)1526-4998(200001)56:1<74::AID-PS96>3.0.CO;2-C
Schmidt FB (2002) Linha básica de suscetibilidade de Spodoptera frugiperda (Lepidoptera: Noctuidae) a lufenuron na cultura do milho. Dissertação, Escola Superior de Agricultura Luiz de Queiroz: Universidade de São Paulo. https://doi.org/10.11606/D.11.2002.tde-04112002-171009
Sokal RR, Rohlf F (1969) Biometry. The principles and practice of statistics in biological research. Freeman and Co., San Francisco
Sonoda S, Tsumuki H (2005) Studies on glutathione S-transferase gene involved in chlorfluazuron resistance of the diamondback moth, Plutella xylostella L. (Lepidoptera: Yponomeutidae). Pestic Biochem Physiol 82:94–101. https://doi.org/10.1016/j.pestbp.2005.01.003
Stará J, Kocourek F (2007) Insecticidal resistance and cross-resistance in populations of Cydia pomonella (Lepidoptera: Tortricidae) in Central Europe. J Econ Entomol. https://doi.org/https://doi.org/10.1603/0022-0493(2007)100[1587:IRACIP]2.0.CO;2
Stone BF (1968) A formula for determining degree of dominance in cases of monofactorial inheritance of resistance to chemicals. Bull World Health Organ 38:325–326. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2554319/. Accessed 12 June 2020
Suzuki Y, Shiotsuki T, Jouraku A et al (2017) Benzoylurea resistance in western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae): the presence of a point mutation in chitin synthase 1. J Pestic Sci 42:93–96. https://doi.org/10.1584/jpestics.D17-023
Tsukamoto M (1983) Methods of genetic analysis of insecticide resistance. Pest Resist to Pestic 71–98. https://doi.org/https://doi.org/10.1007/978-1-4684-4466-7_3
van Daalen JJ, Meltzer J, Mulder R, Wellinga K (1972) A selective insecticide with a novel mode of action. Naturwissenschaften 59:312–313. https://doi.org/10.1007/BF00593360
Van Leeuwen T, Demaeght P, Osborne EJ et al (2012) Population bulk segregant mapping uncovers resistance mutations and the mode of action of a chitin synthesis inhibitor in arthropods. Proc Natl Acad Sci USA 109:4407–4412. https://doi.org/10.1073/pnas.1200068109
Wang X, Chen Y, Gong C et al (2018) Molecular identification of four novel cytochrome P450 genes related to the development of resistance of Spodoptera exigua (Lepidoptera: Noctuidae) to chlorantraniliprole. Pest Manag Sci 74:1938–1952. https://doi.org/10.1002/ps.4898
Wilson TG, Cain JW (1997) Resistance to the insecticides lufenuron and propoxur in natural populations of Drosophila melanogaster (Diptera: Drosophilidae). J Econ Entomol 90:1131–1136. https://doi.org/10.1093/jee/90.5.1131
Yu SJ (2015) The Toxicology and Biochemistry of Insecticides, 2nd ed. CRC Press, Boca Raton. https://doi.org/10.1201/b18164
Zimmer CT, Garrood WT, Singh KS, et al (2018) Neofunctionalization of duplicated p450 genes drives the evolution of insecticide resistance in the brown planthopper. Curr Biol. https://doi.org/10.1016/j.cub.2017.11.060
Acknowledgements
We thank PROMIP (SISBIO License #40380-5) for helping to collect insect samples. Research was carried out using the computational resources of the Center for Mathematical Sciences Applied to Industry (CeMEAI) funded by FAPESP (grant 2013/07375-0).
Funding
We thank the São Paulo Research Foundation (FAPESP) for the fellowship to ARBN (Grants #2016/09159–0 and #2014/26212-7) and the Young Investigator project to KLSB (#2012/16266-7). We also thank the Brazilian National Council for Scientific and Technological Development (CNPq) (#403851/2013-0) and the Brazilian Insecticide Resistance Action Committee (IRAC‐BR) for providing partial financial support for this study. We acknowledge computing resources from the Ohio Supercomputer Center (OSC). The funding bodies had no role in the study design, data collection, analysis and interpretation, or drafting of the manuscript.
Author information
Authors and Affiliations
Contributions
ARBN, FLC, AM, and CO conceived the study. ARBN and JGR collected the data. CO and AM provided reagents and sequencing financing. ARBN, VACP, and KLSB performed the analysis. ARBN and FLC wrote the main manuscript. All authors contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare they have no conflicts of interest.
Ethical approval
This research did not involve humans or vertebrates.
Additional information
Communicated by Emmanouil Roditakis .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
do Nascimento, A.R.B., Pavinato, V.A.C., Rodrigues, J.G. et al. There is more than chitin synthase in insect resistance to benzoylureas: molecular markers associated with teflubenzuron resistance in Spodoptera frugiperda. J Pest Sci 95, 129–144 (2022). https://doi.org/10.1007/s10340-021-01373-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-021-01373-4