
Abstract
In modern ultra-high performance liquid chromatography set-ups, short columns (max. 10 cm) with a narrow ID (2.1 mm) packed with small, (sub-2 µm) fully or superficially porous particles are used. Since the volume and corresponding peak variance arising from these columns are very small, the dispersion contribution from the chromatographic system has a large effect on the overall separation performance. In gradient elution, the on-column focusing of the sample band at the start of the gradient results in the elimination of the pre-column band broadening. However, since gradient elution separations yield very narrow sample peaks at the outlet of the column, any post-column band broadening has a large effect on the obtained separation quality. In this contribution, the main factor of post-column band broadening is investigated, i.e., that from the UV detector, by comparing the peak width measured on capillary directly in front of the UV detector using an LIF detector and the peak widths obtained in the UV detector. These experiments show that there is a clear increase in peak variance with flow rate up to around 0.4–0.6 mL/min (depending on the investigated flow cell). It is found that modern low-dispersion flow cells generate a dispersion contribution around 0.7 µL2 at high flow rates, whereas standard flow cells can have a contribution up to 5.8 µL2. For the investigated nano-flow cell (80 nL), no significant dispersion was observed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The improvements in column and system performance over the past two decades have resulted in significant increase in separation efficiency and an even larger decrease in analysis time [1, 2]. The use of small, down to sub-2 µm, particles and/or superficially porous particles, in combination with the introduction of so-called ultra-high performance liquid chromatography (UHPLC) instrumentation that can operate at pressures up to 1500 bar [3] allows to increase separation performance in a fully kinetically optimized system by a factor of 8 [2, 4, 5]. With the introduction of UHPLC systems, column formats have drastically changed from the standard 4.6 mm ID columns with a length between 15 and 25 cm, to narrow 2.1 or 3 mm ID column with lengths between 2 and 10 cm. For example, a 25 cm-long column (4.6 mm ID) packed with 5 µm fully porous articles (Hmin = 10 µm), or a 5 cm-long column (2.1 mm ID) packed with 1.3 µm superficially porous particles (Hmin = 2 µm [6, 7]), both yield around 25,000 theoretical plates at their optimum velocity. The column void volume V0, assuming a total porosity εT = 0.6 and εT = 0.585 for the fully and superficially porous particle columns, respectively, is, however, a factor 25 lower for the short 2.1 mm column. As a result, the column’s volumetric peak variance \(\sigma _{{V,{\text{col}}}}^{2}\), i.e., the physical parameter describing the band broadening when the analyte band passes through the column, decreases by a factor of 625 as it is calculated according to Eq. (1) (for an isocratic separation):
The total variance of a chromatographic peak is, however, not only determined by its band broadening in the column, but also by the dispersion in the system as a result of the injection system and volume, system tubing, preheaters, connectors, and the detector(s). The overall system contribution is often lumped in a single extra-column (EC) band broadening contribution as \(\sigma _{{V,{\text{EC}}}}^{2}\), giving for the total peak variance \(\sigma _{{V,{\text{tot}}}}^{2}\):
Alternatively, one could also split the extra-column band broadening contribution into a pre-column part (injection system, injection volume, preheaters, tubing) \(\sigma _{{V,{\text{EC}} - {\text{pre}}}}^{2}\) and a post-column part (tubing, post-column coolers, detector) \(\sigma _{{V,{\text{EC}} - {\text{post}}}}^{2}\) as [8,9,10]:
Starting from Eqs. (1)–(2), it is directly clear that a strong reduction in column volume would quickly result in an excessive contribution of the system dispersion to the overall separation quality if no concomitant reduction in extra-column volume is achieved. Modern UHPLC instrumentation, therefore, typically have a much lower \(\sigma _{{V,{\text{EC}}}}^{2}\) contributions (2–15 µL2) than standard HPLC instrumentation (40–100 µL2) [9]. A second point that can be derived from Eqs. (1–2), is that the relative contribution from \(\sigma _{{V,{\text{EC}}}}^{2}\) decreases quadratically with increasing retention factor. In gradient elution chromatography, the column peak variance is given by:
where ke is the retention factor of the analyte at the point of elution. As the value for ke is typically between 2 and 3 for all components in the separation, the value of \(\sigma _{{V,{\text{col}}}}^{2}\) no longer increases with increasing retention time. However, when the analytes enter the column, they are much strongly retained (k = ki) than at point of elution. As a result, the sample plug focusses at the head of the column and Eq. (3) needs to be rewritten as
Since in most cases ki is much larger than ke, Eq. (5) simplifies to
Equation (6) illustrates that any post-column band broadening is especially detrimental in gradient elution as here the peak volumes remain small, even for strongly retained components, as the value of ke does not increase significantly with increasing retention time. Reduction of post-column tubing, both in length and ID, can reduce \(\sigma _{{V,{\text{EC}} - {\text{post}}}}^{2}\). Smaller ID tubing, however, comes at the cost of increased back pressure (~ ID4 at fixed flow rate). The other contributor to post-column EC dispersion is the detector, whose volumetric peak variance contribution for a flow cell-type detector if often described as:
where Vcell is the volume of the detector flow cell and θ is a factor depending on the cell geometry and, often neglected, the flow rate [8, 10,11,12,13,14,15,16]. Values found in literature for θ vary widely from 1 to 12 [17]. The wide variety of values does not only depend on the cell geometry. As already mentioned, dispersion in the detector cell, which can be approximated as a short piece of tubing, will depend on flow rate [10, 11] and thus θ is not a universal constant contrary to some reports [17]. Where often this contribution reaches a maximum plateau value at high flow-rate, this is not always reached in all experimental conditions. In addition, the geometrical volume of the detection cell is most often provided by the vendor, whereas the sample band often passes a non-negligible length of tubing in the whole detector cell before reaching the point of detection, a volume which can vary significantly from one type of cell to another and which is not included in Vcell. Finally, sharp turns or changes in ID in the detector cell can result in fluidic dead zones that contribute significantly to band broadening and increase peak tailing. Whereas the reduction of detector cell volume normally leads to a reduction of dispersion, this often comes at the cost of a reduction in signal-to-noise (S/R) ratio and thus, detection sensitivity. A detailed discussion regarding the trade-off between chromatographic resolution and S/R-ratio and a comparison of modern internal reflection to conventional flow cells can be found in [18].
Determining the EC contribution from the detector is also a difficult task as a second, independent measurement of the peak width before or after the detector cell is needed to compare with the obtained signal from inside the UV detector. Coupling two UV detectors in series and subtracting the peak variances of the two UV traces is, for example, not representative as the obtained \(\Delta \sigma _{V}^{2}\) not only represents the dispersion in the second detector, but is also influenced by the dispersion that occurs while the peak flows out of the first detection cell and through the outlet capillary to the inlet of the second detector. An alternative approach was recently investigated by Dasgupta et al. [11], where a section of the mobile phase-flow rate was diverted to waste prior to the detector cell. By extrapolating to a zero residence time in the detector, the contribution originating from the rest of the system could be obtained and, by subtracting this from the total variance, the contribution from the detector itself was found. The authors also distinguished the contribution from the detector cell itself and from the inlet/outlet, but this required a specially constructed absorbance detection cell that allows a continuous variation of the path length.
In this contribution, the whole detector cell variance will be measured using the on-capillary LIF approach recently used by our group to measure the dispersion directly after the valve of a sample injector [19]. In this methodology, the sample band width will be measured directly before entering (of after exiting) the detector using an on-capillary LIF detector (see Fig. 1). Fitting the latter onto a narrow (50 µm) ID capillary, the dispersion originating from the LIF detection cell (= auxiliary detector) and that of the short (< 8 cm) connection between the detection window and the entrance of the actual detector can be neglected compared to the peak dispersion of the actual detector. The value of 12 corresponds to the ideal case of a rectangular plug entering and leaving the cell without any mixing or dispersion, whereas the value of 1 is often referred to as the behavior of an ideal mixer. It is, however, important to mention, as recently explained by Dasgupta et al. [11], that for that for a poorly swept cell (i.e., containing fluidic dead zones), a value of θ < 1 would be found, so θ = 1 is certainly not the worst case.

a Schematic representation of the experimental set-up from the injector valve, to the stainless steel connection capillary, union, fused silica capillary with LIF detection window and detector cell. b Photo of the experimental set-up when using the MLC-type detector cell with c zoom-in on the short distance between the location of the LIF detector and entrance of the ML-cartridge. d Quartz-type detector cell housing with custom-made inlet and outlet capillary, with LIF detection window and e experimental set-up with the quartz-type flow cell installed in the detector and LIF just prior to the inlet of the flow cell
In the present contribution, our focus will be on UV flow cell-type detectors. Two types of detector cells that are typically employed on current (U)HPLC instrumentation will be investigated, i.e., the so-called Max Light Cartridge (MLC) cell and the more standard quartz-type cell. For the former, two variants were tested, i.e., the standard MLC cell and the ultra-low dispersion variant. Also two quartz cells were tested, respectively, with a cell volume of 500 nL and 80 nL. Most observations can, however, also be transferred to other detectors with a flow cell-type detection unit, such as electrochemical and fluorescence detection [20, 21]. For the dispersion from MS-type detectors, the reader is referred to an extensive study on the extra-column dispersion caused by this type of detectors performed by Spaggiari et al. [22].
Experimental
Instrumentation
The chromatographic system was an Agilent LC 1290 Infinity II (Agilent technologies, Waldbronn, Germany) with a binary pump (G7120A), a DAD detector (G7117B or G1315C) and an autosampler (G7167D). The autosampler was equipped with the dual-needle option that results in two flow paths with two needles, two sample loops and needle-seats (120 µm ID), along with an additional valve. The important aspect of this autosampler for the current investigation is that the sample is pressurized before injection, yielding practically no pressure dip upon injection due to the re-pressurization of the sample loop and needle and thus also no disturbance in the flow rate. Using an Universal Interface Box (UIB, G1390B) the system was interfaced with the LED-Light Induced Fluorescence detector (ZETALIF LED 480) from Picometrics (Picometrics Technologies SAS, Labège, France), allowing data acquisition for both the LIF and UV detector, data handling and instrument control with Agilent Open Lab software. For each of the two different DAD detectors, two different flow cells were used. For the G7117B DAD either the Ultra-Low Dispersion (ULD) Max Light Cartridge (MLC) cell with V(σ) = 0.6 µL (G4212-60038) or the standard MLC with V(σ) = 1.0 µL (G4212-60008) were used. Two cells of each type of MLC cell were tested. The V(σ) -values are a vendor-specified measure for the expected peak-width contribution from the flow cells. As stated in the manual “V(σ) has been experimentally determined by the injection of 50 nl Thiourea in a flow of 10%H2O/90%ACN at a flow rate of 0.5 mL/min. The corresponding chromatographic peak has been evaluated by measuring the tangent width, which is then multiplied by the flow rate and divided by a factor of 4 to achieve the V(σ) value” [23]. The geometrical volume of these cells is given in Table 1. For the G1315C DAD detector, either the nano 80 nL (G1315-68716) or the semi-nano 500 nL (G1315-68724) quartz flow cells were used. The injection valve of the system was connected with a 0.075 × 70 mm nanoViper and a 0.13 × 350 mm Viper (Thermo Fisher, Waltham, MA, USA) joined by a zero dead volume union to the inlet capillary of the detector. For the G7117B detector this was a 0.05 × 120 mm fused silica capillary, for the G1315C, a modified inlet capillary of 0.05 × 120 mm (fused silica) was used. A detection window was burned on these capillaries at 4 cm and 8 cm, respectively, before the entrance of the flow cell. For the LIF measurements after the flow cell, a 0.1 × 120 mm fused silica capillary was used.
Experimental Conditions and Set-up
All experiments with the LIF were performed using a 100 mM Trizma buffer (Trizma base (≥ 99.9%) & Trizma HCl (≥ 99%)) at pH 8 (Sigma–Aldrich, Overijse, Belgium) as the mobile phase and sample solvent, to prevent peak distortion due to solvatochromic shifts [24]. Water was obtained from a MilliQ Purification System from Millipore (Bedford, MA, USA), and fluorescein sodium salt was used as the fluorescent dye (Sigma–Aldrich, Overijse, Belgium), dissolved at a concentration of 100 µg/mL for all experiments. The injection volume was always 1 µL. UV signals were recorded at 254 nm and 160 Hz with a response time of < 0.031 s (G7117B) or 80 Hz with a response time of < 0.031 s (G1315C). The LIF was equipped with a 480 nm LED source and a standard emission filter block (515–760 nm). The photomultiplier high voltage was set to 300 V and the rise time was set to 0 s. The sampling rate (determined by the UIB) was set to 100 Hz with a response time of < 0.031 s. Polyimide-coated fused silica capillaries with an ID of 50 and 100 µm were purchased from Polymicro Technologies (Phoenix, Arizona, USA). To obtain a detection window for the LIF, the coating was burned off over a distance of 3 mm after which the capillary was placed in the detection cell holder. The resulting ‘detection cell’ thus had a volume of around 6 nL (50 µm ID) and as a result had a negligible contribution to the measured dispersion. To keep the cell in place near the injector valve, an arm holder for LIF–LC coupling (12-80CEL/LC) was used. Figure 1a schematically illustrates the flow path for the measurements with the LIF detector placed before the UV detector. Figure 1b, c shows pictures of the experimental configuration for the MLC cells, illustrating the very short distance between LIF detection window and UV cell. Figure 1d, e finally shows the modified quartz flow cell assembly where the custom-made inlet capillary was also used as LIF detection window. All injections were performed in triplicate and the average values reported. For a given set of experimental conditions, RSD values were maximum 3.5%.
Results and Discussion
Comparison of Recorded Peak Profiles in the LIF and UV Detector
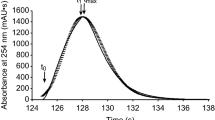
Figure 2a provides an example chromatogram measured at a flow rate of 1.0 ml/min using the LIF detector right before the UV detector (left axis) and the UV signal in the detector (right axis). Figure 2a shows the actual experimental results for the standard MLC cell with the largest volume (V(σ) = 1 µL and Vcell = 2.4 µL) at the maximum flow rate of 1.0 ml/min. As expected, this figure shows that the maximum of the UV signal is observed slightly after that of the LIF-signal one, because the UV detector is located downstream from the LIF. As both detectors have a different scale and units, a double axis was used on this figure. The scale on both axis is adjusted such that their maxima are the same. To better compare the peak widths, Fig. 2b–e represent the normalized signal (signal divided by their maximum) and shifted to make the peak maxima overlap. Figure 2b thus represents the normalized and shifted equivalent of Fig. 2a. It is obvious from Fig. 2b that the peak measured in the detector is significantly broader than that in the tubing just before it and also exhibits more tailing. Similar behavior is seen on Fig. 2c for the ULD-MLC, but less pronounced as this low-dispersion version of the MLC-type cell has a smaller volume (V(σ) = 0.6 µL and Vcell = 0.8 µL). A similar difference in peak width as for the ULC-MLC cell is found for the 500 nL quartz cell (Fig. 2d), whereas the peaks for the 80 nL cell in the LIF and UV detectors are hardly distinguishable, as expected for such a small volume cell (Fig. 2e). The tailed peaks observed for both LIF and UV signals are the typical peak shapes observed when the sample band elutes from a flow through injector (see Fig. 2a in ref. [19]).

a Experimentally measured LIF signals (left axis, blue curve) before the UV detector and inside the UV detector (right axis, red curve) using the standard MLC-type detector cell at a flow rate of 1 mL/min. b Same data as (a) but now normalized (vs. maximum) and shifted in time so both signals overlap. c–e Same as (b) but now for the ULD-MLC cell (c), the 500 nL quartz-type cell (d) and the 80 nL quartz-type cell (e). For (e) the UV signal (red curve) is dashed so it could be distinguished from the LIF signal (blue curve)
Volumetric Variance due to the Detector
The detector responses were subsequently exported and the temporal peak variances \(\sigma _{t}^{2}\) (normalized second order moments) were determined using in-house written software discussed earlier [25]. The data reported in the current report are based on the 5σ-peak-width method (width at 4.4% of peak maximum for a Gaussian peak) as well as the method of moments implemented in such a way that the integration boundaries were set at 1.1% of the peak maximum (6σ). As the values of \(\sigma _{t}^{2}\) vary strongly with the volumetric flow rate F, these were recalculated in the volumetric peak variance \(\sigma _{V}^{2}\) according to Eq. (8):
Using a sampler with loop precompression, the flow F remains constant during the passage of the sample plug at the LIF and UV detector. This is crucial to avoid changes in flow rate affecting the observed peak width. Figure 3a,b show the peak variances as a function of flow rate for the standard MLC-flow cell presented in Fig. 2a,b, illustrating the clear increase in variance experienced by the peaks when passing through this detector. Figure 3a represents the results based on the 5σ peak width (σV,5σ2) data and Fig. 3b the data obtained using the method of moments (σV,mom2). As is often the case, the method of moments results in higher values for the peak variances as the contribution from even a very shallow tail already has a strong effect on the peak’s central 2nd order moment. Figure 3c repeats these results, but now for the largest quartz flow cell (i.e., that of Fig. 2d, with Vcell = 500 nL). It is directly clear that this detector, having a much smaller volume, has a significantly smaller effect on the peak variance since both curves now lie very close together. For small flow rates (0.2–0.3 ml/min) the values are even hardly distinguishable. Similar curves were obtained for the ULD-MLC and quartz-type flow cell (80 nL), albeit with an even smaller difference in peak variance between the LIF and UV signals than their larger counterparts presented in Fig. 3. Figure 4 subsequently represents the difference in peak variance measured in the LIF and the UV -detectors, using both the 5σ peak-width method and the method of moments determined as:

Plot of volumetric peak variance of the LIF signal (diamonds) before the UV detector and inside the UV detector (squares) as a function of flow rate for the standard MLC cell using a the 5-sigma peak-width method and b the method of moments at 1.1% of the maximum peak height (6-sigma). c Same data for the 500 nL quartz-type cell using the 5-sigma method (full symbols) or method of moments (open symbols)

Plot of difference in volumetric peak variance of the LIF signal before the UV detector and inside the UV detector as a function of flow rate (calculated using Eq. 9) for the two standard MLC cells (diamonds), the ULD-MLC cells (circles), the 500 nL quartz-type cell (squares) and the 80 nL quartz-type cell (triangles), using a, c the 5-sigma peak-width method and b, d the method of moments at 1.1% of the maximum peak height (6-sigma). c, d give a zoom-in of (a, b). Curve fits explained in the text. Error bars corresponding to ± 1σ, dashed bars correspond to open symbols and full line bars to full symbols
The curve through the data points was obtained by fitting them to a simple exponential decaying equation, i.e., \(\Delta \sigma _{{V,{\text{det}}}}^{2}=\Delta \sigma _{{V,\infty }}^{2}\,\cdot\,\left( {1 - {e^{ - {\text{b}}\,\cdot\,{\text{F}}}}} \right)\,\) to guide the eye. A physical motivation for this type of fitting equation can be found in [26, 27]. As the error and variance on the obtained data increases because they are obtained by subtraction of the two data series using Eq. (9), there is more scatter on the obtained variances. This is even more pronounced for the smaller detector volumes (i.e., the ULD-MLC cell, and the 500 nL and 80 nL quartz-type flow cells, see also Fig. 4b–d). To illustrate the uncertainty on the \(\Delta \sigma _{{V,{\text{det}}}}^{2}\) values, the ± 1σ error bars were added, calculated as the sum of the standard deviations on the LIF and UV data sets. The individual standard deviations were always smaller than the size of the symbols in Fig. 3 and were therefore not shown. For the standard MLC-type flow cell, the error bars were added in Fig. 4a, b and for the other cells in Fig. 4c, d. For each of the standard and ULD-MLC-type flow cells (Vcell = 2.4 µL and Vcell = 0.8 µL, respectively), two different detector cells per type were tested. Despite being of exactly the same type and volume, differences of around 10% (moment method) to 15% (5σ-method) in \(\Delta \sigma _{{V,{\text{det}}}}^{2}\) (0.6 µL2 to 0.8 µL2) are clearly visible for the standard MLC-type flow cell. For the ULD-MLC flow cell, the differences between the two tested cells are less pronounced in absolute numbers, but still around 0.24 µL2 (5σ) and 0.31 µL2 (moment method). As in this case the total difference is much smaller (< 1 µL2 for the ULD vs. >6 µL2 for the standard cell), these smaller differences do actually correspond to larger relative differences in the obtained values.
Figure 4a, b also show that the three other (lower internal volume) flow cell types exhibit a much lower peak variance than the standard MLC-type flow cell, with variances lying mostly around or below 1.0 µL2. Figure 4c, d zoom in on the lower part of the Y-axis of Fig. 4a, b, readily showing that the calculated detector contribution to band broadening for the smallest quartz-type cell (Vcell = 80 nL) is very small, as the highest difference found was only 0.26–0.27 µL2, a value which is close to the uncertainty on the obtained results due to the subtraction of two variances. The only conclusion that can thus be drawn for this 80 nL cell is that it contributes little to nothing to the band broadening in a typical UHPLC system, in agreement with earlier observations [10]. For the 500 nL quartz-type cell, a more steady increase and plateau around 0.6 µL2 can be observed (when neglecting the two points at 0.2 and 0.3 ml/min). The results for the ULD-MLC flow cell show more scatter, both for the obtained variances for a single cell measured at different flow rates, as between the two different cells of the same type and volume, measured at the same flow rate. An average plateau value of around 0.7 µL2 was found. Using Eq. (7), it is now possible to extract the values for θ from the obtained peak variances, which are listed in Table 1. As the peak variance only drops for the very low rates, we deemed it only worthwhile to discuss the θ-values corresponding to the high flow rate plateau, keeping in mind though that the θ-values can be higher at low flow rates (0.3 ml/min and below). For the 80 nL flow cell, the difference in peak variance between the LIF and UV signals is negligible and it was therefore not attempted to calculate a θ-value. Table 1 summarizes the obtained θ values, calculated using the real geometrical detector cell volume Vcell in Eq. (7) [11]. For the two MLC-type flow cells, very similar θ-values are found, close to 1, for both detector volumes. For the 500 nL quartz-type flow cell, a lower value θ (i.e., higher dispersion) close to 0.5 is obtained, showing that the new generation of flow cells has a 50% lower dispersion contribution relative to its cell volume squared. As the inlet tubing and internal flow path inside the detector cells are however different, part of the observed peak is not due to the cell volume, but rather due to the dispersion in these preceding fluidic channels [11]. This additional tubing and flow path inside the detector cell itself, that is not included in Vcell, might have a large impact on the dispersion due to presence of sharp bends or the presence of fluidic dead zones. Overall, a detector dispersion contribution lower than 1 µL2, obtained for all detectors except the largest flow cell (standard MLC) can be observed. Nevertheless, for weakly retained compounds (k < 4) on short (5 cm or lower) 2.1 mm ID columns, this still accounts for a large fraction of the overall dispersion. Taking for example the 2.1 × 50 mm column with 1.3 µm particles discussed in the introduction, the expected peak variance contribution from the column for a retention factor of 2 is equal to 3.7 µL2. As a result, a 1 µL2 contribution from the detector alone would increase this by 27%. Switching to even narrower ID columns (e.g., 1 mm), required to e.g., alleviate the effects of viscous heating occurring at very high operating pressures, would require even smaller detector dispersion contributions as the reduction from 2.1 to 1 mm ID reduces the column peak variance by a factor of almost 20.
Post and Total Detector Variance
For some experimental conditions, it is preferred to have more than one detector installed in the system, e.g., UV followed by a fluorescence detector or UV + MS. In this case, also the dispersion caused by the sample band flowing out of the detector contributes to the peak width in the second detector. To estimate this contribution, the LIF detector was also placed at the outlet of the different detector cells. The use of the 50 µm detector capillary however introduces a very large backpressure on the flow cell, limiting the experimental set-up. In addition, as was encountered during repeat experiments on the first tested ULD-MLC flow cell, clogging of the fine capillary resulted in a quick pressure build up before the system could shut down due to the overpressure settings, causing the failure of the flow cell. It was therefore decided to switch to a 100 µm ID LIF detector capillary to reduce the backpressure and thus pressure inside the flow cell and to reduce the risk of clogging. As a result, the post detector variance of 80 nL quartz cell flow was not investigated. Figure 5 shows the results for the three other cells. Strangely enough, the contribution seems to decrease with increasing flow rate, although no clear relationship can be found. For the two MLC-type detector cells, the main conclusion that can be drawn from these experiments is that an additional contribution < 1 µL2 (F > 0.5 ml/min) to < 1.5 µL2 (F < 0.5 ml/min) needs to be taken into account for the elution from the detector cell. For the 500 nL quartz-type detector cell, a contribution around 0.2–0.4 µL2 is found below F = 0.5 ml/min, and little to no dispersion at higher flow rates.

Plot of difference in volumetric peak variance measured in the UV detector and the LIF signal (100 µm ID capillary) after the UV detector as a function of flow rate (calculated using Eq. 9) for the standard MLC cells (diamonds), the ULD-MLC cell (circles) and the 500 nL quartz-type cell (squares) using a 5-sigma peak-width method and b the method of moments at 1.1% of the maximum peak height (6-sigma). Error bars corresponding to ± 1σ
Conclusion
The band broadening in four contemporary types of UV detectors (2 different flow cell types combined with two different cell volumes for each) was investigated using a very accurate on-capillary LIF detector positioned just prior or after the UV detection cell. A slight increase of the detector cell dispersion with flow rate was observed, which reaches plateau values around 0.4 ml/min (MLC-type cells)–0.6 ml/min (quartz-type flow cells). For the Max Light Cartridge-type detector cells, the θ factor in the relationship between detector peak variance \(\Delta \sigma _{{V,{\text{det}}}}^{2}~\)and the cell volume \({V_{{\text{det}}}}\), given by Eq. (7), was found to be close to one. This implies the expected volumetric band broadening for such a type of detector cell is close to \(V_{{{\text{det}}}}^{2}\). For the quartz-type detector cell of 500 nL, the θ value was found to be 0.5. The very low dispersion for the 80 nL flow cell was such that the obtained contribution from the cell itself was within the experimental error and thus did not allow for an accurate determination, but is probably insignificant to the overall band broadening when using standard (2.1 mm ID or larger) chromatographic columns.
References
De Vos J, Broeckhoven K, Eeltink S (2016) Advances in ultrahigh-pressure liquid chromatography technology and system design. Anal Chem 88:262–278
Broeckhoven K, Desmet G (2014) The future of UHPLC: Towards higher pressure and/or smaller particles? TrAC Trends Anal Chem 63:65–75
De Vos J, De Pra M, Desmet G, Swart R, Edge T, Steiner F, Eeltink S (2015) High-speed isocratic and gradient liquid-chromatography separations at 1500 bar. J Chromatogr A 1409:138–145
Broeckhoven K, De Vos J, Desmet G (2017) Particles, pressure, and system contribution: the holy trinity of ultrahigh-performance liquid chromatography. LC GC Eur 30:618–625
Majors RE (2015) Future needs of HPLC and UHPLC column technology. LCGC North Am 33:886–887
Fekete S, Guillarme D (2013) Kinetic evaluation of new generation of column packed with 1.3 µm core–shell particles. J Chromatogr A 1308:104–113
Sanchez AC, Friedlander G, Fekete S, Anspach J, Guillarme D, Chitty M, Farkas T (2013) Pushing the performance limits of reversed-phase ultra high performance liquid chromatography with 1.3 µm core–shell particles. J Chromatogr A 1311:90–97
Hupe K-P, Jonker RJ, Rozing G (1984) Determination of band-spreading effects in high-performance liquid chromatographic instruments. J Chromatogr 285:253–265
Fekete S, Fekete J (2011) The impact of extra-column band broadening on the chromatographic efficiency of 5 cm long narrow-bore very efficient columns. J Chromatogr A 1218:5286–5291
Vanderlinden K, Broeckhoven K, Vanderheyden Y, Desmet G (2016) Effect of pre- and post-column band broadening on the performance of high-speed chromatography columns under isocratic and gradient conditions. J Chromatogr A 1442:73–82
Dasgupta PK, Shelor CP, Kadjo AF, Kraiczek KG (2018) Flow-cell-induced dispersion in flow-through absorbance detection systems. True column effluent peak variance. Anal Chem 90:2063–2069
Cohen KA, Stuart JD (1987) A practical method to predict the effect of extra-column variance on observed efficiency in high performance liquid chromatography. J Chrom Sci 25:381–390
Yukuei Z, Miansheng B, Xiouzhen L, Peichang L (1980) High-performance liquid chromatographic columns of small diameter. J Chrom 197:97–108
DiCesare JL, Dong MW, Atwood JG (1981) Very-high-speed liquid chromatography: II. Some instrumental factors influencing performance. J Chrom 217:369–386
Kamahori M, Watanabe Y, Miura J, Taki M, Miyagi M (1989) High-sensitivity micro ultraviolet detector for high-performance liquid chromatography. J Chrom 465:227–232
Scott RPW, Kucera P (1979) Mode of operation and performance characteristics of microbore columns for use in liquid chromatography. J Chrom 169:51–72
Gritti F, Guiochon G (2011) On the minimization of the band-broadening contributions of a modern, very high pressure liquid chromatograph. J Chromatogr A 1218:4632–4648
Kraiczek KG, Rozing GP, Zengerle R (2013) Relation between chromatographic resolution and signal-to-Noise ratio in spectrophotometric HPLC detection. Anal Chem 85:4829–4835
Broeckhoven K, Vanderlinden K, Guillarme D, Desmet G (2018) On-tubing fluorescence measurements of the band broadening of contemporary injectors in ultra-high performance liquid chromatography. J Chromatogr A 1535:44–54
Kok WTh, Brinkman UATh, Frei RW, Hanekamp HB, Nooitgedacht F (1982) H.Poppe, Use of conventional instrumentation with microbore columns in high-performance liquid chromatography. J Chromatogr 237:357–369
Van Schoors J, Maes K, Van Wanseele Y, Broeckhoven K, Van Eeckhaut A (2016) Miniaturized ultra-high performance liquid chromatography coupled to electrochemical detection: Investigation of system performance for neurochemical analysis. J Chromatogr A 1427:69–78
Spaggiari D, Fekete S, Eugster PJ, Veuthey J-L, Geiser L, Rudaz S, Guillarme D (2013) Contribution of various types of liquid chromatography–mass spectrometry instruments to band broadening in fast analysis. J Chromatogr A 1301:45–55
https://www.agilent.com/cs/library/usermanuals/public/G4212-90122_TN_for_Flowcell.pdf, retrieved on 10/07/2018
Raikar US, Renuka CG, Nadaf YF, Mulimani BG, Karguppikar AM, Soudagar MK (2006) Solvent effects on the absorption and fluorescence spectra of coumarins 6 and 7 molecules: Determination of ground and excited state dipole moment. Spectrochim Acta Part A Mol Biomol Spectrosc 65:673–677
Vanderheyden Y, Broeckhoven K, Desmet G (2014) Comparison and optimization of different peak integration methods to determine the variance of unretained and extra-column peaks. J Chromatogr A 1364:140–150
Broeckhoven K, Desmet G (2007) Approximate transient and long time limit solutions for the band broadening induced by the thin sidewall-layer in liquid chromatography columns. J Chromatogr A 1172:25–39
Broeckhoven K, Desmet G (2009) Numerical and analytical solutions for the column length dependent band broadening originating from axisymmetrical trans-column velocity gradients. J Chromatogr A 1216:1325–1337
Funding
This study was funded by a Research Grant from FWO Vlaanderen (1520115N) to KB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Published in the topical collection Rising Stars in Separation Science, as part of Chromatographia’s 50th Anniversary Commemorative Issue.
Rights and permissions
About this article

Cite this article
Vanderlinden, K., Desmet, G. & Broeckhoven, K. Measurement of the Band Broadening of UV Detectors used in Ultra-high Performance Liquid Chromatography using an On-tubing Fluorescence Detector. Chromatographia 82, 489–498 (2019). https://doi.org/10.1007/s10337-018-3622-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-018-3622-1